
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA color-tunable single-benzene fluorophore-based sensor for sensitive detection of palladium in solution and living cells†
Paulina
Takacsova
 a,
Marie
Kudlickova Peskova
b,
Pavel
Svec
a,
Zbynek
Heger
*a and
Vladimir
Pekarik
*bc
a,
Marie
Kudlickova Peskova
b,
Pavel
Svec
a,
Zbynek
Heger
*a and
Vladimir
Pekarik
*bc
aDepartment of Chemistry and Biochemistry, Mendel University in Brno, Zemedelska 1, Brno CZ-613 00, Czech Republic. E-mail: heger@mendelu.cz
bInstitute of Physiology, Faculty of Medicine, Masaryk University, Kamenice 5, CZ-625 00, Brno, Czech Republic. E-mail: pekarik@mail.muni.cz
cCentral European Institute of Technology, Masaryk University, Kamenice 5, CZ-625 00 Brno, Czech Republic
First published on 14th March 2023
Abstract
Single-benzene fluorophores are bright and the smallest fluorochromes known so far. In single-benzene fluorophores, the fluorescence is mediated by the push/pull effect of substituting groups. Despite a plethora of advantageous properties, this group of molecules has not been extensively studied for design of high-performance fluorescent sensors of catalytic or enzymatic activities. Thus, herein, new fluorescent probes based on the Tsuji–Trost reaction were developed for the selective detection of palladium and other transition metals (platinum and gold) in an aqueous/organic mixed solvent with the sensitivity down to 2.5 nM (for palladium). The relative flexibility in the synthesis of these probes allows for facile color tuning of the emitted fluorescence. In this study, we have successfully utilized a yellow emission variant for sensitive detection of palladium under cell-free conditions and in living cells, validating its possible applicability for high-throughput optical sensing of catalysts for bioorthogonal chemistry under physiological conditions.
1. Introduction
Single-benzene fluorophores (SBFs) belong to a group of fluorophores whose fluorescence is not based on a system of conjugated pi-orbitals as is common for many other fluorescent compounds. In contrast, the fluorescence of SBFs is solely dependent on a push–pull system of electron withdrawing groups (EWG) and electron donating groups (EDG).1 The advantages of SBFs over other fluorophores are particularly their substantial quantum yield and very small molecular size, which is of utmost interest for imaging small metabolites/molecules in living cells.2 To date, the most common application of SBFs is in organic light-emitting diodes and other optoelectronic devices. This is also because unlike most luminescent organic materials, SBF fluorescence is not compromised by the aggregation-caused quenching effect.3To date, a plethora of SBF systems have been produced;1,4,5 however, it must be noted that they all suffer from laborious synthesis procedures. The venue for a broader use of these fluorophores was paved by the work by Xiang et al.,6 who devised a general strategy for a facile synthesis of SBFs based on tetrafluoroterephthalonitrile (TFTPN) providing a strongly electron-withdrawing nitrile group. Vicinal fluorine atoms of TFTPN can be easily substituted with electron-donating amino or hydroxyl groups.
In analytical chemistry, many fluorescent compounds have been used to construct sensors sensitive to a variety of analytes of interest. In this sense, the SBFs have been greatly overlooked. To date, to the best of our knowledge, only two known sensors are based on SBFs. The first one utilizes TFTPN to detect and differentiate biological thiols, cysteine, homocysteine and glutathione.7 The second sensor has been developed to detect hypochlorous acid.6
Palladium (Pd) is a transition metal with excellent catalytic properties frequently utilized in the organic synthesis of many industrially and pharmaceutically important compounds.8 In recent years, there has been growing interest in the use of Pd complexes in bioorthogonal reactions, i.e. artificial chemical reactions that can be carried out directly in biological systems. Pd-containing compounds can also be toxic to organisms and their widespread use in the production of many pharmaceutical compounds can represent a potential health and environmental risk.9 This fact prompted the development of many assays to detect residual amounts of Pd in pharmaceutical products or industrial wastewaters. These methods are mostly based on chelatometric reactions,10 or take advantage of the catalytic properties of Pd enabling Tsuji–Trost propargyl ether conversion to alcohols.11 Using these strategies, several probes have been designed to effectively detect Pd complexes in solutions,12,13 in cells14 or in complexes with biomolecules.15
In this work, we have designed and synthesized a color tunable SBF sensor for sensitive detection of Pd in solution. In addition, in this study, we demonstrate that our synthesised SBF sensor can be used for a facile, high-throughput analysis of bioorthogonal reactivity of Pd-based catalysts designed for bioorganometallic reactions in physiological milieu. This was validated with Pd-loaded poly(lactic-co-glycolic acid) (PLGA)-chitosan-based nanoparticles (PLGA-Chit NPs) whose catalytic activity was examined directly under cell culture conditions.
