
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new look at an old classic: implementation of a SERS-based water hardness titration†
Ngoc Mai
Duong
a,
Angélina
Noclain
 a,
Victoria E.
Reichel
a,
Pierre
de Cordovez
a,
Jean-Marc
Di Meglio
a,
Pascal
Hersen
b and
Gaëlle
Charron
*a
a,
Victoria E.
Reichel
a,
Pierre
de Cordovez
a,
Jean-Marc
Di Meglio
a,
Pascal
Hersen
b and
Gaëlle
Charron
*a
aLaboratoire Matière et Systèmes Complexes, UMR 7057, Université Paris Cité, CNRS, 10 rue Alice Domon et Léonie Duquet, 75013, Paris, France
bLaboratoire Physico Chimie Curie, UMR 168, Institut Curie, Université PSL, Sorbonne Université, CNRS, 75005 Paris, France
First published on 7th July 2023
Abstract
The routine use of SERS as an analytical technique has been hindered by practical considerations among which the irreproducibility of its signals and the lack of robustness of its calibration. In the present work, we examine a strategy to perform quantitative SERS without the need for calibration. The method reinvests a colorimetric volumetric titration procedure to determine water hardness but involves monitoring the progression of the titration through the SERS signal of a complexometric indicator. Upon reaching the equivalence between the chelating titrant and the metal analytes, the SERS signal abruptly jumps, which conveniently serves as an end-point marker. Three mineral waters spanning divalent metal concentrations varying by a factor of 25 were successfully titrated in this way, with satisfactory accuracy. Remarkably, the developed procedure can be run in less than an hour, without laboratory-grade carrying capacity and would be relevant for field measurements.
Introduction
Owing to its extreme sensitivity, and to the fingerprint-like character of its spectra, surface-enhanced Raman scattering (SERS) has been highlighted as a potentially ground-breaking tool for chemical analysis for more than 20 years.1–4 However, due to a complex interplay of electromagnetic and physico-chemical effects in the mechanism of SERS, this tool is intrinsically hard to master and results can differ significantly between laboratories, between days or even between nanoparticle (NP) batches or nanostructured substrates.5,6As a quantification technique, SERS has been mostly used in spectrophotometric mode; the simplest implementation involves an analyte generating a distinctive SERS signal that gradually builds up as the concentration grows. Analytes that do not give a sufficient signal, or no SERS signal at all, can alternatively be tracked through their interaction with a chromogenic receptor giving rise to strong SERS signals or with a receptor bound to a SERS tag, thereby leading to turn-on or turn-off sensors (Fig. S0†). One typical case is that of a metal ion interacting with a ligand that acts as a loud SERS proxy for an otherwise silent analyte.7–13 In both cases, direct or receptor-mediated sensing, quantification relies on the dependence of the signal on the concentration through a calibration model. However, owing to inhomogeneities at the nanoscale of the SERS-active material, to ageing of the material or to variations of the concentrations in the stocks (over time or from one batch to another), the validity of the calibration model is short-lived. Any new SERS material batch or any new day will most likely require a new calibration step. This can consume precious SERS-active materials and is time-consuming. We recently demonstrated that calibration is actually the largest driver of the consolidated cost of a metal ion analysis by SERS.14 However, quantification does not need to rely on calibration of a spectral signal.
Before the advent of spectrophotometric techniques, concentrations of metal ions were measured by complexometric volumetric titrations.15 Briefly, these methods rely on three chemical protagonists: the target metal ion, a chelator (titrant) having a strong affinity for it, and a highly absorbing chelating dye having a somewhat weaker affinity and two distinct colours in its free and metal-bound states. A complexometric volumetric titration proceeds as follows. The indicator is introduced in the sample in default, typically a few percents, with regards to a gross estimation of the concentration of the analyte; it binds to the analyte quantitatively and imparts its bound-state colour to the medium. The titrant is then introduced portion-wise into the mixture; it binds to free metal ions. Once virtually all free metal ions have been consumed, any new addition of the titrant steals analytes from the binding cavity of the indicator, thereby releasing free indicator molecules that bring their free-state colour to the solution. The switch between bound and free-state colours of the indicator occurs about the equivalence point and therefore serves as an end-point marker. Complexometric methods are available for the titration of virtually every metallic ion.16 Selectivity is often achieved by adjusting the pH or by using masking agents. However, due to the colorimetric (absorption) nature of the visualisation of the end-point, those methods are often limited to concentrations in the mid-μM to mM range.
Complexometric indicators often bear highly Raman-active chromophores in the form of aromatic rings. Revisiting those molecules as end-point markers in SERS-transduced titration could alleviate the need to build a reliable, robust calibration model by converting the calibration problem into a switch visualisation. Quantification would then stem from the switching of the SERS spectrum of the indicator, and not from absolute or relative intensities. Moreover, this revisiting of the use of complexometric indicators could extend their usefulness to lower concentration ranges due to the giant nature of SERS signals compared to their absorption features.
Here we examine the possibility of implementing an indicator-based metal complexometric titration in SERS on a well-known case example: the determination of water hardness. The developed method is successfully applied to three commercial mineral waters having vastly differing compositions – Évian, Volvic and Contrex – and whose alkaline earth metal ion concentrations span one and a half order of magnitude. It would be of interest to aquatic geochemists tracking calcium and magnesium fluxes, who are largely limited in their field measurements capacities.
