
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a modularized aptamer targeting the nuclear T-cell suppressor PAC1†
Zixi
Hu‡
,
Zhongyu
Jiang‡
,
Zeliang
Yang
,
Liang
Liu
,
Zhenyu
Zhu
,
Yan
Jin
and
Yuxin
Yin
 *
*
Institute of Systems Biomedicine, Department of Pathology, School of Basic Medical Sciences, Peking-Tsinghua Center for Life Sciences, Beijing Key Laboratory of Tumor Systems Biology, Peking University Health Science Center, Beijing 100191, China. E-mail: yinyuxin@bjmu.edu.cn
First published on 16th May 2023
Abstract
Aptamers associated with cancer targeting therapy are commonly focused on cell membrane proteins; however, the study of intracellular, particularly, nuclear proteins is limited. The nuclear phosphatase PAC1 has been reported to be a potential T cell-related immunotherapeutic target. Here, we identified an aptamer, designated as PA5, with high affinity and specificity for PAC1 through the systematic evolution of ligands by exponential enrichment (SELEX) procedure. We then developed a dual-module aptamer PAC1-AS consisting of a cell-internalizing module and a targeting module, which can recognize PAC1 in the nucleus under physiological conditions. This modularized aptamer raises the possibility of manipulating endosomes and provides insights into the exploration and development of an efficient cancer immunotherapy approach.
Introduction
Phosphatase of activated cells 1 (PAC1), also known as DUSP2 (dual-specificity phosphatase 2), is a mitogen-induced nuclear protein threonine/tyrosine phosphatase.1,2 It is the only member of the DUSP family that is exclusively expressed in immune systems.3,4 In a previous study, we illustrated that PAC1 suppresses the transcription of effector T cell genes by recruitment of the nucleosome-remodeling-deacetylase (NuRD) complex, leading to T cell dysfunction and tumor progression. Thus, PAC1 functioned as an immune checkpoint to downregulate the anti-tumor capability of T cells. In a tumor microenvironment, effector T cells expressing high levels of PAC1 switch to “exhausted conditions”, which is correlated with poor prognosis. A study indicated that the depletion of PAC1 could enhance the activity of effector T cells and evidently suppress tumor growth in mice.5 Therefore, PAC1 is a prospective candidate for drug development in cancer immunotherapy. Since there is no effective targeted drug of PAC1, the development of effective molecular probes specifically recognizing and influencing the function of PAC1 is of great significance for reversing T cell exhaustion to promote cancer immunotherapy.Early efforts to develop targeted diagnostic and therapeutic molecular probes were mainly focused on monoclonal antibodies.6,7 However, antibodies are easily inactivated during storage or chemical modification, and often lack satisfactory pharmacokinetics.8 In addition, it is a large obstacle for antibodies (10–15 nm) to cross the cell or even the nuclear membrane in living cells.9 These disadvantages limit the application of monoclonal antibodies to efficiently bind proteins within the cell.
Aptamers, often called chemical antibodies, are short single-stranded oligonucleotides that fold into unique three-dimensional conformations to identify the corresponding targets.8–11 As a promising alternative to antibodies, aptamers have characteristics or advantages of smaller size (∼3 nm), flexible structure, high chemical stability, easy synthesis and modification.12–14 These oligonucleotides can serve as antagonists or agonists of ligands, or as targeted delivery vehicles for imaging agents and drugs for precision medicine.15–18 Usually, aptamers are screened from a random oligonucleotide library by systematic evolution of ligands by exponential enrichment (SELEX), which progressively evolves the sequences with high affinity and specificity.19,20 At present, aptamers have been widely used in sensing, purification, diagnosis, drug delivery and targeted therapy.21–23 Although internalized aptamers can be screened from some special methods,24,25 many excellent aptamers still have some obstacles for internalization.26,27 Due to the electrostatic cell–surface repulsion, the negatively charged phosphate backbone of the aptamers hinders their internalization into cells. Therefore, most aptamers that target a non-membrane component alone cannot be internalized into cells. In particular, the nucleolin aptamer AS1411 can enter the cell by macropinocytosis, while other cell-internalizing aptamers are often taken in via membrane receptor-mediated endocytosis, which is dependent on cell metabolism.28,29 The reason is that the sequence of AS1411 is rich in G, which forms the special structure G-quadruplex for easy internalization. It can freely shuttle between the membrane and cytoplasm, which makes it a great candidate for delivery of therapeutic agents.28–30
Here, we first screened out the aptamer PA5 that specifically recognizes PAC1 by the SELEX system. Furthermore, we developed a modular aptamer PAC1-AS consisting of the optimized sequence PA5c and the cell-internalization module AS1411, with the purpose of increasing the internalization efficiency. The PAC1-AS aptamer could precisely target PAC1 in the nucleus under physiological conditions. This modular aptamer system provides an ideal strategy for the further application of aptamers to regulate the physiological functions of nuclear proteins.
Materials and methods
Cell culture and generation of stable cell lines
Human T lymphocyte leukemia cells (Jurkat) and human embryonic kidney cells (HEK293T) were purchased from American Type Culture Collection (ATCC). The cells were cultured at 37 °C under a 5% CO2 atmosphere in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, PAN Biotech). The insect cell line Sf9 was obtained from Invitrogen (B82501) and cultured in Grace's insect medium (Gibco).For transient transfection, HEK293T cells were seeded in a 6-well plate (5 × 105 cells per well) and after 12 h, they were transfected with pCCL-mCherry-PAC1 or pCCL-PAC1-Flag using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. For a stable transfection, the cells were generated from infection with lentivirus and then sorted using a flow cytometer (BD Biosciences).