2. Results and discussion
2.1. SBF probe design and synthesis
The design of SBF probes has been inspired by the work published by Chaves et al.,16 who showed that terephthalic acid becomes fluorescent upon hydroxylation. Thus, we assumed that a similar compound based on terephthalonitrile-containing cyano groups, which are more inductively withdrawing than carboxyl groups, should yield a bright blue fluorescent compound. Klemes et al.17 demonstrated that TFTPN can be effectively converted into mono- and disubstituted ethers by modulating the speed of base addition during the synthesis. It is known that propargyl ethers and allyl ethers can be effectively cleaved by Pd in the Tsuji–Trost reaction with a subsequent release of alcohol.18 Therefore, we decided to synthesize propargyl ether-substituted TFTPN. TFTPN was reacted with propargyl alcohol in DMSO with sequential addition of K2CO3 (Scheme 1). Unfortunately, we were not able to obtain pure monosubstituted TFTPN. The reaction always yielded a mix of mono- and dipropargyl ethers (SBF-Pg and SBF-2 × Pg), which had to be separated by silica gel chromatography. When using 1.2 equivalents of propynol with 1 equivalent of TFTPN, the most prominent product was monopropargylated SBF-Pg, while a small amount of dipropargylated SBF-2 × Pg and a competing blue fluorescent hydroxylated product (probably SBF-Pg-OH) were visible on the TLC plate exposed to UV. After overnight incubation of the reaction mixture containing K2CO3 base at room temperature, phenolation of the mono-substituted product occurred and the presence of hydroxylated SBF-Pg-OH significantly increased (data not shown). Therefore, it was obvious that the extraction of SBF-Pg and SBF-2 × Pg has to be performed immediately after the synthesis. When the reaction was carried out using 2.4 equiv. of propynol, the monosubstituted SBF-Pg was transformed into SBF-2 × Pg and SBF-Pg-OH impurities. | ||
| Scheme 1 Design and synthesis of propargylated SBF probes for Pd detection. | ||
The deprotected hydroxy- and dihydroxy-terephthalonitrile exhibits bright blue emission (Fig. 1a, c and d and Fig. S2†), which is fully acceptable for in vitro analytical assays; however, it is problematic for any in vivo applications, in which strong autofluorescence is produced at short wavelength illumination. Xiang and co-workers have shown that secondary amine substituents shift the fluorescence of SBFs to the green region.6 Therefore, we have tried to color-tune the monopropargylated SBF-Pg with piperidine, which yielded a hybrid molecule SBF-PgP, which after its catalytic deprotection, emits yellow/green fluorescence (Fig. 1a, c and d and Fig. S3†). As shown in Fig. S4,† all deprotected SBFs are effectively excited at 380 nm. Deprotection (schematized in Fig. 1b) of SBF-Pg and SBF-2 × Pg with Pd leads to a shift of the absorption maximum from 320 nm to 380 nm, while in the case of SBF-PgP, the shift is not as prominent and the absorption maximum moves from 400 nm to 380 nm (Fig. S2†).