Experimental section
Synthesis of nanoparticles
Ag nanoparticles were synthesized on a scale of 100 ml (1.2 mM Ag) through the Lee and Meisel protocol.17 Residual citrate was removed by precipitation–redispersion cycles against 0.2 mM sodium chloride and then against ultra-pure water, at constant volume, according to a procedure reported by Stewart et al.18 The resulting nanoparticles were analysed by TEM.Assembly of SERS-active aggregate (“SERS beads”)
Two types of SERS-active aggregates were assembled from Ag NPs: plain ones, for studying their plasmonic properties and morphologies, and aggregates incorporating Eriochrome Black T (EBT) as a Mg2+ indicator. Plain beads were assembled by first mixing the Ag NP solution with a polyethylene glycol (PEG) solution in Britton and Robinson buffer (BRB),19 at pH 10.5 and then by initiating the cross-linking of nanoparticles through addition of polyethyleneimine (PEI) and further homogenization. The final concentrations of the medium are 5.7 × 10−4 M Ag, 9 μM PEI (expressed as monomers), 8.4 wt% PEG in 2 mM BRB (boron content). The mixtures were aged for 1 h before measurements by UV-vis, SERS and optical imaging (at 100× magnification).Preparation of SERS measurement samples
For each titration measurement sample, a test portion of water sample with a fixed volume was supplemented with EBT, diluted with a fixed volume of a mixture of BRB buffer and ethylenediamine tetraacetic acid (EDTA) in variable proportions, to which constant volumes of chloride-derived Ag NP solution, PEG stock and PEI stock were added (Fig. S5†). The mixture was homogenised after each addition. The final concentrations of the mixture were 4.4 × 10−4 M Ag, 7 μM PEI (as monomers), 6.5 wt% PEG, 3.8 μM EBT in 2.2–4.6 mM BRB (boron content). The volumic fraction of the test portion of sample (pure water, Évian, Volvic or Contrex) in each measurement sample was 7.7%. The mixtures were aged for 1 h before measurements by Raman spectroscopy.Raman acquisitions and processing
Raman/SERS spectra were acquired using portable spectrometers with laser irradiation at 638 and 785 nm and maximal laser powers comprised between 50 and 420 mW. Measurements were acquired using PMMA disposable cuvettes in backscattering mode. Spectra were processed and baselines were removed independently by the lead and corresponding authors using Python or R. Both processing gave consistent results.Speciation analysis was performed using Visual MINTEQ
The full experimental procedures, including a graphical depiction of the composition of the SERS measurement samples can be found in ESI.† The data can be downloaded from a scientific data repository (https://doi.org/10.5281/zenodo.6980619).20Results
Water hardness refers to the total concentration of calcium and magnesium ions in a water sample. The classic complexometric method for measuring it consists in titrating Ca2+ and Mg2+ ions with EDTA, an hexadentate ligand, in the presence of the complexometric indicator Eriochrome Black T (EBT, Fig. S1†).21 The titration is conducted at pH 10 to ensure quantitative complexation. On first approximation, the titration can be considered to proceed as follows. Before titration begins, EBT is added to the sample in default compared to the divalent metal content and binds to a fraction of Mg2+ ions (eqn (1)), thereby turning pink. As the first drop of titrant is added, EDTA binds to free calcium ions (eqn (2)). Once all Ca2+ ions have been consumed, EDTA binds to free Mg2+ ions (eqn (3)). When the reservoir of free Mg2+ ions is exhausted, newly added EDTA displaces Mg2+ ions from the EBT chelating cavity, which then retrieves its characteristic blue colour as a free species (eqn (4)).| HEBT2− + Mg2+ → MgEBT− + H+ (Kex,EBT,Mg) = 10−4.55 | (1) |
| HY3− + Ca2+ → CaY2− + H+ (Kex,Y,Ca) = 100.7 | (2) |
| HY3− + Mg2+ → MgY2− + H+ (Kex,Y,Mg) = 10−1.57 | (3) |
| MgEBT− + HY3− → MgY2− + HEBT2− (Kex,switch,Mg) = 102.98 | (4) |
To visualize the end-point of this classic titration by SERS, we entrapped EBT into SERS-active aggregates made of hundreds of Ag nanoparticles (NPs) that were directly formed in the presence of the water sample and added titrant (Fig. 1).