Protein expression and purification
The sequences encoding the human PAC1-N-terminal (Met1-Arg160) were cloned into a vector pET-28a with six histidine residues at the N-terminal. The proteins were expressed using the Bac-to-Bac Baculovirus expression system (Invitrogen), and then purified by Ni-NTA column (GE Healthcare) chromatography and size exclusion chromatography (Superdex 200).DNA library and primers
The SELEX library and primers were designed according to previous reports.31 The primary library consisted of a 40-nt random sequence flanked by two 18-nt constant regions with the primer sequences for amplification (5′-ATCCAGAGTGACGCAGCA-N40-TGGACACGGTGGCTTAGT-3′). The forward primer labeled carboxyfluorescein (FAM) at the 5′ end is 5′-FAM-ATCCAGAGTGACGCAGCA-3′ and the reverse primer labeled biotin at the 5′ end is 5′-biotin-TGGACACGGTGGCTTAGT-3′. All oligonucleotides were synthesized by Shanghai Biological Technology Co. (Shanghai, China) and purified by high-performance liquid chromatography. The PCR conditions were as follows: starts at 95 °C for 3 min to activate Taq DNA polymerase, then at 95 °C for 30 s, at 56.3 °C for 30 s and at 72 °C for 30 s for each amplification cycle, followed by a final extension at 72 °C for 5 min.Protein-immobilized beads
5.0 μL of stock Ni Sepharose high performance beads (∼2 × 105 Ni-beads, GE Healthcare) was equilibrated with 500.0 μL protein buffer (PBS containing 30 mM imidazole, pH 8.0) three times. The remaining beads were suspended in 100.0 μL of protein buffer containing the designated protein (600.0 μg mL−1 or 300.0 μg mL−1) and incubated for 1 h at 4 °C on a shaker. The prepared protein-immobilized beads (P-beads) were used for aptamer selection as the positive target, while the bare Ni-beads were used as the negative target.Aptamer selection
SELEX was performed using the ssDNA library binding to the P-beads and DNA survivors were collected after several washing steps, followed by amplification by PCR (Fig. 1). For the first round, an initial ssDNA library (5.0 nmol) of randomized sequences was used as a starting pool. For later rounds, 1.0 nmol to 0.2 nmol of survivors from the previous round was used as the secondary library. The selection procedure was performed as described.32 In the first three rounds of selection, we immobilized the PAC1-N protein on Ni-beads (P-beads) as the target for positive selection. This step can absorb more potential ssDNA sequences, and ensure sufficient ssDNA sequences in subsequent selection. Starting from the fourth round, a negative selection only with Ni-beads was added before the positive selection to winnow away non-specific sequences. After negative and positive screening of each round, the eluted DNA was amplified by PCR. The resulting biotin–dsDNA products were bound to streptavidin-beads (GE Healthcare) and separated using 0.2 M NaOH to obtain the ssDNA pool. | ||
| Fig. 1 Scheme of systematic enrichment of aptamers against PAC1. The ssDNA library was incubated with Ni-beads (negative selection) to remove negative binding sequences. The unbound sequences were incubated with PAC1-N protein-immobilized Ni-beads for positive selection. Then the bound DNAs were retained for PCR. During amplification, the dsDNA pool was modified with biotin and the ssDNA pool was prepared using streptavidin-beads. DNA sequencing and screening was performed to identify the potential aptamers (PA pool). | ||
After 8 rounds of consecutive selection, the final DNA pool was amplified and cloned using a previously described method.19 50 clones were randomly picked for sequencing (TSINGKE Biological Technology, China), and the data were analyzed using Clustal X 2.0.3 software for highly conserved regions. The sequences with a high degree of enrichment as the aptamer candidates (PA pool) were used for further study. The entire process is summarized in Table S1.†
Aptamer sequence optimization
The secondary structure of aptamers was predicted using NUPACK software (https://www.nupack.org). The candidate PA5 was partially truncated from the 3′ and 5′ ends based on its secondary structure. The truncated aptamers (Table S3†) were analyzed for target binding capacity by flow cytometry.PA5c with the linker sequence GCGAAAACGC at the 5′ end and AS1411 with the linker sequence GCGTTTTCGC at the 5′ end (Table S4†) were used. PAC1-AS was formed by mixing PA5c and AS1411 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) in annealing buffer (10.0 mM Tris, 50.0 mM NaCl and 1.0 mM EDTA, pH 8.2) to a concentration of 100.0 μM at 95 °C for 5 min, and then cooling to room temperature (RT) slowly.
:1 ratio) in annealing buffer (10.0 mM Tris, 50.0 mM NaCl and 1.0 mM EDTA, pH 8.2) to a concentration of 100.0 μM at 95 °C for 5 min, and then cooling to room temperature (RT) slowly.
To increase the nuclease resistance of PAC1-AS, three bases at the 3′ termini of PA5c or AS1411 were replaced with phosphorothioate oligonucleotides (Table S4†). 3.0 μM (final DNA concentration) of PAC1-AS or phosphorothioated-PAC1-AS (PAC1-AS-p) were incubated in RPMI-1640 medium with 10% FBS for different times at 37 °C. Samples were flashed to −80 °C at the assigned time and run on 3% agarose gels until all samples were harvested. The band density was measured by a molecular imaging analysis system (Bio-Rad).
Aptamer binding assay
Flow cytometry was used to monitor the enrichment of the evolved library in the course of selection and verify the binding specificity of the aptamer candidates. For each sample, 10.0 μL of P-beads or bare beads was washed with 500.0 μL of cold-PBS three times. The prepared beads were incubated with 250.0 nM FAM-aptamer in 100.0 μL of binding buffer (PBS supplemented with 5.0 mM of MgCl2, 0.1 mg mL−1 of yeast tRNA, and 1.0 mg mL−1 of BSA) for 30 min at 4 °C. Then, the beads were washed twice and suspended in 300.0 μL of PBS. The fluorescence intensity was analyzed using a microscope (Nikon) or a flow cytometer (BD Immunocytometry Systems).The equilibrium dissociation constant (Kd) of the aptamer–protein interactions was determined by flow cytometry. The P-beads were incubated with different concentrations of the FAM-labeled aptamer in 100.0 μL of binding buffer at 4 °C for 30 min. After washing three times, the cells were resuspended and analyzed. The Kd was determined by fitting the dependence of the mean fluorescence intensity of specific binding with the equation Y = BmaxX/(Kd + X) using SigmaPlot software.
