
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput Raman spectroscopy allows ex vivo characterization of murine small intestinal intra-epithelial lymphocytes (IEL)†
Rustam R.
Guliev
 a,
Tina
Vogler
be,
Natalie
Arend
a,
Simone
Eiserloh
ac,
Alexander
Wiede
a,
Timo
Kunert
be,
Martin
Dinkel
be,
Jürgen
Popp
acd,
Iwan W.
Schie
af,
Kai
Hildner‡
be and
Ute
Neugebauer‡
*acd
a,
Tina
Vogler
be,
Natalie
Arend
a,
Simone
Eiserloh
ac,
Alexander
Wiede
a,
Timo
Kunert
be,
Martin
Dinkel
be,
Jürgen
Popp
acd,
Iwan W.
Schie
af,
Kai
Hildner‡
be and
Ute
Neugebauer‡
*acd
aLeibniz Institute of Photonic Technology, 07745 Jena, Germany. E-mail: ute.neugebauer@uni-jena.de
bUniversity Hospital Erlangen, Medical Department 1, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91054 Erlangen, Germany
cCenter for Sepsis Control and Care, Jena University Hospital, 07747 Jena, Germany
dFriedrich Schiller University Jena, Institute of Physical Chemistry and Abbe Center of Photonics, 07743 Jena, Germany
eDeutsches Zentrum Immuntherapie (DZI), University Hospital Erlangen, Erlangen, Germany
fUniversity of Applied Sciences Jena, Department for Medical Engineering and Biotechnology, 07745 Jena, Germany
First published on 31st March 2023
Abstract
T cells are considered to be critical drivers of intestinal inflammation in mice and people. The so called intra-epithelial lymphocyte (IEL) compartment largely consist of T cells. Interestingly, the specific regulation and contribution of IELs in the context of inflammatory bowel disease remains poorly understood, in part due to the lack of appropriate analysis tools. Powerful, label-free methods could ultimately provide access to this cell population and hence give valuable insight into IEL biology and even more to their disease-related functionalities. Raman spectroscopy has demonstrated over the last few years its potential for reliable cell characterization and differentiation, but its utility in regard to IEL exploration remains unknown. To address this question experimentally, we utilized a murine, T cell-driven experimental model system which is accepted to model human gut inflammation. Here, we repopulated the small intestinal IEL compartment (SI IELs) of Rag1-deficient mice endogenously lacking T cells by transferring naïve CD4+ T helper cells intraperitoneally. Using multivariate statistical analysis, high-throughput Raman spectroscopy managed to define a cell subpopulation ex vivo within the SI IEL pool of mice previously receiving T cells in vivo that displayed characteristic spectral features of lymphocytes. Raman data sets matched flow cytometry analyses with the latter identifying T cell receptor (TCR)αβ+ CD4+ T cell population in SI IELs from T cell-transferred mice, but not from control mice, in an abundance comparable to the one detected by Raman spectroscopy. Hence, in this study, we provide experimental evidence for high-throughput Raman spectroscopy to be a novel, future tool to reliably identify and potentially further characterize the T cell pool of small intestinal IELs ex vivo.
Introduction
In the last few years, Raman spectroscopy has proven to hold high potential for answering important questions from medicine and life sciences.1–3 Raman spectroscopy is based on inelastic scattering of monochromatic laser light and can provide insights into the overall biochemical composition of biological samples, such as cells and tissue, in a label-free and non-destructive manner. In the last few years, important technological advances have been achieved that enable the acquisition of several hundreds or even thousands of cells in acceptable time (<1 hour) for single cell analysis.4,5 Such high-throughput Raman devices open the door for credible data sets and sample sizes required for single cell analysis.6For the application of Raman spectroscopy in the field of immunology, important progress has been reported.7 Raman spectroscopy has successfully been applied to identify and differentiate major leukocyte subpopulations,5,8 human granulocytes,9,10 lymphocyte subtypes, such as B and T cells11 or different lymphocyte cell lines,12 as well as further subtypes such as CD4+ T cells, CD8+ T cells and CD56+ Natural Killer cells.13 Targeted activation of immune cells is reflected in the Raman spectra, as was demonstrated after in vitro cell stimulation with pathogen-associated molecular patterns (PAMPS),14,15 with whole pathogens,16,17 as well as after in vivo stimulation using an endotoxemic mouse model.18 First results from clinical studies show that Raman spectroscopic signatures of leukocytes from peripheral blood have high potential to stratify sepsis patients.19
The mucosal immune system of the gastro-intestinal tract significantly contributes to the immune cell pool. It contains so-called intraepithelial lymphocytes (IELs) which are defined by their localization within the epithelial layer. While the denomination implies that IELs are exclusively composed of lymphocytes, IEL-resident immune cells are in fact not a uniform population and also embrace non-lymphocyte immune cells. However, under physiological conditions, CD4+ T helper cells represent the predominant T cell population in the lamina propria, i.e. the subepithelial region of the gut, while CD8+ T cell subsets are prevailing within the intra-epithelial lymphocytes (IEL) compartment and represent roughly 85% of all IELs.20 Despite their importance, regulation and function of IELs are only poorly understood. Importantly, IELs are a histopathologic diagnostic marker for distinct intestinal disease states like, e.g., coeliac disease21 and have been pathogenetically implied in the pathogenesis of inflammatory bowel disease (IBD). Over the last 5 years, data from various studies provide strong evidence for a meaningful role of gut resident CD8+ T cells, both as pathogenetic drivers as well as potential biomarkers of IBD. A recent high-profile publication, for example, aimed to unravel the molecular and functional complexity of CD8+ T cells in ulcerative colitis22 and more studies on this topic are summarized here.23 Lastly, in mouse models of TNF-driven small intestinal inflammation, the so-called TNFΔARE mouse models, CD8+ T cells have been identified to play a key role in promoting tissue inflammation, overall shedding important light beyond lamina propria dominating CD4+ T cell biology on CD8+ T cells mostly residing within the intra-epithelial layer compartment in the gut.24,25 T cells are assumed to be key promotors of intestinal inflammation both in human patients suffering from intestinal inflammation as in IBD and in many rodent experimental systems modelling human gut inflammation.26 However, novel methodologies, such as Raman spectroscopy, that aim to detect and characterize putatively inflammation-promoting T cells within the IEL compartment have not been evaluated so far.
One established model system of T cell-driven intestinal inflammation is elicited in mice carrying a targeted inactivation of the gene recombination activating 1 or 2 (Rag1 or Rag2).27 Rag1 or Rag2 redundantly encode for proteins that are critically involved in the regulation of the recombination of the V-D-J locus within immunoglobulins. In the absence of either protein, T and B cells cannot fully develop so that resulting mice are essentially lymphopenic with few immature, residual lymphocytes remaining.28 Hence, in Rag1−/− mice, the IEL niche immunologically consists almost exclusively of myeloid cell-derived cell pools. However, upon intra-peritoneal (i.p.) transfer of naïve CD4+ T helper cells into lymphopenic Rag1−/− recipient mice, T cells are systemically activated, expand and repopulate both lymphoid as well as non-lymphoid tissues resulting especially in gut tissue inflammation affecting the colon while the small intestine shows only moderate signs of inflammation.29 Consecutively, all T cells found in the gut of these mice are essentially derived from the transferred T helper cell pool.
Due to the absence of endogenous T cells, Rag1−/− mice with versus without T cell complementation represent a suitable model system to evaluate novel technologic modalities, such as Raman spectroscopy, for their ability to characterize T cells within the pool of other tissue residing immune cells at distinct anatomic sites as e.g. the intestinal tract. Hence, in this study we sought to evaluate high-throughput Raman spectroscopy for its utility to provide insight into the IEL compartment by correlating Raman spectroscopy-generated readouts with standard flow cytometric analysis.
Experimental
Mice
This study was carried out in accordance with the recommendations of the government of Lower Franconia in Bavaria, Germany. The protocol was approved by the government of Lower Franconia in Bavaria, Germany (#55.2.2-2532.2-785-14).Rag1−/− mice were purchased from the Jackson laboratory and intercrossed with Batf3−/− mice30 to generate Rag1−/− Batf3−/− mice.