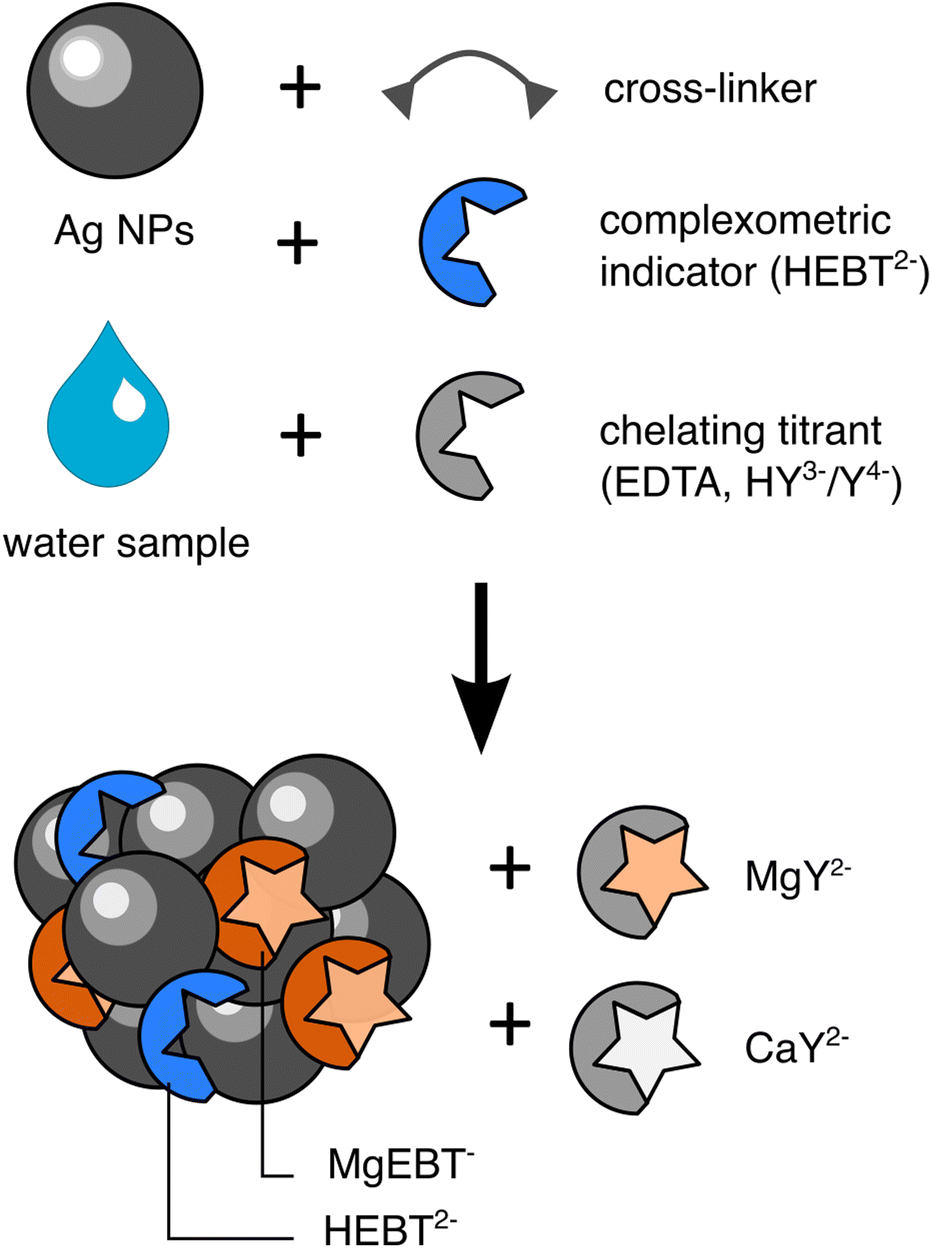 | ||
| Fig. 1 Scheme of the assembly of SERS active aggregates entrapping EBT in the presence of Ca2+, Mg2+ and EDTA. | ||
SERS aggregates
The SERS-active material was designed from simple building blocks so as to avoid being limited in our SERS measurements by the synthesis of the plasmonic aggregates. Details can be found in ESI.† Briefly, Ag nanoparticles were directly aggregated into water samples supplemented with EBT using a polyamine cross-linker (Fig. 1). The Ag NPs were synthesised according to the Lee–Meisel method on a scale of 120 μmoles of Ag. The as-synthesised colloidal solution contains about 88 μM residual citrate ligands, a fraction of which on their surface (see ESI†).22 Those strongly coordinate to Mg2+ and Ca2+ ions and therefore interfere with their titration. Hence we attempted to replace the native surface ligands with chloride ions after synthesis using a simple precipitation-redispersion protocol.18 This procedure removes 95% of the total citrates (based on dilution), most likely the unbound ones, but we had no mean to characterize a full removal of the surface-bound ones. The Cl-derived NPs have a diameter of 80 ± 23 nm (Fig. S2†). The chloride derived NPs are significantly larger than what is traditionally observed for Lee–Meisel NPs. This is due to the purification process which tends to discard smaller NPs. Aggregation of NPs is then launched through addition of polyethyleneimine (PEI) as a cross-linking agent and PEG as a screening agent. The incorporation of PEG (20 kDa) slows down the aggregation through increased medium viscosity and steric crowding of the NP surface. It enables the formation of discrete, spherical, sub-micrometric (788 ± 225 nm in diameter as measured by optical microscopy) NP aggregates, hereafter referred to as SERS-beads, instead of large objects that precipitate out of the solution (Fig. S3†). The aggregates are negatively charged (zeta potential = −7 mV). To monitor the plasmonic properties of the SERS-active aggregates, we recorded the extinction spectra of SERS beads that were assembled without EBT. The extinction spectrum of the SERS-beads exhibits a broad resonance with a maximum at 650 nm that overlaps well with the 638 nm and 785 nm excitation wavelengths of the used lasers and should therefore warrant acquisition of good quality SERS spectra (Fig. S4†).SERS spectra of aggregate-entrapped EBT
In order to check the suitability of EBT as a SERS-based endpoint marker, we acquired the SERS spectra of SERS-active aggregates which were assembled in the presence of EBT in water samples having extreme compositions: no calcium and magnesium (pure water), Ca2+ and Mg2+ brought by Évian water, and Ca2+ and Mg2+ fully bound to EDTA, all other conditions being fixed (3.7 μM EBT, pH = 10, Fig. S5†).The SERS spectrum of EBT exposed to pure water recorded under 638 nm irradiation displays very intense features between 1100 and 1400 cm−1 (Fig. 2, solid orange trace). We were unable to link them to spectra from published literature since the spectral features are highly dependent on the pH and pH was not reported in said studies.23–25 These features do not stem from the ligands of the NPs or the crosslinker and are not the result of normal Raman transitions (Fig. S6†).