Confocal microscopy imaging
The glass slides were placed in 6-well plates and the cells were seeded on the glass at a density of 1.0 × 106 cells per well for 12 h. For suspension cells, poly-D-lysine (Sigma) pretreatment was performed on the glass slides to promote cell adhesion. The cells were fixed with 4% polyoxymethylene (Sigma) for 15 min after washing with PBS twice, and then permeabilized for 10 min with 0.5% Triton X-100 (Sigma) in PBS. The slides were blocked with 1% BSA in PBS for 1 h after rinsing twice with PBS. The cells were incubated with the FAM-labeled aptamer or library (250.0 nM) for 1 h at RT and stained with 4′,6-diamidino-2-phenylindole (DAPI, Life Co.). After washing twice, the slides were removed from plates and mounted with coverslips. The cells’ fluorescence imaging analysis was performed with a confocal microscope (A1; Nikon, Tokyo, Japan). For internalization analysis, the FAM-labeled aptamer or library (250.0 nM) was incubated with the cells for 3 h before fixing.Western blot and the aptamer-pull down assay
The expression of PAC1 in cells was measured by western blot. The cell lysates were resolved by 10% SDS-PAGE and transferred onto a nitrocellulose filter. The immunoblots were incubated with the anti-PAC1 antibody from our laboratory or the tag antibody (anti-flag, Sigma; anti-HA, Sigma; anti-GFP, Sigma), and then incubated with the peroxidase-conjugated secondary antibody. The signals were detected with enhanced chemiluminescence (Santa Cruz).For the pulldown experiments, the biotinylated aptamer (250.0 nM) was incubated with the cell lysates for 4 h at 4 °C. Then the mixture was incubated with streptavidin-beads for 3 h at 4 °C. After washing three times, the beads were collected and the samples were subjected to SDS-PAGE for immunoblot analysis.
Quantitative RT-PCR
Jurkat cells expressing a control vector (J-Mock) were stimulated with 0.1 μg mL−1 PMA and 0.5 μg mL−1 ionomycin for 3 h as described.5 mRNA was isolated from cells with Trizol reagent (Invitrogen) and cDNA was obtained with the Reverse Transcription System (Promega).33 The SYBR Green Supermix (TransGen Biotech) was used for real-time PCR. The reverse-transcription product was analyzed with gene-specific primers on an ABI 7500 system (Applied Biosystems). The sequences of the PCR primers are listed in Table S5.† The data of each gene were normalized to the control gene encoding GAPDH and the relative expression of each sample was calculated by the change-in-cycling-threshold (2−ΔΔCt) method.Statistical analyses
All data were analyzed using Prism GraphPad software v5.0 and expressed as mean ± SEM. Statistical significance was determined by the Student's t-test and P < 0.05 was considered to be significant differences between different groups. Each experiment was performed in triplicate.Results and discussion
Screening of the PAC1 binding aptamer
The C-terminus of PAC1 is a conserved region of the DUSP family, while the N-terminus is a unique region required for chromatin remodeling in effector T cells.34 In order to obtain the specific aptamer of PAC1, we purified the N-terminal protein of PAC1 (1–160 amino acids; PAC1-N) as a positive target, and tested the optimal incubation ratio of protein and Ni-beads at 12 μg:1 μL (Fig. S1†). The N-terminal of the PAC1 protein, as the target protein, not only improves the specificity of screening but also avoids the region of the phosphorylation activity of the C-terminal of PAC1. As the screening rounds increase, the selective pressure increased gradually (Table S1†). After 8 rounds of screening, the enrichment processes of the third, fifth, seventh and eighth rounds were monitored by flow cytometry. As shown in Fig. 2A, the third round of P-beads showed a significant right shift in the fluorescence peak compared to the initial library, indicating that the ssDNA sequences targeting PAC1 began to be enriched. As the selection progressed, the fluorescence peak showed a stable right shift, while there was no further significant change from the 7th to the 8th round. These data suggested that the enrichment of the 8th pool reached saturation. For Ni-beads, there was no obvious change in the fluorescence peaks of the first to the eighth round. Based on the above results, the 8th round library was considered to be a successful enriched pool of PAC1 for further determination.
 | ||
| Fig. 2 Monitoring of the screening process and binding ability of aptamer candidates. (A) The binding capacity of the initial library or the 3rd, 5th, 7th and 8th rounds of secondary pools with the N-terminal of PAC1 protein coated Ni-beads (P-beads) and bare Ni-beads. Flow cytometric assay (B) and fluorescence microscopy assay (C) inspected the binding ability of the candidate aptamers PA2, PA4, PA5, PA12 with P-beads and Ni-beads. Scale bars: 100 μm. (D) Binding ability of these candidates with protein DUSP4, the N-terminal of PAC1 (PAC1-N) or full-length of PAC1 (PAC1-F) coated beads, respectively. Ni-beads coated with BSA were used as the control (Ctrl). | ||
The 8th round library was subjected to PCR amplification and cloning, and then 50 clones were selected for sequencing. Based on the homology and secondary structure of the sequence, we selected four candidate aptamers (PA2, PA4, PA5, PA12; Table S2†). After being modified with FAM fluorescent labeling, the binding ability of these aptamers to the target P-beads and the control Ni-beads was determined by flow cytometry. The results showed that all four candidates had stronger fluorescence intensity with P-beads compared with the original library (Fig. 2B). In contrast, the candidate aptamers with Ni-beads had almost no positive fluorescent signal, illustrating that these four aptamers could specifically bind to the PAC1 protein. The fluorescence microscopy imaging results were consistent with the flow cytometry results (Fig. 2C). All candidates showed high binding affinity to PAC1 with the Kd value in the nanomolar range (PA2: 86 ± 25 nM, PA4: 118 ± 14 nM, PA5: 61 ± 17 nM and PA12: 93 ± 41 nM) and the Kd value of PA5 was the lowest (Fig. S2†). Since DUSP4 and PAC1 have high homology and both are located in the nucleus, we further immobilized the PAC1 full-length protein (PAC1-F) or DUSP4 on Ni-beads as binding targets, and then their binding capacity was determined by flow cytometry. The fluorescence intensity of PAC1-F with FAM-labeled candidate aptamers was similar to the N-terminal protein of PAC1 (PAC1-N), but the fluorescence signal of DUSP4 was significantly weakened, manifesting that these four aptamers can specifically recognize PAC1 (Fig. 2D). Based on the best binding capacity, PA5 was chosen for further evaluation.