Induction of T cell transfer mediated intestinal inflammation
Rag1−/− Batf3−/− mice were divided into two groups: group 1 (three animals, mice M1–M3) received T cells (intestinal inflammation group) while mice of group 2 (three animals, M4–M6) were left untreated, i.e. did not receive T cells intraperitoneally (control). For the isolation of T cells, single suspensions of splenocytes from wildtype mice on a C57Bl/6 mouse background were generated by mincing spleens followed by red blood cell removal with ACK lysis buffer (0.15 M NH4Cl, 1 M KHCO3, 0.8 M Na2EDTA in H2O, pH 7.2). CD4+ CD25− T cells were further isolated from the splenic cell suspension using magnetic cell sorting (MACS, Miltenyi Biotec). Here, T cells were first enriched for CD4+ T cells (negative selection) followed by positive depletion of CD25+ T cells resulting in a CD4+ CD25− naïve T cell pool. This procedure routinely yields a >95% pure T cell population as confirmed by flow cytometry. To repopulate T and B cell-deficient Rag1−/− mice with T cells, 106 CD4+ CD25− T cells were administered by intra-peritoneal injection (day 0).IEL isolation
Twelve days after the start of the experiment, experimental mice were sacrificed and small intestinal IELs were isolated from either group 1, i.e., T cell receiving (intestinal inflammation) mice or group 2, i.e. untreated (control) Rag1−/− Batf3−/− mice. First, the small intestine (SI) was removed from the experimental mice. Then, the lumen of the SI was rinsed and Peyer's patches were removed. SI was opened up longitudinally and then cut into small pieces. SI pieces were incubated twice in predigestion solution (accordingly to manufacturer's instruction, Lamina Propria Disscociation Kit, mouse, Miltenyi Biotec), each for 20 min while shaking at 37 °C. IELs were released from the tissue by vortexing for 10 s and collected in a new tube by filtration through a 100 μM cell strainer (BD Falcon). To fully remove all IELs from the tissue, intestine pieces were once again incubated in HBSS with HEPES for 20 min while shaking at 37 °C. IELs were collected in the tube together with the first isolates after filtration through a 100 μM cell strainer. Subsequently, IELs were purified by density centrifugation (Percoll, GE Healthcare). Total number of isolated cells was determined using standard procedures including a Neubauer hematocytometer. Total numbers of live cells isolated from SI ranged between 3 × 106 and 6 × 106 per animal. 2 × 106 cells were used for Raman analysis, i.e. centrifuged, chemically fixed in Histofix (Carl Roth), washed, resuspended in PBS and stored at 4 °C until Raman analysis was performed. The remaining cells were used for flow cytometry analysis.Characterization of isolated IELs using flow cytometry
Isolated SI IELs were washed in FACS buffer (PBS/3% FCS, VWR/PanBiotech). FACS buffer also served as staining solution upon adding 7AAD-PerCP-Cy5.5 (Biolegend) and the following fluorochrome-coupled antibodies directed against the indicated antigens for 15 min at 4 °C protected from light: EpCAM (biotinylated clone G8, Biolegend); T cell receptor beta chain (Pacific blue, clone H57-597, Biolegend); T cell receptor gamma delta chain (APC, clone GL3, Biolegend); CD4 (FITC, clone GK1.5, Biolegend); and CD8alpha (APC-Cy7, clone 53-6.7, Biolegend). After washing, cells were stained with diluted Streptavidin-PE (Biolegend). After extensive washing, cells were resuspended in FACS buffer (VWR/PanBiotech) and analyzed with the BD instrument Fortesssa.The data were analyzed using Flowjo Software version 10.8.1. The following gating strategy was utilized: first of all, doublets were excluded and then only live, EpCAM− singlets were further analyzed. Here, TCRαβ+ T cells were identified and further evaluated for CD4+vs. CD8α+ expression profiles.
Characterization of isolated IELs using Raman spectroscopy
Cells were placed into an in-house fabricated measuring cartridge and placed under an upright microscope of an in-house developed high-throughput (HT) Raman device described in earlier studies.5 The device is equipped with a 785 nm single mode laser (MatchBox, Integrated Optics) with 400 mW nominal output power, a spectrometer (IsoPlane160, Princeton Instruments, USA) with a 400 g mm−1 grating and a back-illuminated deep-depletion charge-coupled device (PIXIS-400BReXcelon, Princeton Instruments, USA). Light was focused through a 60×, NA 1.0 water immersion objective (Nikon, Japan) onto the sample. With a widened laser focus the focal spot covered a diameter of ∼10 μm, so that spectral information of the whole cell was collected at once. The Raman scattered light was collected through the same objective.IELs were automatically selected using image recognition in the bright field image developed for an earlier application.31 Automated location and auto-focus using motorized x–y translational stages (CONEX MFA-Series; Newport) mounted on an automated z-positioning stage (MTS25-Z8, Thorlabs), enabled collecting more than 2000 cells per mouse in less than 1.5 hours per mouse. Integration time per cell was set to 1 s.
Analysis of Raman data
Processing of Raman data was done using R (version 4.1),32 R Studio (version 1.4) IDE and the following packages: ggplot2,33 GGally,34 and ggpubr35 for visualizations, MASS36 for LDA calculation, hyperSpec37 and dplyr38 for data import and manipulation, and mdatools39 for MCR-ALS.The initial dataset consisted of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 313 spectra acquired by the HT Raman device. Spectra with low signal-to-noise ratio (mostly due to misfocus of the automated system), outliers and spectra that showed high background were removed during spectral pre-processing. As can be seen in Table 1, high background was mostly observed in cells from mice after T cell transfer. Cosmic ray spikes were corrected by the algorithm described in Ryabchykov et al.,40 baseline was corrected by Statistics-sensitive Non-linear Iterative Peak-clipping (SNIP)41 algorithm, and spectrum area was normalized to be equal to one. After preprocessing, 11127 spectra were kept for further analysis.
313 spectra acquired by the HT Raman device. Spectra with low signal-to-noise ratio (mostly due to misfocus of the automated system), outliers and spectra that showed high background were removed during spectral pre-processing. As can be seen in Table 1, high background was mostly observed in cells from mice after T cell transfer. Cosmic ray spikes were corrected by the algorithm described in Ryabchykov et al.,40 baseline was corrected by Statistics-sensitive Non-linear Iterative Peak-clipping (SNIP)41 algorithm, and spectrum area was normalized to be equal to one. After preprocessing, 11127 spectra were kept for further analysis.
| Condition | Group 1 (with T cell transfer) | Group 2 (control) | ||||
|---|---|---|---|---|---|---|
| Mouse # | M1 | M2 | M3 | M4 | M5 | M6 |
| Measurement day 1 | 2102 | 2431 | 0 | 0 | 0 | 2570 |
| Measurement day 2 | 0 | 0 | 2035 | 2587 | 2588 | 0 |
| After pre-processing | 1003 | 1870 | 1201 | 2362 | 2442 | 2249 |
| # of cells removed due to high background | 25 | 39 | 151 | 22 | 12 | 82 |
| Low PC1 cluster (from HCA) (% after pre-processing) | 434 (43.3%) | 655 (35.0%) | 466 (38.8%) | 613 (26.0%) | 574 (23.5%) | 635 (28.2%) |
| High PC1 cluster (from HCA) (% after pre-processing) | 569 (56.7%) | 1215 (65.0%) | 735 (61.2%) | 1749 (74.0%) | 1868 (76.5%) | 1614 (71.8%) |
First, the variations in the preprocessed Raman data were analyzed by principal component analysis (PCA). Here, PCA analysis was applied separately to Raman spectra of cells from (a) only group 1 (after T cell transfer), (b) only group 2 (control), and (c) combined spectra of both treatment groups. All three datasets (i.e. a, b, and c) were centered and scaled before applying PCA. Then, it was found that the first two principal components (when applied to combined spectra from both groups) contained practically all the relevant information. Therefore, in further analysis only these two components were used.
In addition, multivariate curve resolution alternating least square (MCR-ALS) was used for better biochemical interpretation of variations. In this work, MCR-ALS was applied using non-negativity constrains for both contributions and pure components spectra. Manually selected spectra were used for initialization of the algorithm. The number of pure components was chosen by varying and selecting the best in terms of residuals and interpretability. The calculated pure component spectra were assigned to biochemical groups (nuclei, lipids, cytoplasm) by characteristic Raman bands (Table 2) and comparison with reference water spectrum.

|
Although the analysis of PCA scores, combined with visualization of corresponding spectra, already allowed revealing some important information, a formal analysis with statistical tools was also applied. First, the data was clustered using hieratical cluster analysis (HCA) into two clusters. The distance between points was measured using Euclidean distance and Ward's algorithm was applied for clustering. Cluster separation was found to highly correlate with PC1 scores values. For subsequent analysis, only the cluster corresponding to low PC1 values (i.e. smaller cells) was kept. Then, a linear discriminant analysis (LDA) model differentiating cells extracted from the small intestine of mice with T cell transfer and control animals (i.e. differentiating cells from treatment group 1 and group 2, respectively) was trained. In order to avoid overfitting, a nine-fold cross validation was applied. The cross-validation folds were prepared in a way that the training set always consisted of spectra from four mice (two mice with T cell transfer and two control mice) and, correspondingly, the remaining two mice for the test set (i.e., one mouse with T cell transfer and one control mouse, for further details see ESI Table 1†). In addition, principal components were calculated using only data of the training set. Number of used principal components was fixed to 2.