 | ||
| Fig. 1 (a) Digital images of wells containing probes in 50% ethanol or 50% DMSO without or with activation by Pd(OAc)2 in the presence of sodium borohydride (BH). (b) Schematic representation of depropargylation mediated by Pd. Normalized emission spectra of probes excited using the λexc of 380 nm in (c) 50% EtOH or (d) 50% DMSO. | ||
2.2. Response of SBF probes to Pd in various solvents and the specificity of sensing
The optical response of the probes to Pd was evaluated in two different mixed solvents: EtOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and DMSO/H2O (1:1). This was because SBF-Pg and SBF-2 × Pg were almost insoluble in pure water, and in previous experiments, we have noticed that the response to Pd and other metals is strongly influenced by a solvent.15 All synthesized SBF probes are virtually non-fluorescent, but upon contact with Pd in the presence of sodium borohydride (BH), the probes are rapidly deprotected into corresponding phenolates (Fig. 1b), yielding blue or yellow fluorescence (Fig. 1a). The Tsuji–Trost reaction is predominantly catalyzed by Pd, but it has been shown that other transition metals can also effectively cleave propargyl ethers. In order to determine the specificity of the synthesized SBF probes, we further analyzed the response of monopropargylated SBFs to various metals (Fig. 2). It is worth noting that SBF-Pg and SBF-PgP selectively responded to Pd and also to Pt in 50% DMSO. However, when the assay was carried out in 50% EtOH, detection capabilities of probes extended to Au and Ir and to a certain extent to Cu. These results corroborate the observation that the correct choice of solvent can dramatically improve the specificity of the assay. Quantitative measurements also revealed that the reaction sensitivity is markedly improved in 50% EtOH over DMSO (Fig. 2) despite its slightly decreased specificity.
:1) and DMSO/H2O (1:1). This was because SBF-Pg and SBF-2 × Pg were almost insoluble in pure water, and in previous experiments, we have noticed that the response to Pd and other metals is strongly influenced by a solvent.15 All synthesized SBF probes are virtually non-fluorescent, but upon contact with Pd in the presence of sodium borohydride (BH), the probes are rapidly deprotected into corresponding phenolates (Fig. 1b), yielding blue or yellow fluorescence (Fig. 1a). The Tsuji–Trost reaction is predominantly catalyzed by Pd, but it has been shown that other transition metals can also effectively cleave propargyl ethers. In order to determine the specificity of the synthesized SBF probes, we further analyzed the response of monopropargylated SBFs to various metals (Fig. 2). It is worth noting that SBF-Pg and SBF-PgP selectively responded to Pd and also to Pt in 50% DMSO. However, when the assay was carried out in 50% EtOH, detection capabilities of probes extended to Au and Ir and to a certain extent to Cu. These results corroborate the observation that the correct choice of solvent can dramatically improve the specificity of the assay. Quantitative measurements also revealed that the reaction sensitivity is markedly improved in 50% EtOH over DMSO (Fig. 2) despite its slightly decreased specificity.