Sequence optimization of the aptamer PA5
Previous studies demonstrated that truncated aptamers could reduce the cost of chemical synthesis, increase tissue penetration, and maintain the structural stability of the original sequence.13 Therefore, we further optimized the full-length aptamer PA5 (76 nt, 1–76) via gradually removing nucleotides from the fixed primer sequence at both ends. Table S3† lists four truncated sequences, PA5a (58 nt, 1–58), PA5b (58 nt, 19–76), PA5c (40 nt, 19–58) and PA5d (28 nt, 25–52). The binding affinity of PAC1 was detected by flow cytometry. As shown in Fig. 3A, compared with the original aptamer PA5, the binding ability of PA5a, PA5b and PA5d to PAC1 was weakened, while the fluorescence intensity of PA5c was similar to that of PA5. We use NUPACK to predict the most likely secondary structure of these truncated aptamers under a lower free energy (ΔG) (Fig. 3B). PCA1-A5c was further assessed with a higher binding affinity (Kd = 51 ± 20 nM, Fig. 3C) and it maintains specificity (Fig. 3D). By analyzing the recognition ability and specificity of these aptamers, we obtained the optimized truncated version PA5c of PA5. Meanwhile, the binding ability between PA5c at different concentrations and P-beads was evaluated by fluorescence microscopy. As shown in Fig. S3,† PA5c had strong fluorescence intensity with P-beads, proving the efficient recognition ability of PA5c. | ||
| Fig. 3 Truncation of PA5 and characterization of PA5c. (A) Binding ability of different truncated versions of PA5 with the target PAC1 protein. (B) Secondary structure of PA5 and the corresponding truncated versions were simulated with NUPACK. (C) Dissociation constant (Kd) of PA5c for PAC1. (D) Binding assay of PA5c to DUSP4, PAC1-N or PAC1-F protein immobilized beads. Ni-beads coated with BSA were used as the control (Ctrl). | ||
PA5c detects PAC1 in cytochemistry
To assess the binding of the aptamer PA5c to cellular PAC1, we stably overexpressed mock (J-Mock), mCherry (J-mCherry), PAC1-Flag (J-PAC1-Flag), mCherry-PAC1 (J-mCherry-PAC1) in the Jurkat T cell leukemia cell line (Fig. S4A and B†). In order to determine the target identification of PA5c, we used the lysate of the stably transfected Jurkat cells incubated with agarose beads, an agarose-conjugated library or agarose-conjugated PA5c, respectively. Proteins in the pull-down mixture were analyzed by western blot using the flag antibody. As expected, PAC1 was pulled down by the aptamer PA5c, rather than agarose beads or the library, implying the interaction between PA5c and PAC1 (Fig. 4A). The aptamers labeled with FAM were used for cytochemical analysis. After immobilizing and permeabilizing, the stably transfected Jurkat cells were incubated with the FAM-labeled library or PA5c, respectively. Confocal microscopy results showed that PA5c and the mCherry-PAC1 fusion protein had co-localization and aggregation in the nucleus, but not in the FAM-library (Fig. 4B). And there was no fluorescence signal of aptamers in J-mCherry cells which did not express PAC1 (Fig. 4B). Together these results suggest that the aptamer PA5c can specifically recognize PAC1.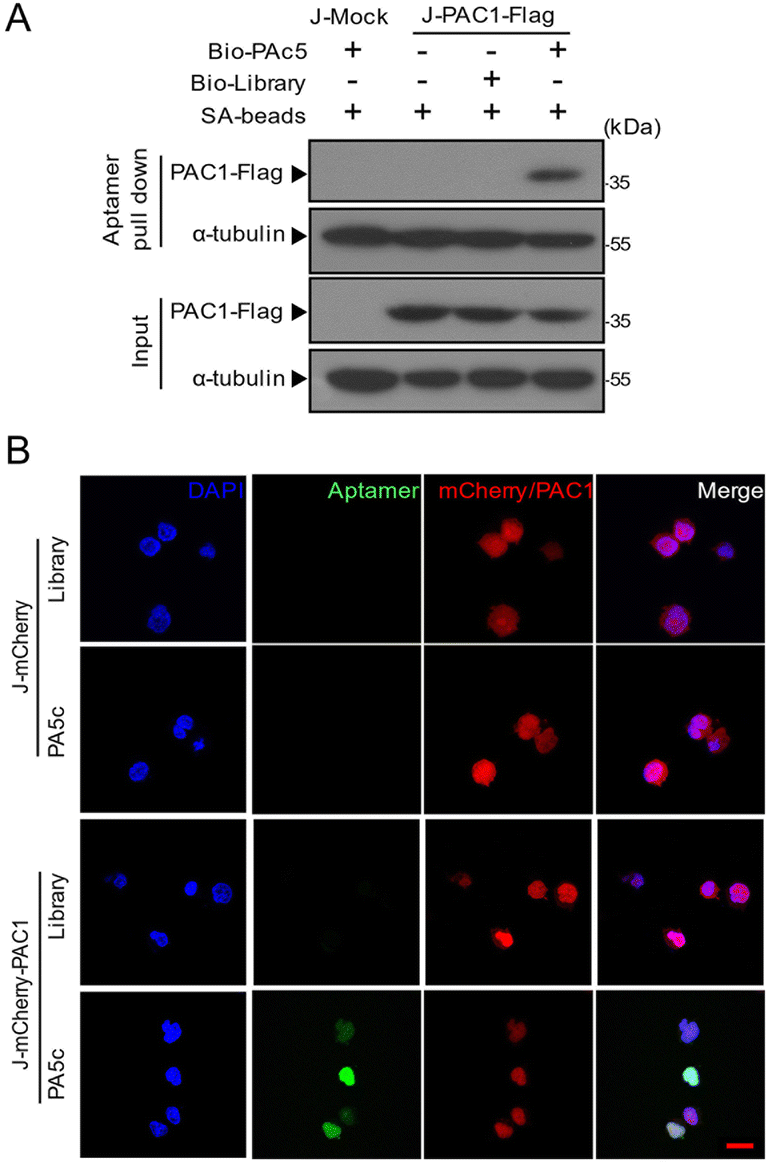 | ||
| Fig. 4 The specificity of PA5c binding to PAC1. (A) Binding of PA5c to PAC1 was analysed by the aptamer pull-down assay. Biotin–aptamers (PA5c or library) were added into the Jurkat cells stably expressing the control vector (J-Mock) or the flag-tagged PCA1 (J-PAC1-Flag) cell lysate and then incubated with streptavidin-agarose beads (SA-beads). After washing, the bound flag-tagged PAC1 protein was denatured and confirmed by western blotting with the anti-flag antibody. (B) After being fixed and permeabilized, the Jurkat cells stably expressing mCherry (J-mCherry) or mCherry-tagged PAC1 (J-mCherry-PAC1) were incubated with the FAM labeled library or PA5c, and then imaged by confocal microscopy. Cell nuclei were stained with DAPI (blue), FAM emits green fluorescence and mCherry is shown in red. Scale bars: 20 μm. | ||
Internalization of PA5c into cells