Results
To evaluate Raman spectroscopy for its utility to characterize small intestinal IEL populations ex vivo, we generated two groups of experimental mice: group 1 consisted of Rag1−/− Batf3−/− mice that received naïve CD4+ T cells, while Rag1−/− Batf3−/− mice of group 2 remained untreated (to simplify the denomination, recipient mice are termed Rag1−/− mice throughout the study). 12 days after T cell transfer into group 1, SI IELs from both groups were isolated from the small intestines of Rag1−/− mice. Of note, in our model system and in line with published data,42 colonic inflammation was expectedly absent in group 2 while it was detectable at this time point in animals of group 1 (data not shown), while the small intestine was macroscopically indistinguishable between both groups and hence lacked visible signs of inflammation.Flow cytometric analysis identifies transferred CD4+ T cells within the SI IEL compartment of group 1 mice
Results of flow cytometric analysis are depicted in Fig. 1. Since the isolation procedure also unavoidably results in the recovery of cell clumps, dead or dying cells and intestinal epithelial cells by definition not belonging to the IEL compartment, we applied a vigorous gating strategy to eliminate those confounding factors. First, SI IELs from T cell receiving mice (i.e., group 1) displayed relatively fewer so-called doublets events compared to control mice (group 2): with 74 ± 3% singlets in group 1 vs. 64 ± 3% in group 2 (Fig. 1A). Therefore, for further analysis, doublets were excluded and only so-called singlets, i.e. single cells were further included in consecutive gatings. Moreover, we employed live/dead cell discrimination and intestinal epithelial cell-centered EpCAM stainings (Fig. 1A). With around 66 ± 7% live+, EpCAM− (negative) cells in group 1 and 68 ± 7% in group 2, no significant differences between group 1 and 2 were detected. Furthermore, the T cell composition of live+ EpCAM− IELs was assessed by broadly distinguishing between TCRαβ+ and TCRγδ+ T cell pools (Fig. 1A). Singlet+ live+ EpCAM− SI IELs from T cell receiving Rag1−/− mice (group 1) contained around 20% (20 ± 9%) TCRαβ+ T cells. In the SI IEL pool of the control Rag1−/− mice (group 2) TCRαβ+ T cells were essentially not detectable (Fig. 1A) as expected, since Rag1−/− mice lack mature T cells and only group 1 mice initially received TCRαβ+ T cells. We did not identify TCRγδ+ T cells in the SI IEL pools of either experimental group: while control Rag1−/− mice (group 2) are naturally lacking this population (data not shown), mice of group 1 were only treated with spleen-derived CD4+ T cells naturally consisting of TCRαβ+ but essentially lacking TCRγδ+ T cells. Within the TCRαβ+ CD4+ T cell pool of group 1, some were co-expressing CD8α (Fig. 1B). This finding is in line with a previous study showing that T cells with a cytotoxic T cell phenotype derived from the transferred CD4+ T cell pool are developing under lymphopenic conditions in the SI IEL compartment.42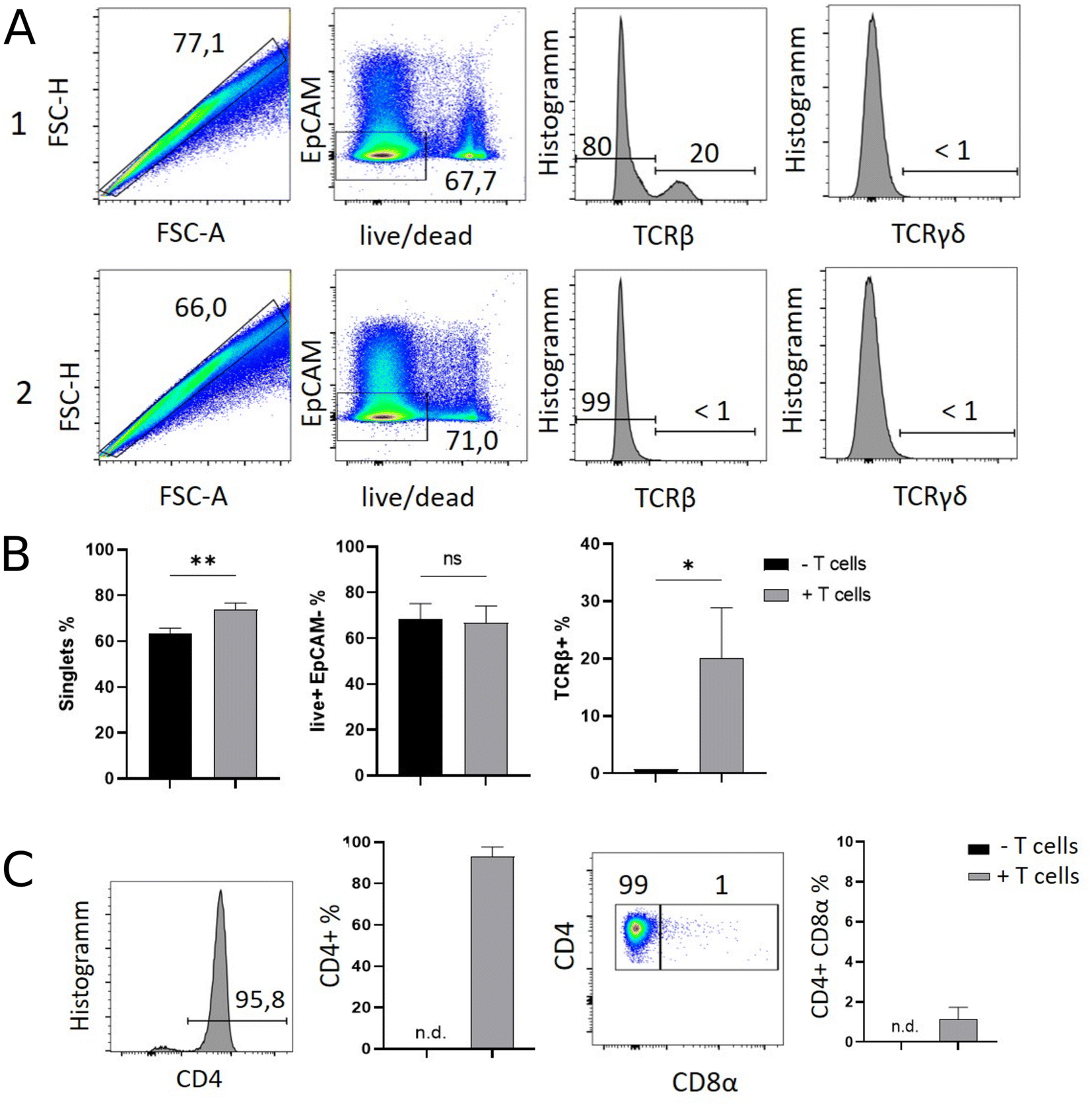 | ||
| Fig. 1 Flow cytometry allows the ex vivo identification and phenotypical classification of T cells within the SI IEL compartment of endogenously T lymphocyte-deficient Rag1−/− deficient recipient mice previously complemented with initially naïve, splenic CD4+ T cells. Rag1−/− Batf3−/− mice either received 106 CD4+ CD25− spleen-derived T cells i.p. (+ T cell transfer, group 1) or were left untreated (− T cell transfer, group 2). Twelve days later, SI IELs were isolated and flow cytometrically assessed using subsequent gating strategies as illustrated. In (A) gating strategy exemplarily display flow cytometry data of group 1 (top row) and group 2 (bottom row) shown as pseudocolour blot or histogram. First column: doublet exclusion, i.e. identification of singlets; second column: live/dead and EpCAM staining of singlets. Third column: Singlet+ live+ EpCAM− cells were assessed for the frequency of TCRβ+ and in forth column for TCRγδ+ T cells. (B) Bar graphs of the flow cytometry analysis shown in A using data of all n = 3 mice per group. (C) Singlet+ live+ EpCAM− TCRβ+ cells were assessed for CD4 expression (first column: representative histogram; second column: bar graph reflects n = 3 mice per group). Then singlet+ live+ EpCAM− TCRβ+ CD4+ T cells assayed for the frequency of CD8α co-expressing T cells within SI IELs (third column: representative pseudocolor blot; fourth column: bar graph with n = 3 mice per group). For statistical analysis, student's t-test was applied. (n.d. = not detectable). | ||
Overall, our flow cytometric data show that transferred, exogenous CD4+ T cells are repopulating the SI IEL compartment of lymphopenic mice thereby partially adopting phenotypical features as e.g. upregulation of CD8α expression in a fraction of CD4+ IELs and hence mimicking the SI IEL CD4+ T cell phenotype found in lympho-sufficient mice in the steady state. Moreover, our data indicate that the majority of the immune cells in the SI IEL compartment after T cell transfer and induction of intestinal inflammation is composed of non-T cells. This non-T cell pool is presumably of largely myeloid cell-derived origin (e.g. innate lymphoid cells, macrophages, dendritic cells etc.); however, the nature and composition of the non-T cell population were not the focus of this study and hence have not been further assessed.
Characterization of SI IELs using Raman spectroscopy
Concomitantly to the flow cytometry analysis, isolated SI IELs were characterized by means of Raman spectroscopy using a high-throughput Raman device. Cells were automatically selected using image recognition in the bright field images. Fig. 2 shows a representative bright field image of cells from each mouse. It can be seen that smaller cells were present in the T cell transfer group (group 1, M1–M3) compared to the cells from the control mice (group 2, M4–M6). | ||
| Fig. 2 Bright field images of isolated cells under high-throughput Raman device. Top: Images of isolated cells origination from mice with T cell transfer (group 1, M1–M3). Bottom: Images of cells isolated from control mice (group 2, M4–6). Scale bar is 10 μm in all images. Colored circles indicate positions where Raman spectra were recorded. White circles correspond to spectra that were excluded during preprocessing, red circles – cells with spectra in high PC1 cluster, blue circles – cells with spectra in low PC1 cluster. | ||
First, the variations in the pre-processed Raman data were analyzed by unsupervised principal component analysis. The analysis was applied to three datasets separately: (a) Raman spectra of cells from mice of group 1 (after T cell transfer, M1–M3); (b) spectra of cells originating only from group 2 mice (control, M4–M6) and (c) combining cells from mice of both treatment groups (M1–M6). PCA scores plots for the first ten principal components of those three models are presented in ESI Fig. S1.† Upon first inspection, it seems that no distinct clusters were formed and the spectral data points were distributed as one cloud. Considering more principal components also led to the same results. However, a closer look at the scores using density estimates in form of histogram plots revealed that the first principal component scores had a mixture distribution which was most pronounced when group 1 data was included (ESI Fig. S1-A and S1-C†). The following analysis used only the first two components of PCA applied to both groups (i.e. the data presented in Fig. 3). It is also shown later, in LDA analysis part, that adding more components did not improve the discrimination.