 | ||
| Fig. 2 Response of (a) SBF-Pg and (b) SBF-PgP to various metal ions in mixed solvents in the presence of BH. | ||
2.3. Evaluation of the sensitivity of SBF probes
In order to determine the sensitivity of SBF probes, we performed the reaction with various amounts of Pd(OAc)2. Fig. 3 shows that the detection limit of SBF-Pg and SBF-PgP lies in the range of 5 nM, which proved them extremely sensitive.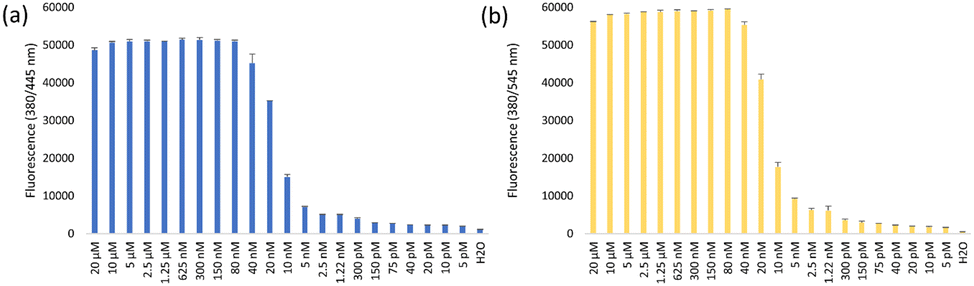 | ||
| Fig. 3 The sensitivity of (a) SBF-Pg and (b) SBF-PgP to various concentrations of Pd. The reaction mixture contained 100 μM SBF probes and annotated concentrations of Pd(OAc)2 in 50% EtOH. | ||
2.4. Response of SBF probes to various concentrations of BH
The Tsuji–Trost reaction relies on the cycling of Pd oxidative species between Pd0 and Pd2+. After the release of the activated product of SBF probes, the regeneration of Pd0 from Pd2+ is essential for its functioning in the next catalytic cycle. That is why reducing agents such as BH are used. However, an excess of BH might prevent the oxidation step of the reaction, which would interfere with the following cycle and SBF probe depropargylation. This could subsequently lead to a decrease in the sensitivity. Hence, we have further evaluated the sensitivity of the assay in the presence of various concentrations of BH. Notably, the previously obtained results (Fig. 2) revealed that Pt could activate the SBF probes too. Therefore, we included several therapeutically relevant Pt-based anticancer compounds (cisplatin, carboplatin and oxaliplatin) to examine possible applicability of our assay for their analysis.Indeed, we found out that Pd complexes effectively activated the SBF-2 × Pg probe at 200 μM BH, and according to the expectations, the increasing BH concentrations dramatically reduced the sensitivity of the assay (Fig. 4). From the data, it was obvious that the reaction was effectively completed in 5 min, and extended incubation for up to 45 min did not improve the performance of the assay. The reduced fluorescence at elevated BH concentrations can be explained by that either the conversion of Pd2+ to Pd0 was prevented, therefore reducing the catalyst recovery efficiency or alternatively the product of the reaction was further catalytically converted into a non-fluorescent species. Interestingly, this was not the case for Pt compounds. K2PtCl4 effectively activated the probe in 500 μM BH and even at higher BH concentrations (at 5 min incubation). The activated fluorescence was reduced upon extended reaction time in a similar manner as observed for Pd compounds. On the other hand, the Pt-based anticancer compounds required much higher concentrations of BH, i.e. up to 2 mM and extended reaction time to activate the probe. Therefore, the obtained data underpin that the reduction of Pt2+ to Pt0 in organometallic complexes requires higher concentrations of BH and longer reaction time. Taken together, these results confirmed that our synthesized SBF probes could be effectively used not only for determination of Pd but also for quantitative measurements of clinically important anticancer Pt-based drugs.
 | ||
| Fig. 4 Effect of the BH concentration on the sensitivity of the SBF assay. Various Pd and Pt compounds were used to activate the SBF-2 × Pg sensor in the presence of different concentrations of BH in 50% EtOH. The microtitration plate was incubated at ambient temperature and fluorescence was evaluated after (a) 5 min and (b) 45 minutes. | ||
2.5. The use of SBF-PgP for the detection of intracellular bioorthogonal activity of Pd
Pd complexes have shown their extensive potential in bioorthogonal reactions that might be used for site-specific prodrug activation not only in anticancer therapy.19,20 Hence, we were curious if a yellow fluorescent probe SBF-PgP could be used to detect catalytically active Pd species directly in cells. For this purpose, we encapsulated bis(trifurylphosphine)Pd (Pd-TFP) in PLGA-Chit NPs.21 PLGA nanoparticles are widely used biocompatible nanocarriers, and chitosan incorporation into the shells of these particles is frequently utilized to improve their cellular uptake. Due to slow hydrolysis, PLGA also protect the bulk of the catalyst from thiol poisoning, which is a common problem in metal-catalyzed bioorthogonal reactions. The empty PLGA-Chit NPs and PLGA-Chit NPs loaded with Pd-TFP were produced as a relatively homogeneous population with a mean diameter of 300 nm (Fig. 5a–c). For experiments, HEK-293 cells were incubated with PLGA-Chit NPs overnight. The following day, the cells were detached with trypsin, split on a new 24-well plate and allowed to attach. After attachment, the cells were incubated with 30 μM SBF-PgP overnight. | ||
| Fig. 5 PLGA-Chit NP characterization and bioorthogonal activation of SBF-PgP in living cells. SEM micrographs of (a) empty PLGA-Chit NPs and (b) PLGA-Chit NPs loaded with Pd-TFP. (c) PLGA-Chit NPs were also characterized using dynamic light scattering. HEK-293 cells were treated with (d–f) empty PLGA-Chit NPs or (g–i) Pd-TFP loaded PLGA-Chit NPs. (d and g) Bright field and (e and h) Hoechst 33258 nuclei counterstaining demonstrate cellular morphology and location. (f) PLGA-Chit-NPs did not activate SBF-PgP in cells, while (i) Pd-TFP-loaded PLGA-Chit-NPs efficiently activated the SBF-PgP probe, and the activation of the probe is directly detectable as an increase in green fluorescence. | ||
The subsequent microscopy analysis of living cells revealed a distinct mostly cytoplasmic green fluorescence increase in cells treated with Pd-TFP loaded PLGA-Chit NPs, while no detectable fluorescence was observed in cells treated with empty PLGA-Chit NPs only (Fig. 5d–i). These results clearly demonstrated that SBF-PgP can be effectively used to detect Pd catalytic activity in living cells.
3. Experimental
3.1. Chemicals
Unless otherwise stated, all reagents and solvents were purchased from commercial suppliers and used without further purification.3.2. Synthesis of SBF-Pg
To a solution of TFTPN (100 mg, 0.05 mmol, 1 equiv.) and 2-propyn-1-ol (33.6 mg, 0.6 mmol, 1.2 equiv.) in 3 mL of dried dimethyl sulfoxide (DMSO) at 80 °C, K2CO3 was successively added in 10 mg aliquots in 30 minute intervals (71 mg, 0.51 mmol, 1.02 equiv.). Reaction progress was monitored by thin layer chromatography (TLC) (Polygram SIL G – 0.2 mm Silica gel 60) with toluene as the mobile phase. The products were visualised by exposure of the TLC plate to diethylamine vapors (Fig. S1†), producing yellow, dim green fluorescent spots detectable under UV light. After cooling, 20 mL of water was added to the reaction mix and the product was extracted 4 times with 3 mL of diethylether, dried using Na2SO4 and evaporated. The dried products were solubilized in toluene and the residue was purified using a silica gel column, using toluene to afford compound SBF-Pg as a pale yellow/green oil (42 mg, 35%). 1H NMR (300 MHz, CDCl3) δ 5.00 (d, J = 2.5 Hz, 2H), 2.64 (t, J = 2.4 Hz, 1H). 19F NMR (282 MHz, CDCl3) δ −123.08.3.3. Synthesis of SBF-2 × Pg
This compound was prepared by the same procedure as that for SBF-Pg; however, with an increased amount of 2-propyn-1-ol (67 mg, 1.2 mmol, 2.4 equiv.) and 15 mg aliquots of K2CO3 added in 20 minute intervals (138 mg, 1 mmol, 2 equiv.). The residue was purified using a silica gel column, using toluene/petrolether (v/v, 4:1) as a white solid (28 mg, 31%). 13C NMR (126 MHz, CDCl3) δ ppm: 62.16 (t, 2 C); 75.84 (2 C); 79.00 (2 C); 103.86 (dd, 2 C); 108.85 (d, 2 C); 142.49 (dd, 2 C); 151.69 (dd, 2 C). 19F NMR (471 MHz, CDCl3) δ ppm: −123.06 (s, 2 F). 1H NMR (500 MHz, CDCl3) δ ppm: 2.64 (t, 2 H); 5.00 (d, 4 H).
3.4. Synthesis of SBF-PgP
SBF-Pg (236 mg, 1 mmol, 1 equiv.) synthesized in the previous step was dissolved in 5 mL of acetonitrile. Piperidine (255 mg, 3 mmol, 3 equiv.) was added and the reaction mixture was mixed at room temperature for 3 hours. After that, the solvent was evaporated and the compound was purified through a silica gel chromatography column, using toluene as the eluent (262 mg, 87%). 1H NMR (500 MHz, CDCl3) δ ppm 1.65 (br t, J = 5.34 Hz, 2 H), 1.74 (quin, J = 5.53 Hz, 4 H), 2.63 (t, J = 2.44 Hz, 2 H), 3.25–3.34 (m, 3 H); 4.89–5.01 (m, 2 H). 13C NMR (126 MHz, CDCl3) δ ppm 6.01 (s, 1 C); 6.14 (s, 1 C); 6.31 (s, 1 C); 6.58 (s, 1 C); 6.70 (s, 1 C); 7.17 (s, 1 C); 7.47 (s, 1 C); 7.82 (s, 1 C); 8.07 (s, 1 C); 8.88 (br dd, J = 48.26, 27.12 Hz, 1 C), 10.16 (br dd, J = 74.92, 58.37 Hz, 1 C), 10.43 (s, 1 C); 10.84 (s, 1 C); 11.64 (br dd, J = 87.32, 37.69 Hz, 1 C), 11.62 (s, 1 C); 12.33 (s, 1 C); 12.76 (s, 1 C); 12.90 (s, 1 C); 13.28 (s, 1 C); 13.96 (br dd, J = 56.07, 19.30 Hz, 1 C), 14.76 (s, 1 C); 15.39 (s, 1 C); 16.18 (br dd, J = 94.22, 43.66 Hz, 1 C), 16.59 (s, 1 C); 16.84 (s, 1 C); 17.27 (s, 1 C); 17.44 (s, 1 C); 17.67 (s, 1 C); 18.07 (s, 1 C); 18.44 (s, 1 C); 18.63 (s, 1 C); 20.03 (br dd, J = 182.92, 91.00 Hz, 1 C), 19.43 (s, 1 C); 19.77 (s, 1 C); 19.90 (s, 1 C); 20.10 (s, 1 C); 20.78 (s, 1 C); 21.45 (s, 1 C); 21.86 (s, 1 C); 22.02 (s, 1 C); 22.17 (s, 1 C); 22.56 (s, 1 C); 23.14 (s, 1 C); 23.32 (s, 1 C); 23.60 (s, 1 C); 23.99 (s, 1 C); 24.44 (s, 1 C); 24.68 (s, 1 C); 26.34 (s, 1 C); 52.92 (s, 1 C); 52.96 (s, 1 C); 61.96 (s, 1 C); 62.01 (s, 1 C); 75.81 (s, 1 C); 76.32 (s, 1 C); 77.84 (s, 1 C); 78.39 (s, 1 C); 78.52 (s, 1 C); 103.00 (s, 1 C); 109.60 (s, 1 C); 111.27 (s, 1 C); 125.28 (s, 1 C); 128.21 (s, 1 C); 129.02 (s, 1 C); 137.87 (s, 1 C); 152.61 (s, 1 C); 153.49 (s, 1 C).3.5. Preparation of Pd-loaded PLGA-Chit NPs
PLGA-Chit NPs were prepared using a diffusion emulsification method. Briefly, 30 μL of PLGA dissolved in acetonitrile (50 mg mL−1) was added to 40 μL of Pd(TFP)2Cl2 catalysts (25 mM) dissolved in dimethylformamide (DMF) to form an organic phase. 1 mL of aqueous solution of low molecular weight chitosan polymer (0.1%) and polyvinyl alcohol as a stabilizer (1%) dissolved in 0.5% acetic acid was continuously injected into the organic phase. The emulsion was homogenized by vortexing at the maximum speed and then incubated 10 min at room temperature. The newly formed particles were harvested from the homogenized emulsion using centrifugation (15 min, 10000 rpm) and resuspended in 200 μL of sucrose (250 mM). This suspension was further centrifuged (5 min, 1000 rpm) to remove large chitosan aggregates.
3.6. Physicochemical characterization of PLGA-Chit NPs
The size and zeta potential of PLGA-Chit NPs was measured using a Zetasizer Nano ZS instrument (Malvern Instruments Ltd, Worcestershire, UK). For the size measurement, samples of nanoparticle suspensions were diluted 200 times in water. All measurements were taken at 25 °C with an equilibration time of 120 seconds. Measurements were taken in quintuplicate. The light scattering data were interpreted using Gaussian analysis. Particle sizes are expressed as number-weighted diameters. For zeta potential determination, the measurements were performed using a Smoluchowski model. The number of measurements was in the range from 20 to 60 runs. Measurements were performed in triplicate. Further, the PLGA-Chit NPs were examined for their size and morphology using scanning electron microscopy (SEM). For all samples, 10 μL of particle suspension was diluted with 300 μL of water and washed 3 times with 500 μL of water using centrifugation (15 minutes at 10000 rpm). The final pellet was recovered in 100 μL of water. The sample was applied on a silicon wafer purchased from Siegert Wafer Company and allowed to dry at laboratory temperature (20–25 °C). This wafer was adhered using carbon tape to the stub that was inserted into the SEM MAIA 3 instrument equipped with a field emission gun (Tescan, Brno, Czech Republic).
3.7. Analysis of probe activation properties under cell-free conditions
The synthesized probes were prepared as 10 mM stock solutions in DMF. The activation reactions were carried out in a 96-well plate in a total volume of 200 μL with 100 μM probe and 10 μM metal ions, Pd catalysts or anticancer platinum (Pt)-based drugs. The reactions were carried out in a mixed solvent of 50% ethanol or DMSO in water at room temperature for 30–60 min in the presence of 200 μM BH unless stated otherwise.3.8. Evaluation of SBF-PgP activation in cells
HEK-293 cells were cultivated in complete media (DMEM/F12 supplemented with 10% FBS, 1× pen/strep) in a humidified incubator with 5% CO2. For the probe activation experiments, the HEK-293 cells were seeded in a 6-well plate at 50% confluence. After 4 h, the cultivation media were replaced with 500 μL of complete cultivation media containing 50 μL of PLGA-Chit NPs with or without Pd(TFP)2Cl2 acting as a bioorthogonal catalytic payload. After overnight incubation, the cells were split to 50% confluence in a 24-well plate and were further allowed to attach for 6 h. The cultivation media were replaced again with 500 μL of a mixture of complete cultivation media containing Hoechst 33258 (2 μg mL−1) and SBF-PgP probe diluted from 50 mM DMF stock solution to a final concentration of 30 μM in the cell culture media. After overnight incubation, the micrographs were captured using a fluorescence microscope Olympus IX53 equipped with an Olympus U-HGLGPS lamp (Olympus, Tokyo, Japan).4. Conclusions
Three new fluorescent probes based on SBF were designed, synthesized and characterized. All probes can be used for a rapid and specific detection of Pd and Pt ions and also organometallic complexes in vitro with an excellent sensitivity of 5 nM. We show that the sensitivity and also selectivity are influenced by the solvent system used in the assay. We have also successfully evaluated the BH concentration that is optimal for the reaction outcomes. It was found out that elevated concentrations of BH exert inhibitory effects on the detection of Pd compounds, while the detection of anticancer Pt complexes (cisplatin, carboplatin and oxaliplatin) requires higher BH concentrations. Finally, it was found that the green fluorescent SBF-PgP probe can be effectively used to detect Pd catalytic activity in living cells, making it a useful sensor for facile, high-throughput evaluation of new catalysts with presumed bioorthogonal activity.Author contributions
PT, MKP and PS performed all experiments. ZH and VP contributed to the planning and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Health Research Council (NU21J-08-00043) and the Internal Grant Agency of Mendel University in Brno (project no. AF-IGA2021-IP059). We also acknowledge CIISB research infrastructure (project LM2018127) funded by MEYS CR for the measurements at the Josef Dadok National NMR Centre.References
- T. Beppu, K. Tomiguchi, A. Masuhara, Y. J. Pu and H. Katagiri, Angew. Chem., Int. Ed., 2015, 54, 7332–7335 CrossRef CAS PubMed.
- S. Benson, A. Fernandez, N. D. Barth, F. de Moliner, M. H. Horrocks, C. S. Herrington, J. L. Abad, A. Delgado, L. Kelly, Z. Y. Chang, Y. Feng, M. Nishiura, Y. Hori, K. Kikuchi and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 6911–6915 CrossRef CAS PubMed.
- J. Kim, J. H. Oh and D. Kim, Org. Biomol. Chem., 2021, 19, 933–946 RSC.
- C. H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed.
- B. Tang, H. Liu, F. Li, Y. Wang and H. Zhang, Chem. Commun., 2016, 52, 6577–6580 RSC.
- Z. Xiang, Z. Y. Wang, T. B. Ren, W. Xu, Y. P. Liu, X. X. Zhang, P. Wu, L. Yuan and X. B. Zhang, Chem. Commun., 2019, 55, 11462–11465 RSC.
- H. T. Zhang, R. C. Liu, J. Liu, L. Li, P. Wang, S. Q. Yao, Z. T. Xu and H. Y. Sun, Chem. Sci., 2016, 7, 256–260 RSC.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zeccal, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- J. Kielhorn, C. Melber, D. Keller and I. Mangelsdorf, Int. J. Hyg. Environ. Health, 2002, 205, 417–432 CrossRef CAS PubMed.
- Y. M. Zhou, Q. Huang, Q. Y. Zhang, Y. H. Min and E. Z. Wang, Spectrochim. Acta, Part A, 2015, 137, 33–38 CrossRef CAS PubMed.
- Z. Y. Xu, J. Li, S. Guan, L. Zhang and C. Z. Dong, Spectrochim. Acta, Part A, 2015, 148, 7–11 CrossRef CAS PubMed.
- W. R. Kitley, P. J. Santa Maria, R. A. Cloyd and L. M. Wysocki, Chem. Commun., 2015, 51, 8520–8523 RSC.
- B. L. Huo, M. Du, A. J. Gong, M. W. Li, L. Q. Fang, A. Shen, Y. R. Lai, X. Bai and Y. X. Yang, Anal. Methods, 2018, 10, 3475–3480 RSC.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Chem. Commun., 2016, 52, 14212–14214 RSC.
- V. Pekarik, M. Peskova, J. Duben, M. Remes and Z. Heger, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- M. R. B. Chaves, M. L. S. O. Lima, L. Malafatti-Picca, D. A. de Angelis, A. M. de Castro, E. Valoni and A. J. Marsaioli, J. Braz. Chem. Soc., 2018, 29, 1278–1285 CAS.
- M. J. Klemes, Y. H. Ling, M. Chiapasco, A. Alsbaiee, D. E. Helbling and W. R. Dichtel, Chem. Sci., 2018, 9, 8883–8889 RSC.
- S. Akkarasamiyo, S. Sawadjoon, A. Orthaber and J. S. M. Samec, Chem. – Eur. J., 2018, 24, 3488–3498 CrossRef CAS PubMed.
- J. T. Weiss, N. O. Carragher and A. Unciti-Broceta, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- M. A. Miller, H. Mikula, G. Luthria, R. Li, S. Kronister, M. Prytyskach, R. H. Kohler, T. Mitchison and R. Weissleder, ACS Nano, 2018, 12, 12814–12826 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00046j |
| This journal is © The Royal Society of Chemistry 2023 |