As shown in Fig. 4B, the aptamer PA5c was allowed to migrate freely to the nucleus for targeting PAC1 after permeabilizing. However, PA5c could not enter the living cells autonomously. Since PAC1 is located in the nucleus, the aptamer needs to pass the physiological barrier of the cell for recognizing the target. Previous works have evidenced that the nucleolin aptamer AS1411 can be efficiently absorbed by cells due to its stable G-quadruplex structures.28,29 Therefore, we designed a compound aptamer, PAC1-AS, which connected PA5c and AS1411 with a complementary linker sequence. The modular structure was formed by annealing these two aptamers via 3′ extensions on each (Fig. 5A). The predicted hybridization and the minimized misfolding of the two modules were both based on NUPACK calculations. Assembly was evaluated using DNA agarose gel that is stained with ethidium bromide to locate all molecules. The bands of aptamer PA5c-L appeared at 50 bp (lane 2), AS1411-L at 36 bp (lane 3) and PAC1-AS at 76 bp (lane 4), indicating that the formation of an aptamer–aptamer structure was assembled efficiently (Fig. 5B). Considering the feasibility of aptamers under physiological conditions, three bases at the ends of PAC1-AS were replaced with phosphorothioate oligonucleotides (PAC1-AS-p) to improve serum stability. The agarose gel electrophoresis showed that the unmodified PAC1-AS was almost completely hydrolyzed at 12 hours, while PAC1-AS-p was not visibly degraded up to 48 hours (Fig. 5C). Flow cytometry analysis also suggested that PAC1-AS-p had similar fluorescence signal strength to PAC1-AS (Fig. 5D), illustrating that the PA5c module still retained the targeting properties with original folding. Overall, these results indicate that PAC1-AS-p possesses better nuclease resistance and also retains the recognition properties of the target module. | ||
| Fig. 5 Design and characterization of PAC1-AS. (A) Secondary structure predictions of two aptamer modules (PA5c and AS1411) annealed via a connector domain to form the complex aptamer PAC1-AS. (B) Agarose gel electrophoresis showed the effective formation of PAC1-AS. (lane 1: marker; lane 2: PA5c-L; lane 3: AS1411-L; lane 4: PAC1-AS) (C) The stability of PAC1-AS and phosphorothioate PAC1-AS (PAC1-AS-p) in cell medium with 10% FBS was tested at different times by agarose gel electrophoresis, and the quantitative degradation curves were plotted. (D) Flow cytometry shows the binding ability of the aptamer with or without phosphorothioate modification to PAC1. (E) Aptamer-mediated internalization properties were assessed after 4 h incubation with J-mCherry-PAC1 cells. Representative images show aptamers FAM-AS1411, FAM-PA5c or FAM-PAC1-AS incubated with target cells under physiological conditions. Nuclei were stained with DAPI (blue), FAM shows a green signal and the mCherry-PAC1 protein is shown in red. The merge of all the channels is shown on the right. Scale bars: 40 μm. | ||
To further observe whether PAC1-AS-p could internalize into cells and target PAC1 under physiological conditions, FAM-labeled AS1411-p, PA5-p and PAC1-AS-p were incubated with J-mCherry-PAC1 cells at 37 °C, respectively, and imaged using a confocal fluorescence microscope. It could be seen that PAC1-AS-p was internalized and co-located with the mCherry-PAC1 fusion protein in the nucleus, while AS1411-p was mainly bound to the cell membrane and cytoplasm (Fig. 5E). In contrast, J-mCherry-PAC1 cells incubated with PA5c-p showed no intracellular fluorescence. These data deduce that PAC1-AS-p can be actively internalized in living cells and can bind PAC1, which is predominantly localized in the nucleus.
PAC1-AS is composed of two aptamer modules via efficient annealing self-assembly, which can be used for nuclear targeting and transmission of oligonucleotides without any other agents. Reducing non-specific binding and minimizing the introduction of other components is essential for the development of aptamer-mediated therapeutic drugs. These results also confirm that the modular aptamer does not interfere with each other's three-dimensional structure, and it can successfully recognize the target in live cells.
Identification of endogenous PAC1