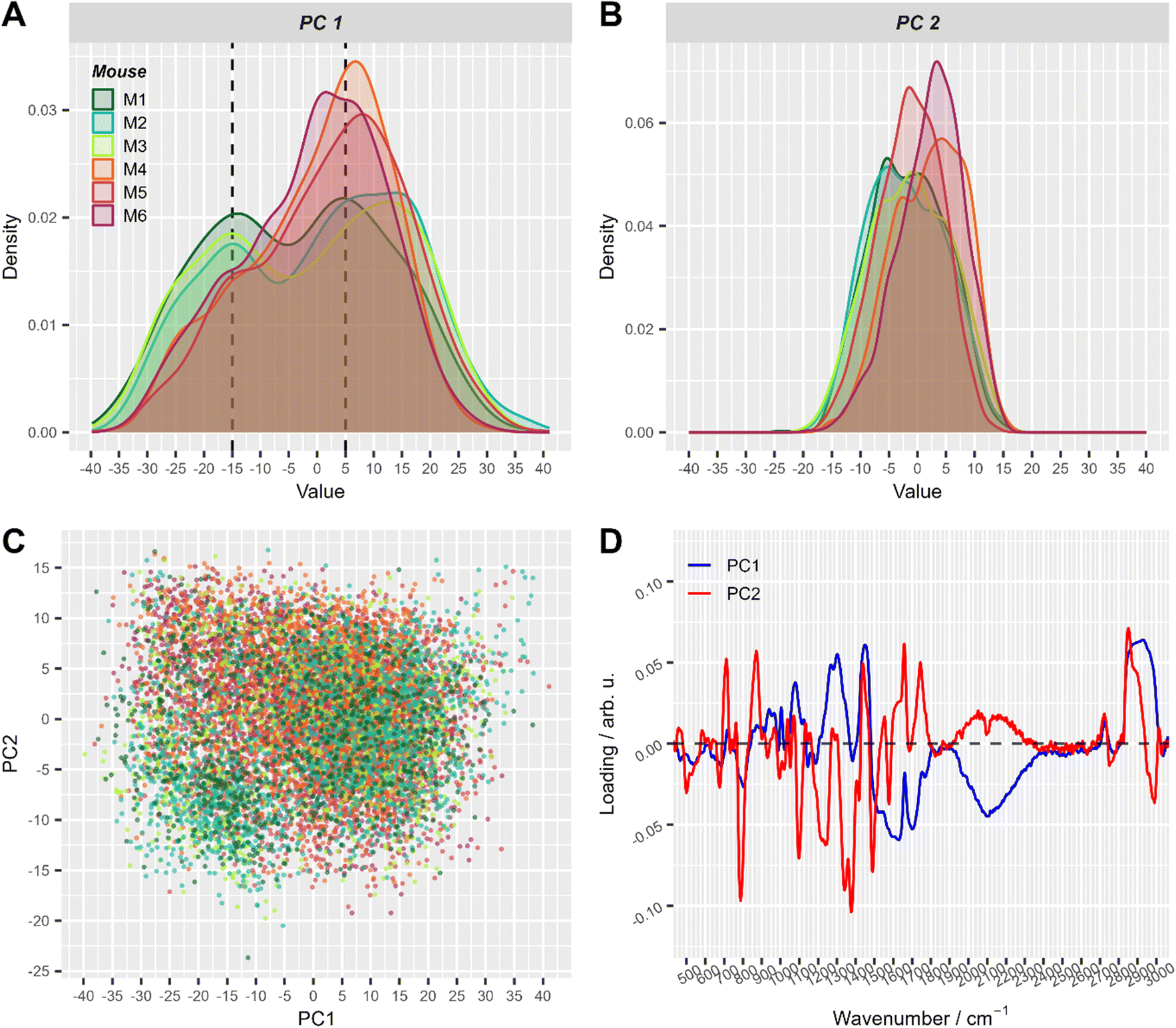 | ||
| Fig. 3 The first two principal components of PCA applied to all pre-processed Raman spectra. (A) and (B) Density plots of PC1 (A) and PC2 scores (B), respectively. This simply represents a smoothed histogram for better comparison between different distributions. (C) Corresponding 2D scatter-plot of PC1 and PC2. (D) Loadings of the first two principal components. Score values in A–C are colored with respect to mouse number (with T cell transfer: M1: dark green, M2: turquoise, M3: green-yellow; control: M4: orange, M5: red, M6: violet). For visual convenience, mice from the same group are colored by close colors: green-like colors for group 1 (with T-cell transfer) and red-like colors for group 2 (control). 2D scatter plots of PC1 and PC2 split per mouse are shown in ESI Fig. S2.† | ||
In the PCA scores plot using spectral data of cells from all mice, differences between the two treatment groups could be seen in the density estimates of the first two principal components (Fig. 3). Raman spectra of cells from mice with T cell transfer (M1–M3) showed a subpopulation with low PC1 values, that was significantly less present in Raman spectra of cells from control mice (M4–M6, Fig. 3A). ESI Fig. S2† provides a detailed view of Fig. 3C where the plots are split by mouse to better demonstrate the two slightly distinguishable cell sub-populations present in treatment group 1. Interestingly, the loadings of PC1 were nearly the same in all three models using the different data sets (ESI Fig. S3†). As can be seen from the loading plot of PC1 (Fig. 3D, ESI Fig. S3†), strong negative bands are found around 2100 cm−1 and around 1640 cm−1. These vibrational bands can be assigned to vibrational bands of water.43 Strong positive bands in PC1 loadings are found around 2900 cm−1, ∼1450 cm−1, ∼1300 cm−1 and ∼1080 cm−1. Those Raman bands can be assigned to C–H stretching and deformation vibrations as well as to amide vibrations and are typically found in organic material. Considering that the laser focus for our single cell high-throughput Raman measurements was 10 μm in diameter, we can assign PC1 to separate small (diameter <10 μm) and large cells (diameter ≥10 μm). Small cells with a diameter of <10 μm will not fill the full laser focus with their cells size and thus, additional water signal is recorded per spectrum.
To further explore the chemical characteristics of the mixed cell distributions visible in PCA analysis, Raman spectra with low (≤−15) and high (≥5) PC1 scores were compared per treatment group. The PC1 values thresholds (≤−15 and ≥5, respectively) were chosen using the maxima of the two subpopulations as could be seen in Fig. 3A where dashed lines show the location of the threshold values with respect to the values distribution. Fig. 4A provides the Raman mean spectra of the measurements corresponding to low and high PC1 scores. All spectra show typical features of eukaryotic cells. An assignment of the most prominent Raman bands to biochemical components is provided in Table 2. As expected from the analysis presented in Fig. 3, mean spectra of cells with low and high PC1 scores differ with respect to relative Raman band intensities of organic material and water signals. The analysis of the loadings and the spectra from two sub-populations shows that the differences between spectra of low and high PC1 scores are mainly due to Raman bands at 788 cm−1 (cytosine and uracil ring breathing), 1094 cm−1 (PO2− phosphate backbone), 1128 cm−1 (C–N stretching vibrations of proteins), 1268 cm−1 (amide III), 1304 cm−1, 1336 cm−1, 1450 cm−1 (CH2 bending in proteins and lipids), 1658 cm−1 (amide I) and at 2894 and 2932 cm−1 (CH2 and CH3 stretching vibrations) which can be assigned do DNA, proteins and lipids.