 | ||
| Fig. 2 SERS spectra of free and bound EBT, in the presence or absence of an excess of EDTA, acquired under 638 nm irradiation. The analytical concentration of EBT is 3.7 μM in each condition. Complexation of the indicator occurs in the presence of 82 μM Mg2+ (and 150 μM of Ca2+). | ||
In the measurement sample prepared from Évian water (containing 232 μM divalent cations), 87.5% of EBT is bound to Mg2+ (92% of EBT is metal-bound) according to speciation analysis. Compared to free EBT, the maximum intensity of the spectrum of bound EBT dramatically drops, by a factor of about 15 (Fig. 2, dashed orange trace). Upon addition of an excess of EDTA compared to the total metal content, the intensity of the SERS spectrum bounces back, consistently with the demetalation of EBT predicted by speciation analysis (Fig. 2, dashed blue trace). However the spectral features do not match those observed for the pure water sample. The first conclusion that can be drawn from this comparison is that the SERS spectra are markedly different depending on the filling of EBT, thereby supporting its trial as a SERS-based indicator. The dramatic variation in SERS intensities between the bound and free state of EBT is likely to stem from the turning on and off of an electronic resonance between the excitation laser and EBT, although different entrapment efficiencies between free and Mg-bound EBT cannot be ruled out. At 638 nm irradiation, the excitation source is almost in perfect resonance with the main electronic absorption band of free EBT. In the case of MgEBT−, the electronic match between the extinction spectrum and the laser source is poorer since the excitation wavelength falls in between the main and secondary absorption bands and the extinction coefficient of the complex at that wavelength is about half that of the free species (Fig. S7†).
We interpret the differences between the spectra of free EBT acquired from pure water or restored after complexation of Mg2+ by EDTA by an interaction between EBT and EDTA on the surface of Ag NPs. Upon addition of the same amount of EDTA to pure water, spectra features similar to those of the Évian sample supplemented with an excess of EDTA are obtained (Fig. 2, solid blue trace). A deeper investigation of the exact speciation of EBT adsorbed onto the SERS-active beads was beyond the scope of the present study, in the frame of which we got satisfied with the spectra of EBT being markedly different when bound or unbound to Mg2+. A tentative link between these spectral features and speciation is nonetheless discussed in ESI (Fig. S8†). Noticeably, the switch in SERS signals is also observed under 785 nm irradiation (Fig. S9†).
SERS-titration of water hardness by EDTA using EBT as an end-point indicator
The switching on and off of the SERS signal of EBT upon demetalation and complexation respectively can be used as a proxy for visualising the equivalence of a complexometric titration. The general procedure to conduct the titration involves preparing a set of spectrophotometric cuvettes containing precursors of the SERS active aggregates, a fixed volume of water sample and a varying volume of titrant so as to sweep the ratio of titrant to divalent metal ions from 0 to 2 while maintaining the total volume constant through appropriate addition of a buffer (Fig. S5†). Évian, Volvic and Contrex mineral waters, which have total alkaline earth metal concentrations differing by a factor of 25 and total mineral contents differing by one order of magnitude, were titrated in this way (Table 1). The molarity of the titrant was adapted for each mineral water to offer the same resolution about the equivalence. The buffer used was Britton & Robinson, an equimolar mixture of sodium acetate, potassium hydrogenophosphate and boric acid.19 To maintain a pH constant at ∼9.8 across the whole set of samples, the buffer molarity in the cuvette had to be increased from about 2.2 mM boron for Évian and Volvic to 4.6 mM boron for Contrex. For each water sample, the titration series were conducted in triplicates using two independent batches of Ag NPs (for a total of 6 titrations per mineral water sample). An additional blank titration was conducted for each target mineral water (and each NP batch) in which ultrapure water was substituted for the mineral water fraction.| Mineral water brand | Mg2+ (mM) | Ca2+ (mM) | Total divalent metal concentration (mM) | Bicarbonate (mM) | Dry residue (mg L−1) |
|---|---|---|---|---|---|
| Évian | 1.1 | 2.0 | 3.1 | 5.90 | 345 |
| Volvic | 0.3 | 0.3 | 0.6 | 1.2 | 130 |
| Contrex | 3.07 | 11.7 | 14.7 | 6.10 | 1121 |
Titration of Évian
Fig. 3a displays the evolution of the SERS spectra acquired under 638 nm irradiation across one typical titration series of Évian water. The spectra neatly aggregate into two groups of distinct shapes before and after equivalence: the first one having its maximum intensity at 1208 cm−1 and the second one having its maximum at 1184 cm−1 (see also replicate measurements in Fig. S10†) The evolutions with titrant addition of those peaks mirror the sigmoidal one of the molar fraction of HEBT2− calculated by speciation simulation (Fig. 3b, S11 & S12†), albeit with a significant lag in the case of the 1208 cm−1 peak tracking. When tracking the ratio of both peaks, an abrupt sigmoid was obtained for each of the titration replicates (Fig. 3c & S13†), in which the height of the jump was maximised compared to the intensity variations observed throughout the blank titration. To read the equivalence from the SERS titration curve, the derivative can be calculated with regards to the number of equivalents of added titrant (Fig. 3d). Alternatively, the experimental points can be fitted to a sigmoid. | ||