As shown in Fig. 6A, the mRNA level of Pac1 in J-Mock stimulated with PMA and ionomycin (P.I.) was higher than that of untreated cells. The protein expression of PAC1 was significantly up-regulated in J-Mock cells after treating with P.I. (Fig. 6B). This is consistent with our previous study that the transcription level and the protein level of PAC1 in J-Mock cells were significantly increased after P.I. stimulation.5 To further investigate whether PAC1-AS-p can identify endogenous PAC1, the cells (J-Mock, J-Mock-P.I.) were incubated with FAM labeled AS1411-p, PA5c-p or PAC1-AS-p. The green fluorescent signal was only observed in the nucleus of J-Mock-P.I. cells which were incubated with FAM-PAC1-AS-p, but not of J-Mock cells (Fig. 6C). And FAM-AS1411-p was mainly displayed on the membrane and cytoplasm of J-Mock and J-Mock-P.I. cells. Besides, the fluorescence signal from FAM-PAC1-AS-p was found on the cell surface of J-Mock, which is possibly due to the AS1411 module. These data confirm that PAC1-AS-p can recognize the endogenous PAC1 protein and implicate its potential function. Moreover, considering the biological application of the aptamer, we verified that PAC1-AS-p was nontoxic to cells by a CCK8 experiment (Fig. S5†). | ||
| Fig. 6 Aptamer PAC1-AS-p can recognize endogenous PAC1. (A) qRT-PCR analysis of Pac1 mRNA levels in J-Mock cells after being stimulated with PMA and ionomycin (P.I.). **** means P < 0.0001. (B) After being treated with P.I., PAC1 expressed in J-Mock cells was tested by western blotting with the antibodies against PAC1, and α-tubulin as the marker for the soluble fraction. (C) Confocal examination of FAM labeled AS1411-p, PA5c-p and PAC1-AS-p localization in J-Mock cells that are untreated or pre-stimulated with P.I. (J-Mock-P.I.) for 3 h. The aptamer signal (green) was only observed in the J-Mock-P.I. cell nucleus when incubated with FAM-PAC1-AS-p. Scale bars: 40 μm. | ||
Influence of PAC1-AS on PAC1 function
We next aimed to determine the effect of PAC1-AS on the function of PAC1. PAC1, as a dual-specificity phosphatase, can mediate significant suppression of Erk1/2 phosphorylation during T cell activation. But this inhibition was not counteracted by the aptamer PAC1-AS (Fig. 7A). The screening target in our work was PAC1-N, which does not contain the phosphatase domain, and our results also confirmed that the binding of PAC1-AS to PAC1 has no effect on PAC1 phosphatase activity. In previous work, we found that PAC1 represses the transcription of effector T cell genes by recruitment of the NuRD complex, thereby dampening T cell antioxidant activity and effector function. However, the western blot analysis showed that the binding of PAC1-AS to PAC1 could not block the interaction between PAC1 and the components of the NuRD complex (Fig. 7B). It is speculated that the size of the aptamer is so small that it is not capable of blocking the interaction between PAC1 and the NuRD complex through the space-occupying effect.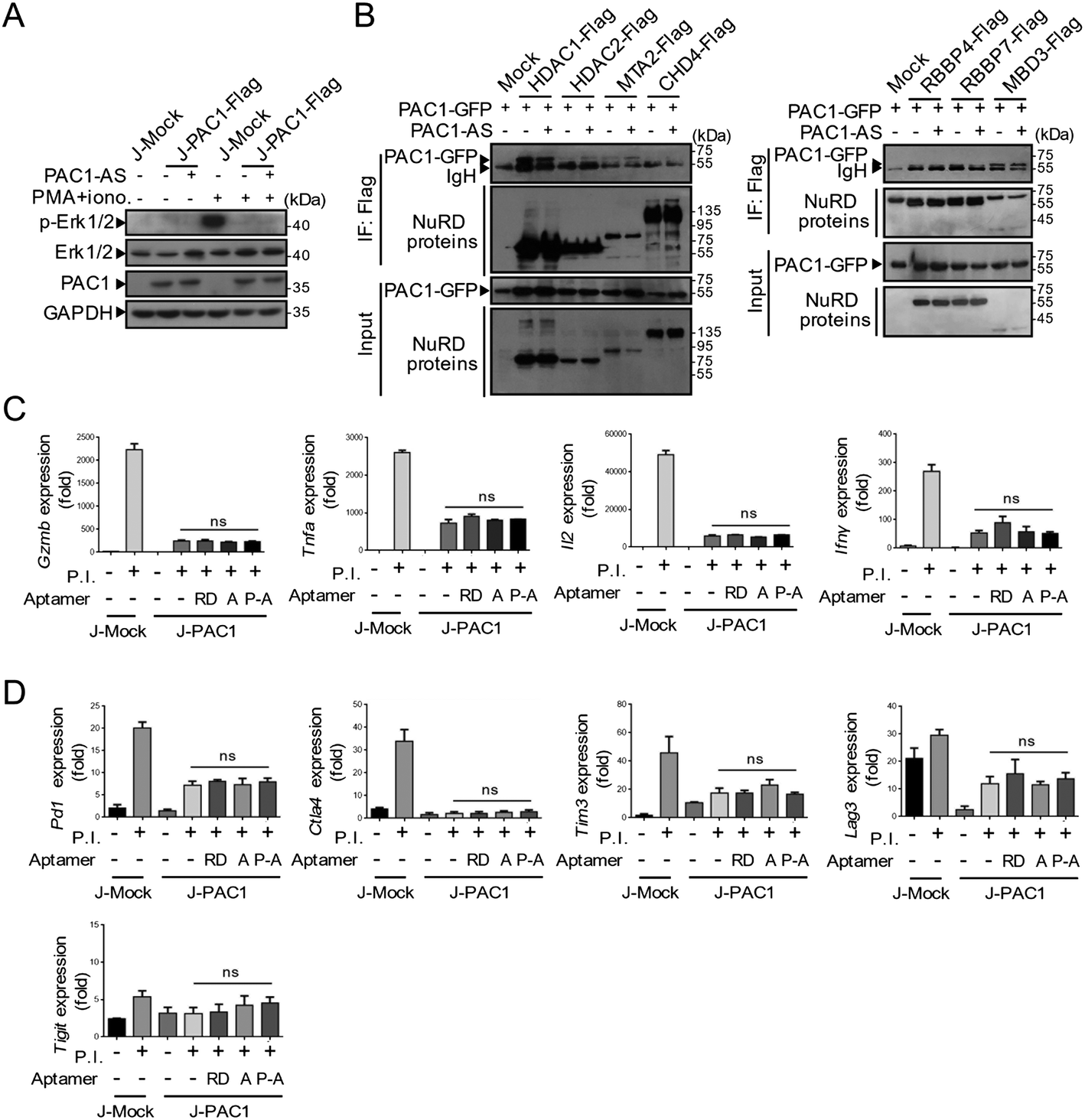 | ||
| Fig. 7 The influence of PAC1-AS on PAC1 function. (A) Cells were stimulated with or without 0.1 μg mL−1 PMA and 0.5 μg mL−1 ionomycin for 3 h. After being treated with PAC1-AS (250 nM) for 4 h, the cell lysates were analyzed by immunoblotting for the availability of phosphorylated Erk1/2 (p-Erk1/2), Erk1/2, PAC1 and GAPDH as an internal control. (B) Co-immunoprecipitation analysis of the interaction between PAC1-GFP and NuRD complexes with or without PAC1-AS treatment. qRT-PCR analysis of the mRNA levels of effector cytokines (Gzmb, Tnfa, Il2 and Ifnγ). (C) Immune checkpoints associated with activated T cells (Pd1, Ctla4, Tim3, Lag3 and Tigit). (D) Jurkat cells stably expressing Mock or PAC1 were untreated or stimulated with PMA plus ionomycin in the presence or absence of indicated aptamers for 6 h. Statistical significance was assessed by the Student's t-test, n = 3, mean ± s.e.m., ns means not significant. (RD as random sequences; A as AS1411; P-A as PAC1-AS.) | ||
On activation by PMA plus ionomycin, J-Mock cells exhibited augmented expression of genes associated with T cell effector function including Gzmb, Ifnγ, Tnfa and Il2. And the overexpression of PAC1 in T cells dramatically suppressed the expression of genes encoding these effector cytokines. After treatment with random sequences, AS1411 or PAC1-AS, the mRNA levels of these effector factors were comparable (Fig. 7C). Exhausted T cells are characterized by loss of effector function and upregulation of the expression of inhibitory receptors. To investigate whether PAC1-AS can inhibit the expression of inhibitory receptors in T cells, we detected the expression of T cell activation-related immune checkpoints including Pd1, Ctla4, Tim3, Lag3, and Tigit, after aptamer treatment (Fig. 7D). The results showed that PAC1-AS had no effect on the inhibitory receptors of T cells. These data thus indicate that PAC1-AS did not affect the phosphatase activity of PAC1, nor did it disturb the inhibition of the T cell effector function of PAC1.