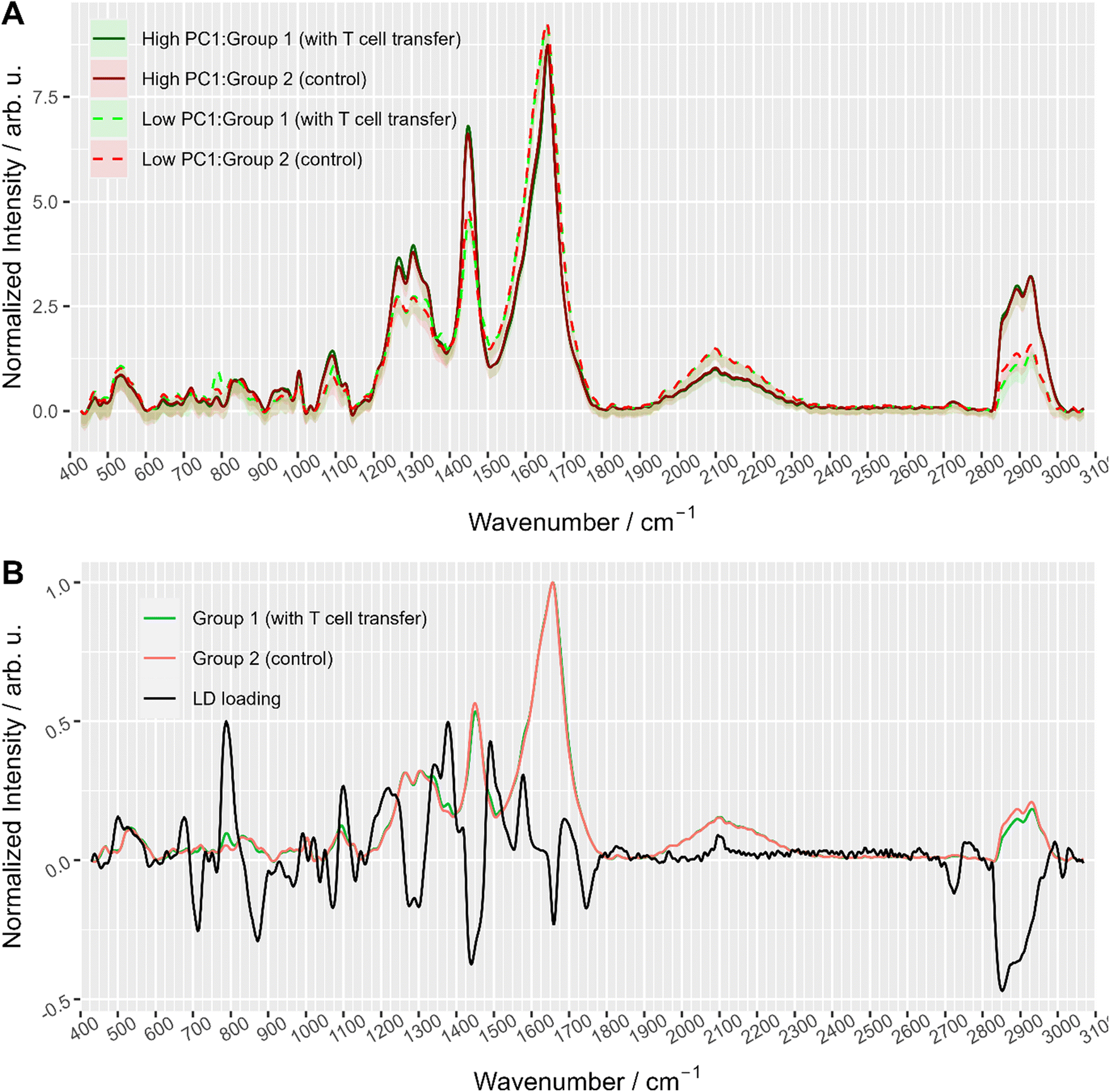 | ||
| Fig. 4 (A) Averaged Raman spectra of measurements with high (≥5) and low (≤−15) PC1 scores colored with respect to treatment group, i.e. with T cell transfer (group 1) and without (group 2, control). The area around the curve illustrates the standard deviation. Dashed lines correspond to spectra with low PC1 values (<−15) and solid lines to spectra with high PC1 score values (≥5). (B) Linear discriminant loading along with mean spectra of the two treatment groups (only smaller cells cluster data were used). | ||
Raman spectra of cells with high PC1 scores (PC1 ≥ 5, solid lines in Fig. 4A) are quite similar between the two treatment groups, i.e. irrespective of T cell transfer. The computed difference between the mean spectra of the two treatment groups reveals small changes in Raman bands that indicate differences in protein structure, in particular in the amide I and amide III region (ESI Fig. S4A†). Due to the relatively small chemical differences of the cells in this high PC1-score population between the two treatment groups, we suggest that these spectra originate from (immune) cells that are present in both treatment groups. Due to the observed spectral features we expect cells with a large cytoplasmic region filled with lipids. Those cells could be, e.g., residual epithelial cells or myeloid cells such as macrophages. Those cells are also expected to be around 10 μm or larger and thus, would confirm the assignment made. Support for this assignment is also found from the flow cytometry data. Fig. 5A and B shows representative flow cytometry scatter plots where at large forward scatter values common cell populations (TCRβ−/EpCAM− cells and EpCAM+ cells) are found in both treatment groups.
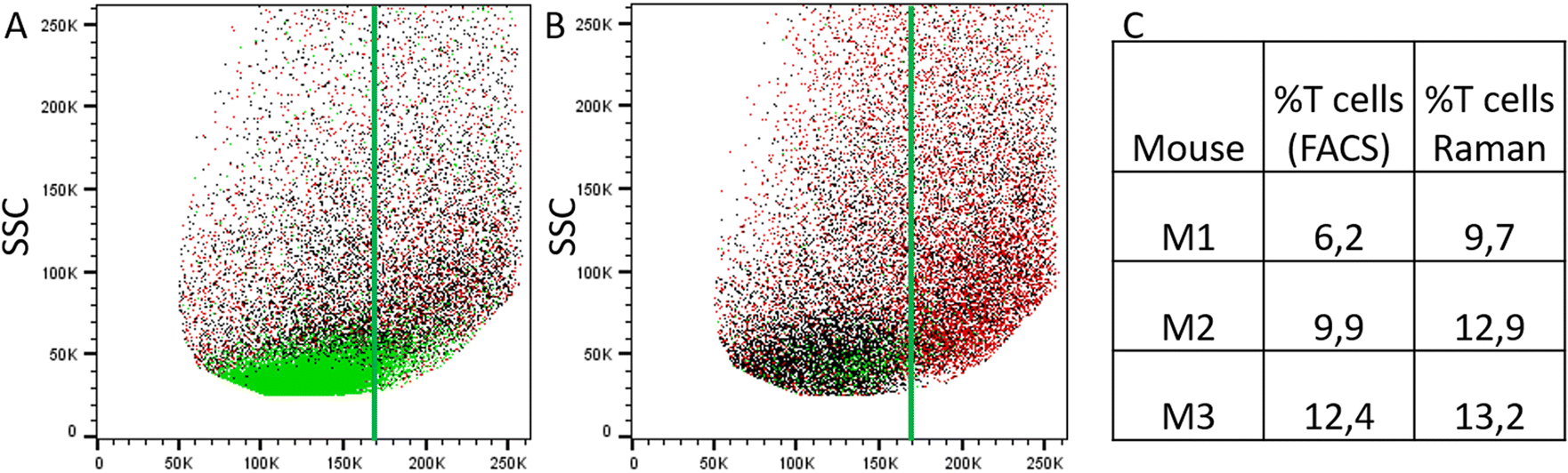 | ||
| Fig. 5 Comparison of results from flow cytometry and label-free Raman analysis. (A) and (B) Representative flow cytometry scatter plots showing forward scatter (FSC) vs. side scatter (SSC) from the isolated IEL fraction from animals with T cell transfer (A, group 1) and without T cell transfer (B, group 2, control) (after pre-gating for singlets). Green: TCRβ-positive cells, black: TCRβ-negative and EpCAM-negative cells and red: EpCAM-positive cells. The vertical green line is drawn to guide the eye. (C) Relative percentage of T cells among the enriched IEL population as determined by flow cytometry (first column) and by Raman analysis (second column). | ||
In contrast, Raman mean spectra with low PC1 score values (≤−15, dashed lines in Fig. 4A) are very different in cells originating from the two different treatment groups (i.e., originating from T cell transfer or control mice, respectively). The computed difference spectrum between the treatment groups (ESI Fig. S4B†) clearly shows prominent spectral features. Spectra originating from cells from T cell treated mice have significantly higher Raman intensities of bands that can be assigned to nucleic acid contributions, i.e. at 788 cm−1 (cytosine and uracil ring breathing), 1094 cm−1 (PO2− phosphate backbone), 1336 cm−1 (nucleic acids), and 1376 cm−1 (thymine, adenine, guanine). On the other hand, spectra of selected cells from the control mice have higher Raman intensities of bands in the high wavenumber region (CH2 and CH3 stretching vibrations at 2894 cm−1 and 2932 cm−1) that can be assigned to protein and lipid contributions. This clearly points to a cell population among cells originating from group 1 mice that possess high nucleic acid contribution. Such high nucleic acid contributions are expected from cells with a large nucleus with respect to the total cell size. Compared to other eukaryotic cells and other immune cells, lymphocytes possess a large nucleus with respect to their cell size, i.e. in lymphocytes the cytoplasm region is only rather small. It is therefore not surprising that in previous work characterizing the spectral features of lymphocytes and other immune cells, nucleic acid bands have always been among the characteristic Raman bands that identify lymphocytes, e.g., when differentiating T-lymphocytes from neutrophils8 or myeloid cells44 or when differentiating lymphocytes from neutrophils and monocytes.5 Thus, the presented loading coefficient agrees with the expected result when isolating IELs from the small intestine of Rag1−/− mice after T cell transfer (experimental group 1) and of Rag1−/− mice without T cell transfer (control, group 2): lymphocytes are expected in mice previously receiving T cells while they are not expected in the controls. Based on the Raman spectroscopic characteristics of lymphocytes in own previous work and literature,5,8,14,44 we propose that the cell population selected by low PC1 spectra from the mice after T cell transfer is rich in lymphocytes (T cells from T cell transfer). For further visualization, the ratio of the two Raman bands at 788 cm−1 and 2894 cm−1 where most pronounced spectral differences were observed was calculated and is presented in ESI Fig. S5.† When all spectra are used a distinct subpopulation shows up with a low 2894 cm−1/788 cm−1 ratio, which is not present when analyzing only spectra with high PC1 score values, but is prominently visible among spectra with low PC1 score values. Comparing with flow cytometry data (Fig. 5A and B) this assignment is also supported as the TCRβ+ cells are found at lower FSC values.