| Fig. 3 Baselined-corrected SERS spectra acquired under 638 nm irradiation over the course of one representative titration of Évian (a), corresponding speciation analysis (the calculations have been run without inclusion of the SERS aggregates) (b), intensity ratio of the 1184 and 1208 cm−1 peaks as a function of added EDTA (c), and derivative of that ratio (d). The dashed lines in speciation, titration and derivative plots serve as visual guides of the theoretical equivalence according to metal concentrations indicated on the bottle label. The shaded area in (b) corresponds to the switching range of the indicator when exploiting it in classic colorimetric titration. | ||
The number of titrant equivalents at which the end-point of the reaction is experimentally read relatively to the theoretical equivalence inferred from the knowledge of the reference analyte concentration gives an estimate of the trueness of the quantification method. While speciation analysis predicts an abrupt increase in HEBT2− molar fraction with an inflexion point at 95% of equivalence (Fig. 3b), the sigmoidal fit of the ratio displays its inflexion point at 94% of the theoretical equivalence on average over the intra and inter-batch replicates. This indicates that the interaction of the indicator with the surface of the NPs does not introduce a prohibitive bias; the SERS-derived end-point is almost as good as the chemical speciation allows it. When monitoring the titration under 785 nm irradiation in SERS, similar results are obtained. Monitoring the maximum peak at 1279 cm−1 yields sigmoidal titration curve with a mean inflexion point at 90% of equivalence (Fig. S14 & S15†). Both irradiation wavelengths are therefore suitable to monitor the titration, albeit with a small difference in trueness.
Remarkably, the SERS-based titrations are 90 to 94% true without any calibration of the spectroscopic signal, nor any attempt at controlling the topologies of the gaps within the aggregates, the insertion rate of EBT within them or the average enhancement factor over the whole sample. Because quantification is here based on tracking an abrupt switch in the spectral properties and not on a mathematical relationship between intensities and concentration, the methodology is permissive towards uncontrolled intensity variations. For instance, the two batches of NPs used for running the Évian titrations give rise to free-EBT signals differing by about 40%, which could reflect different aggregate concentrations, gap topologies or EBT insertion rates, yet lead to the same trueness and precision.
Comparison of SERS-based and classic water hardness titrations
When performing the classic colorimetric water hardness titration monitored by eye, the end-point is pinpointed by the colour transition from pink to blue. That transition actually occurs over a range of numbers of titrant equivalent, which is formalized as the range over which the concentration ratio of the two species of the indicator changes from 0.1 to 10 (shaded area in Fig. 3b).15 That range depends on the ratio of indicator to metal within the titration medium: the larger that ratio, the more progressive that transition (Fig. S16†). To ensure a sharp colour switch, it is therefore recommended to work with indicator concentrations within a few % of the target metal concentration in the reaction vessel, provided the colours are not too faint to be tracked. In the shown Évian SERS titrations, the EBT concentration was 1.7% of the reference metal concentration in the cuvette mix, leading to a SERS-switch that is 90% complete over just 0.1 eq. Under those conditions, colorimetric or spectrophotometric titrations would have been much less sharp (switching over 0.35 to 0.20 eq. respectively) and nearly impossible to track in disposable cuvettes due to low absorption variations (see Fig. S17 and discussion in ESI†). This evidences that EBT is at least as good an indicator in SERS as it is in absorption spectrometry. Knowing that dyes can be detected in SERS at concentration much lower than the μM range, this gives a taste of the reservoir of sensibility that could be tapped when reinvestigating complexometric titrations at lower indicator and metal concentrations in SERS.Titration of Volvic
The main advantage of running a complexometric titration with an indicator is that there is no need to recalibrate the spectroscopic signal when the target analyte concentration changes by one order of magnitude. One just needs to adjust the molarity of the titrant. Volvic water has a total divalent metal contents 1/5th that of Évian. Over its titration, the SERS spectra acquired under 638 nm irradiation aggregate into 3 groups (Fig. S18†): one with a maximum intensity at 1208 cm−1 and overall low intensity (before equivalence), one with a maximum intensity at 1208 cm−1 and overall high intensity (about equivalence) and one with a maximum intensity at 1184 cm−1 and overall high intensity (after equivalence). Again, the ratio of intensities at 1184 and 1208 cm−1 was exploited to track the progress of the titration (Fig. S19†). The evolution of the ratio closely mirrors the build-up of free EBT as HEBT2− (Fig. S16†), albeit with an inflexion point shifted beyond the theoretical equivalence point. For both batches of NPs, estimates of trueness were 113 and 102% respectively, suggesting, by comparison with the Évian results, that some of the titrant could be consumed by adsorption to the surface of NPs. However, when monitoring the titration under 785 nm, the trueness estimates fall at 97 and 88% depending on the NP batch, in closer agreement with speciation analysis (Fig. S20 & S21†). This disqualifies adsorption of EDTA on the surface of the NPs as a source of positive bias in the 638 nm-monitored titration. Remarkably, colorimetric monitoring would have been of poor quality with a colour switching range extending over 0.6 EDTA eq. (Fig. S16†). SERS would also have outperformed spectrophotometric monitoring, since the trueness of the latter would have fallen to 85% (Fig. S17†). The sharpness and trueness achieved in the SERS titration are unmatched.Titration of Contrex
Contrex, having a divalent cation concentration 5 times that of Évian, was chosen as an extreme hardness water sample. Over its titration under 638 nm irradiation, the spectral evolution is similar to that observed for Volvic, with a transitory spectral contribution of high intensity arising about the equivalence (Fig. S22†). Monitoring the ratio of 1184 and 1208 cm−1 peaks give rise to sigmoidal titration curves, in line with speciation analysis, with positively biased trueness estimates of 103 and 117% respectively (Fig. S23†). Under 785 nm, the spectral evolution is exactly similar to those observed for Évian and Volvic, with trueness estimates of 105 and 96% depending on the NP batch (Fig. S24 & S25†).Discussion
Analytical performances
SERS titrations were successfully implemented for mineral waters having hardnesses spanning one and a half order of magnitude (differing 25 fold in total divalent metal cation content). Fig. 4 displays the observed analytical performances aggregated per batch of NP. Through monitoring the ratio of peaks at 1184 and 1208 cm−1 under 638 nm irradiation or monitoring the sole peak at 1279 cm−1 under 785 nm irradiation, the trueness never falls below 89% and never exceeds 117%. The relative standard deviation of trueness, which we use here as a proxy for precision, does not exceed 2% when using a single batch of NPs (over 3 replicate titrations), or at most 7% over 6 titrations performed using two batches of NPs. Noticeably, such accuracies ad precisions have been obtained from modest experimental efforts regarding the synthesis of the SERS-active material and the sample preparation procedure. Since the quantification power does not rely on reproducible signal variations, poorly controlled but cheap and easily scalable NP building blocks can be used, regardless of the batch. Moreover, the worst-case figures of merit reported here are similar to the best ones we obtained in a similar study from the same chemical components (Lee Meisel NPs, a polyamine cross-linker) but using the complexometric metal probe in spectrophotometric mode, i.e. through building a signal calibration model.14 The significant difference is, however, that it took much less time to achieve them using the complexometric probe in indicator mode: where it took 80 samples to build the calibration model and 6 replicate measurements to achieve 5% precision in spectrophotometric mode, one water hardness titration required only 18 samples, in a single titration series. Moreover, the calibration model in the previous study was valid over just one decade of metal analyte concentration, whereas the present detection scheme works over a concentration range extended 2.5 folds. Practically, a titration can be run in one hour, or a little less than two hours when performed in triplicate, from a batch of NPs that can be synthesised easily on a scale of 18 titrations. We see no limitation to the preparation and measurement of titration samples outside of conventional laboratory settings as the preparation steps involve only pipetting and mixing (through shaking) in hemolysis tubes, and measurements were run on two portable Raman spectrometers. | ||
| Fig. 4 Overview of mean trueness estimates and their standard deviation per NP batch given as an estimation of precision. (*) Sigmoidal fitting of intensities at 1208 cm−1 under 638 nm over the course of the titration of Volvic was inappropriate for the data. | ||
Relevance to the issue of water hardness monitoring
Water hardness is critical to the management of pipes and cooling circuits as Ca2+ and Mg2+ react with dissolved carbonates to lead to carbonate rocks thereby causing clogging problems. Sulfuric acid is added to water feeding the cooling pipes of power plants to avoid that problem, with no dynamic adjustment of the acid concentration to the water hardness in general.26 The reverse reaction of Ca or Mg rich carbonates with CO2 is also a very important one to monitor: this reaction is responsible for most of the 0.3 GtC per year pumped from the atmosphere by rock weathering.27 However, real-time monitoring of dissolved alcaline earth is both a necessary and an unresolved issue. Because of microbial activity, partial pressures of CO2 in rivers are commonly 10 times what is expected at equilibrium.28 For groundwaters, they build up to 10–100 fold compared to atmospheric pressure.29 As soon as water is sampled, it starts degassing and its composition drifts towards equilibrium, leading to the precipitation of carbonate phases. To access the dissolved metal fraction, water samples are filtered and then acidified to prevent precipitation. This implies that acid stocks and acid-washed sampling tubes have to be brought on field before bringing the properly conditioned measurement samples back to the lab for analysis by ICP-AES or ion chromatography. This is a strong limitation where the local analytical carrying capacity is weak: in the case of remote measurement campaigns, or when there are no instrumental facility, and samples need to be shipped away. Our SERS titration method offers an alternative: that of direct measurements on the field, that can be performed with moderate chemical expertise, on instruments costing half to 1/4th of the golden standard instruments (∼20 k€-portable battery operated Raman spectrometer vs. 40 to 80 k€ for an ion chromatograph or an ICP-AES respectively).Conclusion
Practical implementation of SERS in routine analytical chemistry has been hindered by the need to calibrate SERS signals. To alleviate this problem in the case of metal cation analytes, we suggested reinvesting tried and tested complexometric volumetric methods which were popular before the advent of modern instrumental analysis. We successfully implemented a SERS-monitored complexometric titration of water hardness, in which the calibration is built-in through the use of a standardised titrant and the SERS signal abruptly switches when the equivalence is reached. Mean trueness estimates between 88 and 117% were obtained, with a precision of maximum 2%, without any external calibration of the spectroscopic signals, and for mineral waters having hardnesses differing by a factor of 25, from 0.6 mM to 15 mM. The full titration can be run in an hour (or a little less than two hours when run in triplicate), with nothing more than pipettes, tubes and a portable Raman spectrometer. The combined advantages of this practicality, of the inferred analytical figures of merit and of their robustness across about one decade and a half of concentrations demonstrate the merit of the complexometric volumetric titration strategy to turn SERS into a practical and reliable analytical technique. In particular, the present system has the potential to alleviate limitations in field measurements of Ca and Mg for clogging management purposes or geochemical monitoring of rock weathering. Given the breadth of complexometric methods and indicators that were employed in the past for titrating virtually every metal cation, but also many anions, and the sensitivity of SERS, we hope that these results will pave the way for implementing practical SERS-based titrations of minor or trace elements and will foster the acquisition of water quality data directly on the field.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We graciously acknowledge the support of the Agence Nationale de la Recherche under the grant ANR-17-CE04-0009-01 (UnivSERS), that of Ville de Paris under the grant Emergences Métal'Eau and of Région Île de France under the grant 13012334 (Scorpions).References
- D. Cialla, A. März, R. Böhme, F. Theil, K. Weber and M. Schmitt, et al., Surface-enhanced Raman spectroscopy (SERS): progress and trends, Anal. Bioanal. Chem., 2011, 403(1), 27–54 CrossRef PubMed.
- R. A. Alvarez-Puebla and L. M. Liz-Marzán, Traps and cages for universal SERS detection, Chem. Soc. Rev., 2011, 41(1), 43–51 RSC.
- R. S. Golightly, W. E. Doering and M. J. Natan, Surface-Enhanced Raman Spectroscopy and Homeland Security: A Perfect Match?, ACS Nano, 2009, 3(10), 2859–2869 CrossRef CAS PubMed.
- R. A. Halvorson and P. J. Vikesland, Surface-Enhanced Raman Spectroscopy (SERS) for Environmental Analyses, Environ. Sci. Technol., 2010, 44(20), 7749–7755 CrossRef CAS PubMed.
- EC Le Ru and P. G. Etchegoin, Basic Electromagnetic Theory of SERS, in Surface Enhanced Raman Spectroscopy, ed. S. Schlücker, Wiley-VCH, Weinheim, Germany, 1st edn, 2011, pp. 1–37 Search PubMed.
- S. E. J. Bell, G. Charron, E. Cortés, J. Kneipp, M. D. L. Chapelle and J. Langer, et al., Towards Reliable, Quantitative Surface-Enhanced Raman Scattering (SERS): From Key Parameters to Good Analytical Practice, Angew. Chem., Int. Ed., 2020, 59(14), 5454–5462 CrossRef CAS PubMed.
- D. Jimenez de Aberasturi, J. M. Montenegro, I. Ruiz de Larramendi, T. Rojo, T. A. Klar and R. Alvarez-Puebla, et al., Optical Sensing of Small Ions with Colloidal Nanoparticles, Chem. Mater., 2012, 24(5), 738–745 CrossRef CAS.
- V. M. Zamarion, R. A. Timm, K. Araki and H. E. Toma, Ultrasensitive SERS Nanoprobes for Hazardous Metal Ions Based on Trimercaptotriazine-Modified Gold Nanoparticles, Inorg. Chem., 2008, 47(8), 2934–2936 CrossRef CAS PubMed.
- S. Sarkar, M. Pradhan, A. K. Sinha, M. Basu and T. Pal, Selective and Sensitive Recognition of Cu2+ in an Aqueous Medium: A Surface-Enhanced Raman Scattering (SERS)-Based Analysis with a Low-Cost Raman Reporter, Chem. – Eur. J., 2012, 18(20), 6335–6342 CrossRef CAS PubMed.
- D. Tsoutsi, L. Guerrini, J. M. Hermida-Ramon, V. Giannini, L. M. Liz-Marzán and A. Wei, et al., Simultaneous SERS detection of copper and cobalt at ultratrace levels, Nanoscale, 2013, 5(13), 5841–5846 RSC.
- L. Guerrini, I. Rodriguez-Loureiro, M. A. Correa-Duarte, Y. H. Lee, X. Y. Ling and F. d. Abajo, et al., Chemical speciation of heavy metals by surface-enhanced Raman scattering spectroscopy: identification and quantification of inorganic- and methyl-mercury in water, Nanoscale, 2014, 6(14), 8368–8375 RSC.
- W. Tang, D. B. Chase, D. L. Sparks and J. F. Rabolt, Selective and Quantitative Detection of Trace Amounts of Mercury(II) Ion (Hg2+) and Copper(II) Ion (Cu2+) Using Surface-Enhanced Raman Scattering (SERS), Appl. Spectrosc., 2015, 69(7), 843–849 CrossRef CAS PubMed.
- G. Xu, P. Song and L. Xia, Examples in the detection of heavy metal ions based on surface-enhanced Raman scattering spectroscopy, Nanophotonics, 2021, 10(18), 4419–4445 CrossRef CAS.
- G. Brackx, D. Guinoiseau, L. Duponchel, A. Gélabert, V. Reichel and S. Zrig, et al., A frugal implementation of Surface Enhanced Raman Scattering for sensing Zn 2+ in freshwaters – In depth investigation of the analytical performances, Sci. Rep., 2020, 10(1), 1–11 CrossRef PubMed.
- D. A. Skoog, D. M. West, F. J. Holler and S. R. Crouch. Fundamentals of Analytical Chemistry, Brooks Cole, 8th edn 2003 Search PubMed.
- T. Imamura, K. L. Cheng and K. Ueno, CRC Handbook of Organic Analytical Reagents, Boca Raton, Fla: CRC Press, 2nd edn, 1992, p. 626 Search PubMed.
- P. C. Lee and D. Meisel, Adsorption and surface-enhanced Raman of dyes on silver and gold sols, J. Phys. Chem., 1982, 86(17), 3391–3395 CrossRef CAS.
- A. Stewart, S. Murray and S. E. J. Bell, Simple preparation of positively charged silver nanoparticles for detection of anions by surface-enhanced Raman spectroscopy, Analyst, 2015, 140(9), 2988–2994 RSC.
- H. T. S. Britton and R. A. Robinson, CXCVIII.—Universal buffer solutions and the dissociation constant of veronal, J. Chem. Soc., 1931, 1456–1462 RSC.
- N. M. Duong, A. Noclain, V. E. Reichel, J. M. Di Meglio, P. Hersen and G. Charron. Datasets of titrations of mineral water hardness monitored using SERS [Internet]. Zenodo; 2022 [cité 23 janv 2023]. Disponible sur: https://zenodo.org/record/6980619.
- A. E. Harvey, J. M. Komarmy and G. M. Wyatt, Colorimetric Determination of Magnesium with Eriochrome Black T, Anal. Chem., 1953, 25(3), 498–500 CrossRef CAS.
- C. H. Munro, W. E. Smith, M. Garner, J. Clarkson and P. C. White, Characterization of the Surface of a Citrate-Reduced Colloid Optimized for Use as a Substrate for Surface-Enhanced Resonance Raman Scattering, Langmuir, 1995, 11(10), 3712–3720 CrossRef CAS.
- K. Carron, K. Mullen, M. Lanouette and H. Angersbach, Selective-Ultratrace Detection of Metal Ions with SERS, Appl. Spectrosc., 1991, 45(3), 420–423 CrossRef CAS.
- R. W. Gauldie, K. Xi and S. K. Sharma, Developing a Raman Spectral Method for Measuring the Strontium and Calcium Concentrations of Fish Otoliths, Can. J. Fish. Aquat. Sci., 1994, 51(3), 545–551 CrossRef CAS.
- L. Szabó, K. Herman, N. Leopold, C. Buzumurgă and V. Chiş, Surface-enhanced Raman scattering and DFT investigation of Eriochrome Black T metal chelating compound, Spectrochim. Acta, Part A, 2011, 79(1), 226–231 CrossRef PubMed.
- POWER. Strategies to Reduce Sulfuric Acid Usage in Evaporative Cooling Water Systems [Internet]. POWER Magazine. 2010 [cité 23 janv 2023]. Disponible sur: https://www.powermag.com/strategies-to-reduce-sulfuric-acid-usage-in-evaporative-cooling-water-systems/.
- AR4 Climate Change 2007: The Physical Science Basis—IPCC [Internet]. [cité 23 sept 2019]. Disponible sur: https://www.ipcc.ch/report/ar4/wg1/.
- A. Marescaux, V. Thieu, A. V. Borges and J. Garnier, Seasonal and spatial variability of the partial pressure of carbon dioxide in the human-impacted Seine River in France, Sci. Rep., 2018, 8(1), 13961 CrossRef PubMed.
- G. L. Macpherson, CO2 distribution in groundwater and the impact of groundwater extraction on the global C cycle, Chem. Geol., 2009, 264(1), 328–336 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed chemical protocols, descriptions of instrumental set-ups and spectral processing, and additional SERS results as figures and raw data. See DOI: https://doi.org/10.1039/d3an00189j |
| This journal is © The Royal Society of Chemistry 2023 |