To determine which biological function changes were induced by the binding of the aptamer PAC1-AS to PAC1 in activated T cells, we conducted proteomic mass spectrometry of the lysates of activated Jurkat T cells imposed with or without PAC1-AS and performed GO pathway enrichment analysis using differentially expressed proteins (Fig. S6†). The analysis showed that the up-regulated differential proteins in Jurkat T cells cultured with PAC1-AS were mainly associated with antigen presentation, gene transcription, nuclear protein transport and localization, and nucleic acid transport, while the down-regulated proteins were significantly involved in cytoplasmic translation. Consistent with previous data, there were no T cell activation or effector function related pathways in the GO enrichment results by these differential proteins. This also provides clues for further study of other potential functions of PAC1.
Conclusions
Despite the fact that aptamers binding plasma membrane proteins have been well studied, much less is known about aptamers targeting intracellular, especially nuclear proteins. In this study, we developed an appropriate aptamer specifically recognizing PAC1, which is predominantly localized in the nucleus and exerts immunosuppressive function dependent on its nuclear accumulation.5 Notably, aptamer PAC1-AS can recognize nuclear PAC1 in live cells under physiological conditions via our modularized design. This strategy overcomes the hurdle of oligonucleotide-based cellular delivery and provides insights to allow for the development of aptamers targeting other promising cancer immunity-associated nuclear proteins such as ADAR1 and TOX.35–37 Furthermore, integrated application of modular aptamers with an E3 ligase-based proteolysis-targeting chimeric molecule (PROTAC)38,39 or an autophagy-based protein degradation system40 would have the potential to implement aptamer-based protein engineering for precise treatment of multiple diseases. Our study opens the door for targeting nuclear proteins which are involved in the functions of endosomal biology and also provides clues for discovering new biological functions of PAC1. Furthermore, this design is expected to further develop a multifunctional assembly system for intracellular protein editing.Author contributions
Zixi Hu: methodology, investigation, validation, and writing – original draft. Zhongyu Jiang: methodology, writing – review and editing, and funding acquisition. Zeliang Yang: investigation, Liang Liu: methodology. Zhenyu Zhu: validation. Yan Jin: visualization and investigation. Yuxin Yin: conceptualization, methodology, writing – review and editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors contributed to the manuscript and approved the final version of the manuscript. This work was supported by grants including the National Key R&D Program of China (Grant 2021YFA1300601), the National Natural Science Foundation of China (Key Grants 82030081 and 81874235), the China Postdoctoral Science Foundation (Grant 2022M720281), and the Lam Chung Nin Foundation for Systems Biomedicine.References
- P. J. Rohan, P. Davis, C. A. Moskaluk, M. Kearns, H. Krutzsch, U. Siebenlist and K. Kelly, Science, 1993, 259, 1763–1766 CrossRef CAS PubMed.
- Y. X. Yin, Y. X. Liu, Y. J. Jin, E. J. Hall and J. C. Barrett, Nature, 2003, 422, 527–531 CrossRef CAS PubMed.
- D. Lu, L. Liu, X. Ji, Y. N. Gao, X. Chen, Y. Liu, Y. Liu, X. Y. Zhao, Y. Li, Y. Q. Li, Y. Jin, Y. Zhang, M. A. McNutt and Y. X. Yin, Nat. Immunol., 2015, 16, 1263–1273 CrossRef CAS PubMed.
- W. Wei, Y. Jiao, A. Postlethwaite, J. M. Stuart, Y. Wang, D. Sun and W. Gu, Genes Immun., 2013, 14, 1–6 CrossRef CAS PubMed.
- D. Lu, L. Liu, Y. Z. Sun, J. Song, Q. Yin, G. Z. Zhang, F. Qi, Z. X. Hu, Z. L. Yang, Z. Zhou, Y. Hu, L. H. Zhang, J. F. Ji, X. Y. Zhao, Y. Jin, M. A. McNutt and Y. X. Yin, Nat. Immunol., 2020, 21, 287–297 CrossRef PubMed.
- S. Folli, P. Westermann, D. Braichotte, A. Pelegrin, G. Wagnieres, H. Vandenbergh and J. P. Mach, Cancer Res., 1994, 54, 2643–2649 CAS.
- D. Neri, B. Carnemolla, A. Nissim, A. Leprini, G. Querze, E. Balza, A. Pini, L. Tarli, C. Halin, P. Neri, L. Zardi and G. Winter, Nat. Biotechnol., 1997, 15, 1271–1275 CrossRef CAS PubMed.
- P. F. Ma, H. Ye, J. Y. Deng, I. M. Khan, L. Yue and Z. P. Wang, Talanta, 2019, 205, 120119 CrossRef CAS PubMed.
- J. Wang, Q. Wang, Y. Luo, T. Gao, Y. Zhao and R. Pei, Talanta, 2019, 204, 424–430 CrossRef CAS PubMed.
- B. D. Wilson, A. A. Hariri, I. A. P. Thompson, M. Eisenstein and H. T. Soh, Nat. Commun., 2019, 10, 5079 CrossRef PubMed.
- W. J. Ma, Y. T. Yang, J. W. Zhu, W. Q. Jia, T. Zhang, Z. Q. Liu, X. Y. Chen and Y. F. Lin, Adv. Mater., 2022, 34, 2109609 CrossRef CAS PubMed.
- J. D. Munzar, A. Ng and D. Juncker, Chem. Soc. Rev., 2019, 48, 1390–1419 RSC.
- T. Wang, C. Y. Chen, L. M. Larcher, R. A. Barrero and R. N. Veedu, Biotechnol. Adv., 2019, 37, 28–50 CrossRef CAS PubMed.
- Z. H. Xu, C. N. Wang, R. Ma, Z. Sha, F. X. Liang and S. Q. Sun, Analyst, 2022, 147, 3350–3359 RSC.
- M. Panigaj, M. B. Johnson, W. N. Ke, J. McMillan, E. A. Goncharova, M. Chandler and K. A. Afonin, ACS Nano, 2019, 13, 12301–12321 CrossRef CAS PubMed.
- M. Mahmoudpour, S. C. Ding, Z. Y. Lyu, G. Ebrahimi, D. Du, J. E. N. Dolatabadi, M. Torbati and Y. H. Lin, Nano Today, 2021, 39, 101177 CrossRef CAS.