To gain further insights into the biochemical origin of the observed spectral variations, multivariate curve resolution – alternating least squares (MCR-ALS) was performed. Assuming that each spectrum is a linear combination of some unknown beforehand pure component spectra, MCR-ALS is a technique that allows to retrieve pure spectra and their contribution profiles.45 This approach is often used for visualization of hyperspectral images46 where each pixel is colored according to contribution of pure components. Although this is similar to decomposition of PCA, MCR-ALS provides results that are typically easier to interpret. ESI Fig. S6† shows MCR-ALS decomposition of the spectra into three main components: lipids, nuclei, and water/cytoplasm. Although the distinction is not so prominent, it is worth noting that the common subpopulation of cells had higher contribution from lipids which supports the idea that they can originate from myeloid cells. On the other hand, the other subpopulation (with more prominent differences between the treatment groups) had higher contribution from nucleic acids and water/cytoplasm which can be explained with larger nucleus and an overall smaller cell size (<10 μm, as discussed above).
In summary, it can be concluded that the measured cells can be categorized by Raman signal into two subpopulations: a subpopulation with Raman spectra with high PC1 score values, presumably myeloid and epithelial cells, which shows only small differences between the two treatment groups. And a second subpopulation of smaller cells with Raman spectra exhibiting low PC1 values. For the latter, significant differences in nucleic acid Raman bands were found, indicating a subpopulation with a large nucleus (T cells) among the cells originating from mice with T cell transfer (group 1, mice M1–M3). These Raman spectroscopic findings agree with the fact that Rag1−/− control mice lack lymphocytes while in the treated animals, T cells have been transferred and are now present in the small intestine. In the Rag1−/− mice, non-T cell immune cells populate in the absence of lymphocytes the intestinal niche.
In order to better formalize the findings, further data analysis tools were applied. First, the data was clustered using hierarchical cluster analysis (HCA) into two clusters as is depicted in ESI Fig. S7.†Fig. 6 shows in a 2D scores plot how the spectra of the two HCA clusters are distributed in the PCA space. It is clear from the figure that clustering results are mostly based on PC1 values. Therefore, for simplicity, the clusters will be referenced as low and high PC1 clusters. In addition, Table 1 provides number of spectra in each cluster and in Fig. 2 measurement positions on bright field images are colored with respect to the cluster membership of the spectrum.
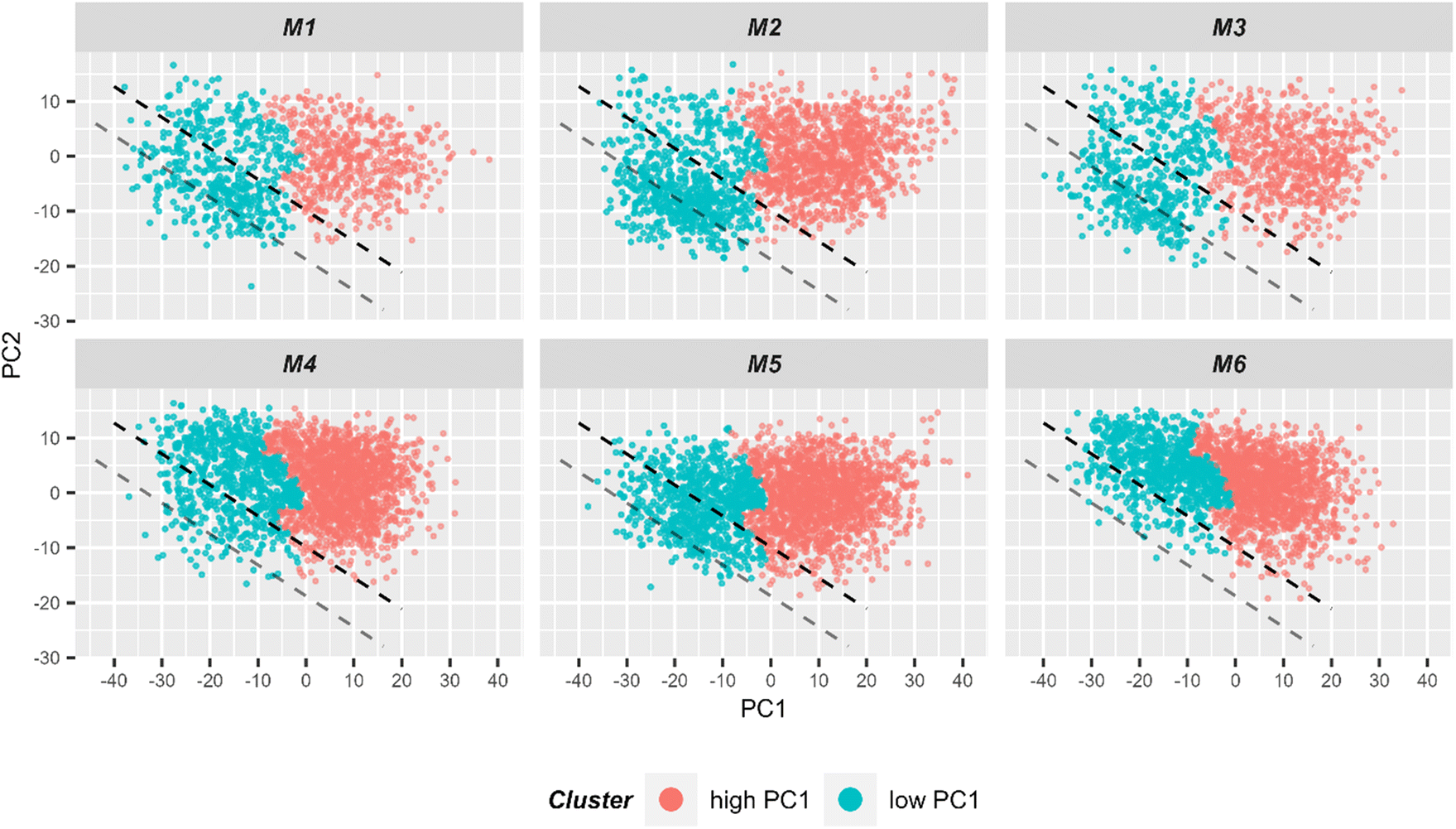 | ||
| Fig. 6 2D scatter plot of Raman spectra for the first two principal components. Six individual scatter plots are shown, one for each mouse to allow better visualization. The color codes the cluster assignment in HCA (ESI Fig. S7†). It can be seen that HCA cluster separation correlates very well with PC1 values. Light red points correspond to spectra in the HCA cluster corresponding to spectra with high PC1 values, while blue points correspond to spectra from the other HCA cluster where spectra with low PC1 values are clustered. For subsequent LDA analysis, only spectra from the low PC1-cluster (i.e. blue points) were used for the model training. The black dashed lined demonstrates the linear discriminant calculated by LDA and separating two treatment groups. The gray line demonstrates linear discriminant with improved centering. | ||
Then, only the cluster corresponding to lower PC1 values was kept and a LDA model differentiating cells extracted from the small intestine of mice with T cell transfer and of control animals (i.e. group 1 and group 2, respectively) was trained. In total, there were 3377 spectra in the cluster with low PC1 values which is around 24% of the initial dataset (i.e. before pre-processing). The model was trained using 9-fold cross-validation in a way that two mice (one control and one with T cell transfer) were always in the test set, i.e. leave-two mice-out. The details of cross-validation performance are provided in ESI Table S1 and Fig. S8.† Based on test sets of the cross-validation, sensitivity and specificity to identify cells from group 1 (with T cell transfer) mice were 67% and 64%, respectively. The area under Receiver-Operating-Characteristics (AUC ROC) curve yielded 71%. This means that, although far from ideal, the LDA model is able to discriminate the two treatment groups to some degree. Fig. 4B depicts the LD coefficient together with the Raman mean spectra of the two treatments groups used in the LDA analysis. It is clear from mean spectra and LD loading that the differences between the two treatment groups after clustering are the same as was shown before in dashed lines in Fig. 4A. From the discussion above, it can be stated that in the cluster of small PC1 values, rather small cells are present. From prior knowledge of the mouse models, it is expected that some of the cell population present in mice of the control group, is also present in mice receiving T cells. Together with the classification results, it is assumed that the cluster with small PC1 values contains T cells and another common cell population that is also present in the control mice.
Using the prior knowledge about common subpopulation of cells, the linear discriminant was further improved by using better centering. The linear discriminant was shifted in order to improve precision, i.e. to exclude more spectra from control mice under the LD. Fig. 6 illustrates both the shifted LD (gray line) and the initial LD (black line). It can be seen that points originating from group 1 are better isolated after the correction. Without the improved centering, 60% of 2000 points under the LD were from group 1 whereas after the shift of LD it increased to 81% (612 points). Such separation can be used for estimating the amount of IELs, since they are the only cell subpopulation that is expected to be significantly different in the two treatment groups. A relative percentage out of all measured cells of 9.7%, 12.9%, and 13.2% could be determined for mouse M1, M2 and M3, respectively. These numbers are in good agreement with the values obtained from flow cytometry (Fig. 5C).
In order to make sure that no important information was missed by considering only the first two principal components in the data analysis, similar models were trained using up to 20 principal components. The results in terms of ROC AUC are provided in ESI Fig. S9A.† Also, similar PCA-LDA analysis was applied to the other (high PC1) cluster data (ESI Fig. S9B†). Adding more principal components into the PCA-LDA model did not improve the accuracy in both cases. The best AUC ROC of the model trained on high PC1 cluster reached only 58% which suggests, that high the PC1 cluster corresponds to a common cell population between the two treatment groups. Overall, the results of LDA agree with the findings and suggestions made in PCA analysis.
Discussion
In our current study, we have applied two complementary cell analysis methods to characterize the intra-epithelial lymphocyte pool within the small intestine of untreated vs. T cell-transferred, endogenously T cell-deficient Rag1−/− mice. Flow cytometry analysis confirmed the expected constellation with T cells detectable within the SI IEL compartment of mice previously receiving T cells while missing in untreated Rag1−/− mice. The finding that a substantial fraction of the isolated and flow cytometrically evaluated cell types is either of non-immune cell origin or not viable, reflects the fact that involved cell types from this specific site are sensitive to rapid cell death ex vivo52 underscoring that the applied procedure is rather an IEL enrichment than IEL isolation step.However, the flow cytometric analysis ex vivo revealed that the IEL compartment is repopulated by T helper cells in vivo upon transfer of splenic and initially naïve CD4+ T cells. This is remarkable since the steady state population of SI IELs predominately consists of antigen-experienced, so called tissue-resident CD8+ T cell subsets. However, transferred CD4+ T cells partially upregulated CD8α expression on their cell surface thereby acquiring a cytotoxic T helper cell phenotype that can be observed in SI IEL steady state populations in vivo and which is in line with other reports describing its IEL presence upon transfer of peripheral T helper cells into lympho-deficient mice.42 Hence, the used model system recapitulates certain aspects of in vivo SI IEL T cell pools while it allows the generation and consecutive ex vivo characterization of T cell-containing vs. T cell-deficient SI IELs compartments.
In line with our flow cytometric studies, using label-free Raman spectroscopy we found clear signs of mixed cell populations when using unsupervised PCA analysis. One subpopulation (exhibiting high PC1 score values) showed only small differences between the two treatment groups, while the other subpopulation (with low PC1 score values) showed significant differences when comparing cells from the control mice and from mice after T cell transfer. Based on the Raman spectral features (and the technical characteristics of our system), we concluded that the subpopulation with high PC1 score values is enriched with larger cells (cell diameter ≥10 μm). Those could be e.g., epithelial cells or myeloid cells (e.g. macrophages). Using flow cytometry data, we find indeed that epithelial cells (EpCam+) are among the larger cells, while the T cells are among the smaller cells (Fig. 5A and B).
Subsequent Raman analysis focused on the Raman spectra that are most distinct between the treatment groups, i.e. spectra showing up in the subpopulation with low PC1 values. These could be also selected when applying hierarchical cluster analysis. Raman analysis revealed typical spectral features of T cells (lymphocytes) in the cell population originating from mice receiving T cell transfer. Those prominent nucleic acid Raman signals that point to a large cell nucleus with respect to total cell size where missing in the cell population isolated from the control cells. Using a linear discriminant analysis (LDA) model and a priori knowledge, we could quantify the relative number of lymphocytes among the totally measured isolated cells to be around 12 ± 2%. This is in excellent agreement with the numbers obtained from flow cytometry (9 ± 3%).
Comparing the Raman spectra of the common cell populations found in both treatment groups (with Raman spectra showing high PC1 score values), very minute variations were also observed. Transferred T cells are expanding in the lymphopenic host and become activated. Shaped by microbial signals, they differentiate into cytokine-secreting effector cells eliciting inflammation both, within tissues like in the intestinal compartment, but probably systemically as well. Thereby also non-T cell host cells and here both immune and non-immune cells (as e.g. epithelial and stromal cell types) become regulated by inflammatory T cell-derived signals and thereby display an altered Raman signal pattern compared to SI IELs isolated from non-inflammatory, non T cell-receiving mice. Thus, Raman analysis can reveal spectral differences between the cell populations as well as of their functional state (activated in T cell transfer model and resting/naive in untreated control mice).
To correctly identify and characterize the non-T cell populations already present in control mice, additional studies beyond the scope of this manuscript are necessary and foreseen for future study designs.
Summary and conclusions
In this study, we performed comparative ex vivo analyses of small intestinal IELs from T cell-deficient Rag1−/− mice and Rag1−/− mice previously complemented with CD4+ T cells using the complementary techniques of flow cytometry and high-throughput Raman spectroscopy. Both techniques reveal a mixed cell population after cell isolation and enrichment for IELs, with some subpopulations sharing high similarities between the treatment groups and other very distinct populations. Flow cytometry expectedly identified the transferred T lymphocyte population (TCRαβ+ CD4+ T cells) in the T cell transfer treatment group only. Importantly, Raman spectroscopy was able to describe a cell subpopulation that clearly displayed spectral characteristics of lymphocytes as major difference between cells isolated from Rag1−/− mice after T cell transfer and cells isolated from control mice without T cell transfer. Morphological features of those cells in bright field image further supported this assignment. The results are in excellent agreement with expectations from the applied mouse model.Overall, Raman spectroscopy holds high potential for advanced and novel immunological characterization of IEL populations ex vivo. Hence, Raman spectroscopy may help to ultimately gain so far undisclosed, specific insights into the regulation and biology of IELs during the course of gut inflammation.
Author contributions
UN, KH: Conceptualization, investigation, methodology, supervision; TV, TK, MD, KH: mouse model, IEL preparation and flow cytometry; SE, NA, AW: Raman measurement; IS, JP: Raman instrumentation; NA, RRG: Raman data analysis; all authors: discussion of results; UN, RRG, KH, NA: writing-original draft; all authors: reviewing and editing of manuscript. All authors have read and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the European Union (Grant No. 861122: “ImageIN”) and the BMBF via Photonics Research Germany (FKZ: Leibniz-IPHT LPI-BT4 13N15708) is acknowledged. We acknowledge support by the BMBF via the CSCC (FKZ 01EO1502), the Leibniz Association via the Leibniz ScienceCampus InfectoOptics (SAS-2015-HKI-LWC), the Jena Biophotonic and Imaging Laboratory (JBIL) and the Free State of Thuringia via ThIMEDOP (FKZ IZN 2018 0002). This work has also been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – TRR 241–375876048 (A08).References
- M. Paraskevaidi, B. J. Matthew, B. J. Holly, B. J. Hugh, C. P. V. Thulya, C. Loren, C. StJohn, G. Peter, G. Callum, K. G. Sergei, K. Kamila, K. Maria, L. M. G. Kássio, M.-H. L. Pierre, P. Evangelos, P. Savithri, A. A. John, S. Alexandra, S. Marfran, S.-S. Josep, T. Gunjan, W. Michael and W. Bayden, Appl. Spectrosc. Rev., 2021, 56, 804–868 CrossRef CAS.
- A. Silge, K. Weber, D. Cialla-May, L. Müller-Bötticher, D. Fischer and J. Popp, TrAC, Trends Anal. Chem., 2022, 153, 116623 CrossRef CAS.
- K. S. Lee, Z. Landry, F. C. Pereira, M. Wagner, D. Berry, W. E. Huang, G. T. Taylor, J. Kneipp, J. Popp, M. Zhang, J.-X. Cheng and R. Stocker, Nat. Rev. Methods Primers, 2021, 1, 80 CrossRef CAS.
- I. Pence and A. Mahadevan-Jansen, Chem. Soc. Rev., 2016, 45, 1958–1979 RSC.
- I. W. Schie, J. Ruger, A. S. Mondol, A. Ramoji, U. Neugebauer, C. Krafft and J. Popp, Anal. Chem., 2018, 90, 2023–2030 CrossRef CAS PubMed.
- C. Beleites, U. Neugebauer, T. Bocklitz, C. Krafft and J. Popp, Anal. Chim. Acta, 2013, 760, 25–33 CrossRef CAS PubMed.
- N. Chaudhary, C. Wynne and A. D. Meade, Biomed. Spectrosc. Imaging, 2020, 9, 23–31 Search PubMed.
- A. Ramoji, U. Neugebauer, T. Bocklitz, M. Foerster, M. Kiehntopf, M. Bauer and J. Popp, Anal. Chem., 2012, 84, 5335–5342 CrossRef CAS PubMed.
- G. J. Puppels, H. S. Garritsen, G. M. Segers-Nolten, F. F. de Mul and J. Greve, Biophys. J., 1991, 60, 1046–1056 CrossRef CAS PubMed.
- A. Dorosz, M. Grosicki, J. Dybas, E. Matuszyk, M. Rodewald, T. Meyer, J. Popp, K. Malek and M. Baranska, Cells, 2020, 9(9), 2041 CrossRef CAS PubMed.
- A. Borek-Dorosz, A. M. Nowakowska, P. Leszczenko, A. Adamczyk, A. Pieczara, J. Jakubowska, A. Pastorczak, K. Ostrowska, M. Ząbczyńska, K. Sowinski, W. I. Gruszecki, M. Baranska, K. M. Marzec and K. Majzner, J. Adv. Res., 2022, 41, 191–203 CrossRef CAS PubMed.
- A. J. Hobro, Y. Kumagai, S. Akira and N. I. Smith, Analyst, 2016, 141, 3756–3764 RSC.
- M. Chen, N. McReynolds, E. C. Campbell, M. Mazilu, J. Barbosa, K. Dholakia and S. J. Powis, PLoS One, 2015, 10, e0125158 CrossRef PubMed.
- N. Chaudhary, T. N. Q. Nguyen, D. Cullen, A. D. Meade and C. Wynne, Spectrochim. Acta, Part A, 2021, 248, 119118 CrossRef CAS PubMed.
- N. Topfer, M. M. Muller, M. Dahms, A. Ramoji, J. Popp, H. Slevogt and U. Neugebauer, Integr. Biol., 2019, 11(3), 87–98 CrossRef PubMed.
- N. Arend, A. Pittner, A. Ramoji, A. S. Mondol, M. Dahms, J. Ruger, O. Kurzai, I. W. Schie, M. Bauer, J. Popp and U. Neugebauer, Anal. Chem., 2020, 92, 10560–10568 CrossRef CAS PubMed.
- A. Pistiki, A. Ramoji, O. Ryabchykov, D. Thomas-Ruddel, A. T. Press, O. Makarewicz, E. J. Giamarellos-Bourboulis, M. Bauer, T. Bocklitz, J. Popp and U. Neugebauer, Int. J. Mol. Sci., 2021, 22, 10481 CrossRef CAS PubMed.
- A. Ramoji, O. Ryabchykov, K. Galler, A. Tannert, R. Markwart, R. P. Requardt, I. Rubio, M. Bauer, T. Bocklitz, J. Popp and U. Neugebauer, ImmunoHorizons, 2019, 3, 45–60 CrossRef CAS PubMed.
- A. Ramoji, D. Thomas-Ruddel, O. Ryabchykov, M. Bauer, N. Arend, E. J. Giamarellos-Bourboulis, J. Eugen-Olsen, M. Kiehntopf, T. Bocklitz, J. Popp, F. Bloos and U. Neugebauer, Crit Care Explor., 2021, 3, e0394 CrossRef PubMed.
- H. Cheroutre, F. Lambolez and D. Mucida, Nat. Rev. Immunol., 2011, 11, 445–456 CrossRef CAS PubMed.
- F. Chang, U. Mahadeva and H. Deere, APMIS, 2005, 113, 385–399 CrossRef PubMed.
- D. Corridoni, A. Antanaviciute, T. Gupta, D. Fawkner-Corbett, A. Aulicino, M. Jagielowicz, K. Parikh, E. Repapi, S. Taylor, D. Ishikawa, R. Hatano, T. Yamada, W. Xin, H. Slawinski, R. Bowden, G. Napolitani, O. Brain, C. Morimoto, H. Koohy and A. Simmons, Nat. Med., 2020, 26, 1480–1490 CrossRef CAS PubMed.
- R. Casalegno Garduño and J. Däbritz, Front. Immunol., 2021, 12, 738762 CrossRef PubMed.
- D. Kontoyiannis, G. Boulougouris, M. Manoloukos, M. Armaka, M. Apostolaki, T. Pizarro, A. Kotlyarov, I. Forster, R. Flavell, M. Gaestel, P. Tsichlis, F. Cominelli and G. Kollias, J. Exp. Med., 2002, 196, 1563–1574 CrossRef CAS PubMed.
- M. Apostolaki, M. Manoloukos, M. Roulis, M. A. Wurbel, W. Müller, K. A. Papadakis, D. L. Kontoyiannis, B. Malissen and G. Kollias, Gastroenterology, 2008, 134, 2025–2035 CrossRef CAS PubMed.
- M. F. Neurath, Nat. Immunol., 2019, 20, 970–979 CrossRef CAS PubMed.
- P. Kiesler, I. J. Fuss and W. Strober, Cell. Mol. Gastroenterol. Hepatol., 2015, 1, 154–170 CrossRef PubMed.
- P. Mombaerts, J. Iacomini, R. S. Johnson, K. Herrup, S. Tonegawa and V. E. Papaioannou, Cell, 1992, 68, 869–877 CrossRef CAS PubMed.
- Y. L. Chen, Y. T. Chen, C. F. Lo, C. I. Hsieh, S. Y. Chiu, C. Y. Wu, Y. S. Yeh, S. H. Hung, P. H. Cheng, Y. H. Su, S. T. Jiang, H. J. Chin and Y. C. Su, Sci. Rep., 2016, 6, 35635 CrossRef CAS PubMed.
- K. Hildner, B. T. Edelson, W. E. Purtha, M. Diamond, H. Matsushita, M. Kohyama, B. Calderon, B. U. Schraml, E. R. Unanue, M. S. Diamond, R. D. Schreiber, T. L. Murphy and K. M. Murphy, Science, 2008, 322, 1097–1100 CrossRef CAS PubMed.
- A. S. Mondol, S. F. El-Mashtoly, T. Frick, K. Gerwert, J. Popp and I. W. Schie, Analyst, 2019, 144, 6098–6107 RSC.
- R Core Team, R: A Language and Environment for Statistical Computing, Foundation for Statistical Computing, Vienna, Austria, 2021, https://www.R-project.org/ Search PubMed.
- H. Wickham, ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag, New York, 2016, https://ggplot2.tidyverse.org Search PubMed.
- B. Schloerke, D. Cook, J. Larmarange, F. Briatte, M. Marbach, E. Thoen, A. Elberg and J. Crowley, GGally: Extension to 'ggplot2', R package version 2.1.2, 2021, https://CRAN.R-project.org/package=GGally Search PubMed.
- A. Kassambara, ggpubr: ‘ggplot2′ Based Publication Ready Plots, R package version 0.5.0, 2022, https://CRAN.R-project.org/package=ggpubr Search PubMed.
- W. N. Venables and B. D. Ripley, Modern Applied Statistics with S, Springer Science+Business Media New York, 2002 Search PubMed.
- C. Beleites and V. Sergo, hyperSpec: a package to handle hyperspectral data sets in R, R package version 0.100.0, 2022, https://github.com/r-hyperspec/hyperSpec Search PubMed.
- H. Wickham, R. Francois, L. Henry and K. Müller, dplyr: A Grammar of Data Manipulation, R package version 1.0.10, 2022, https://CRAN.R-project.org/package=dplyr Search PubMed.
- S. Kucheryavskiy, Chemom. Intell. Lab. Syst., 2020, 198, 103937 CrossRef CAS.
- O. Ryabchykov, T. Bocklitz, A. Ramoji, U. Neugebauer, M. Foerster, C. Kroegel, M. Bauer, M. Kiehntopf and J. Popp, Chemom. Intell. Lab. Syst., 2016, 155, 1–6 CrossRef CAS.
- C. G. Ryan, E. Clayton, W. L. Griffin, S. H. Sie and D. R. Cousens, Nucl. Instrum. Methods Phys. Res., Sect. B, 1988, 34, 396–402 CrossRef.
- P. J. Morrissey, K. Charrier, D. A. Horovitz, F. A. Fletcher and J. D. Watson, J. Immunol., 1995, 154, 2678–2686 CrossRef CAS.
- Y. Tominaga, A. Fujiwara and Y. Amo, Fluid Phase Equilib., 1998, 144, 323–330 CrossRef CAS.
- A. Maguire, I. Vega-Carrascal, J. Bryant, L. White, O. Howe, F. M. Lyng and A. D. Meade, Analyst, 2015, 140, 2473–2481 RSC.
- S. Piqueras, L. Duponchel, R. Tauler and A. de Juan, Anal. Chim. Acta, 2011, 705, 182–192 CrossRef CAS PubMed.
- J. Felten, H. Hall, J. Jaumot, R. Tauler, A. de Juan and A. Gorzsás, Nat. Protoc., 2015, 10, 217–240 CrossRef CAS PubMed.
- V. V. Pully, A. T. M. Lenferink and C. Otto, J. Raman Spectrosc., 2011, 42, 167–173 CrossRef CAS.
- A. Weselucha-Birczynska, M. Kozicki, J. Czepiel and M. Birczynska, Analyst, 2013, 138, 7157–7163 RSC.
- V. Caprettini, J. A. Huang, F. Moia, A. Jacassi, C. A. Gonano, N. Maccaferri, R. Capozza, M. Dipalo and F. De Angelis, Adv. Sci., 2018, 5, 1800560 CrossRef PubMed.
- K. Czamara, K. Majzner, M. Z. Pacia, K. Kochan, A. Kaczor and M. Baranska, J. Raman Spectrosc., 2015, 46, 4–20 CrossRef CAS.
- C. Krafft, T. Knetschke, A. Siegner, R. H. W. Funk and R. Salzer, Vib. Spectrosc., 2003, 32, 75–83 CrossRef CAS.
- K. Enderle, M. Dinkel, E. M. Spath, B. Schmid, S. Zundler, P. Tripal, M. F. Neurath, K. Hildner and C. Neufert, Int. J. Mol. Sci., 2021, 22(10), 5148 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00074e |
| ‡ Shared senior authorship. |
| This journal is © The Royal Society of Chemistry 2023 |