- Y. T. Niu, Y. Yang, Z. Yang, X. Wang, P. Zhang, L. W. Lv, Y. Liu, Y. S. Liu and Y. S. Zhou, Nano Today, 2022, 45, 101529 CrossRef CAS.
- W. M. Li, T. Bing, R. Wang, S. H. Jin, D. H. Shangguan and H. Chen, Analyst, 2021, 147, 187–195 RSC.
- K. Sefah, D. Shangguan, X. L. Xiong, M. B. O'Donoghue and W. H. Tan, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS PubMed.
- C. Lyu, I. M. Khan and Z. P. Wang, Talanta, 2021, 229, 122274 CrossRef PubMed.
- E. M. McConnell, I. Cozma, D. Morrison and Y. F. Li, Anal. Chem., 2020, 92, 327–344 CrossRef CAS PubMed.
- X. Q. Wu, H. L. Liu, D. M. Han, B. Peng, H. Zhang, L. Zhang, J. L. Li, J. Liu, C. Cui, S. B. Fang, M. Li, M. Ye and W. H. Tan, J. Am. Chem. Soc., 2019, 141, 10760–10769 CrossRef CAS PubMed.
- J. R. Xu, J. H. Xiang, J. L. Chen, T. Wan, H. L. Deng and D. R. Li, Analyst, 2022, 147, 634–644 RSC.
- G. Ranches, M. Lukasser, H. Schramek, A. Ploner, T. Stasyk, G. Mayer, G. Mayer and A. Huttenhofer, Mol. Ther.–Nucleic Acids, 2017, 8, 198–210 CrossRef CAS PubMed.
- M. Iaboni, R. Fontanella, A. Rienzo, M. Capuozzo, S. Nuzzo, G. Santamaria, S. Catuogno, G. Condorelli, V. Franciscis and C. L. Esposito, Mol. Ther.–Nucleic Acids, 2016, 5, e365 CrossRef CAS PubMed.
- L. Y. Wan, W. F. Yuan, W. B. Ai, Y. W. Ai, J. J. Wang, L. Y. Chu, Y. Q. Zhang and J. F. Wu, Expert Opin. Drug Delivery, 2019, 16, 207–218 CrossRef CAS PubMed.
- D. Porciani, L. N. Cardwell, K. D. Tawiah, K. K. Alam, M. J. Lange, M. A. Daniels and D. H. Burke, Nat. Commun., 2018, 9, 2283 CrossRef PubMed.
- P. J. Bates, D. A. Laber, D. M. Miller, S. D. Thomas and J. O. Trent, Exp. Mol. Pathol., 2009, 86, 151–164 CrossRef CAS PubMed.
- E. M. Reyes-Reyes, Y. Teng and P. J. Bates, Cancer Res., 2010, 70, 8617–8629 CrossRef CAS PubMed.
- N. Q. Do, W. J. Chung, T. H. A. Truong, B. Heddi and A. T. Phan, Nucleic Acids Res., 2017, 45, 7487–7493 CrossRef CAS PubMed.
- K. Sefah, Z. W. Tang, D. H. Shangguan, H. Chen, D. Lopez-Colon, Y. Li, P. Parekh, J. Martin, L. Meng, J. A. Phillips, Y. M. Kim and W. H. Tan, Leukemia, 2009, 23, 235–244 CrossRef CAS PubMed.
- I. T. Teng, X. W. Li, H. A. Yadikar, Z. H. Yang, L. Li, Y. F. Lyu, X. S. Pan, K. K. Wang and W. H. Tan, J. Am. Chem. Soc., 2018, 140, 14314–14323 CrossRef CAS PubMed.
- D. Lu, J. Song, Y. Z. Sun, F. Qi, L. Liu, Y. Jin, M. A. McNutt and Y. X. Yin, J. Autoimmun., 2018, 94, 156–165 CrossRef CAS PubMed.
- C. Y. Huang and T. H. Tan, Cell Biosci., 2012, 2, 24 CrossRef CAS PubMed.
- A. C. Scott, F. Dundar, P. Zumbo, S. S. Chandran, C. A. Klebanoff, M. Shakiba, P. Trivedi, L. Menocal, H. Appleby, S. Camara, D. Zamarin, T. Walther, A. Snyder, M. R. Femia, E. A. Comen, H. Y. Wen, M. D. Hellmann, N. Anandasabapathy, Y. Liu, N. K. Altorki, P. Lauer, O. Levy, M. S. Glickman, J. Kaye, D. Betel, M. Philip and A. Schietinger, Nature, 2019, 571, 270–274 CrossRef CAS PubMed.
- O. Khan, J. R. Giles, S. McDonald, S. Manne, S. F. Ngiow, K. P. Patel, M. T. Werner, A. C. Huang, K. A. Alexander, J. E. Wu, J. Attanasio, P. Yan, S. M. George, B. Bengsch, R. P. Staupe, G. Donahue, W. Xu, R. K. Amaravadi, X. W. Xu, G. C. Karakousis, T. C. Mitchell, L. M. Schuchter, J. Kaye, S. L. Berger and E. J. Wherry, Nature, 2019, 571, 211–218 CrossRef CAS PubMed.
- R. de Reuver, S. Verdonck, E. Dierick, J. Nemegeer, E. Hessmann, S. Ahmad, M. Jans, G. Blancke, F. Van Nieuwerburgh, A. Botzki, L. Vereecke, G. van Loo, W. Declercq, S. Hur, P. Vandenabeele and J. Maelfait, Nature, 2022, 607, 784–789 CrossRef CAS PubMed.
- M. Schapira, M. F. Calabrese, A. N. Bullock and C. M. Crews, Nat. Rev. Drug Discovery, 2019, 18, 949–963 CrossRef CAS PubMed.
- S. P. He, F. Gao, J. H. Ma, H. Q. Ma, G. Q. Dong and C. Q. Sheng, Angew. Chem., Int. Ed., 2021, 60, 23299–23305 CrossRef CAS PubMed.
- Z. Y. Li, C. Wang, Z. Y. Wang, C. G. Zhu, J. Li, T. Sha, L. X. Ma, C. Gao, Y. Yang, Y. M. Sun, J. Wang, X. L. Sun, C. Q. Lu, M. Difiglia, Y. N. Mei, C. Ding, S. Q. Luo, Y. J. Dang, Y. Ding, Y. Y. Fei and B. X. Lu, Nature, 2019, 575, 203–209 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Tables S1–S5. See DOI: https://doi.org/10.1039/d3an00011g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |