
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From mouth to gut: microfluidic in vitro simulation of human gastro-intestinal digestion and intestinal permeability†
Miguel
Xavier‡
 a,
Patrícia M.
Rodrigues‡
ab,
Mafalda D.
Neto
a,
Maria I.
Guedes
a,
Victor
Calero
a,
Lorenzo
Pastrana
a and
Catarina
Gonçalves
*a
a,
Patrícia M.
Rodrigues‡
ab,
Mafalda D.
Neto
a,
Maria I.
Guedes
a,
Victor
Calero
a,
Lorenzo
Pastrana
a and
Catarina
Gonçalves
*a
aInternational Iberian Nanotechnology Laboratory, Avenida Mestre José Veiga, 4715-330 Braga, Portugal. E-mail: catarina.goncalves@inl.int
bUniversity of Minho, Gualtar Campus, 4710-057 Braga, Portugal
First published on 9th May 2023
Abstract
Reproducible in vitro studies of bioaccessibility, intestinal absorption, and bioavailability are key to the successful development of novel food ingredients or drugs intended for oral administration. There is currently a lack of methods that offer the finesse required to study these parameters for valuable molecules typically found in small volumes – as is the case of nanomaterials, which are often used to carry and protect bioactives. Here, we describe a modular microfluidic-based platform for total simulation of the human gastro-intestinal tract. Digestion-chips and cell-based gut-chips were fabricated from PDMS by soft lithography. On-chip digestion was validated using a fluorescently labelled casein derivative, which followed typical Michaelis–Menten kinetics and showed temporal resolution and good agreement with well-established bench-top protocols. Irreversible inhibition of serine proteases using Pefabloc® SC and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 dilution was sufficient to mitigate the cytotoxicity of simulated digestion fluids. Caco-2/HT29-MTX co-cultures were grown on-chip under a continuous flow for 7 days to obtain a differentiated cell monolayer forming a 3D villi-like epithelium with clear tight junction formation, and with an apparent permeability (Papp) of Lucifer Yellow closely approximating values reported ex vivo (3.7 × 10−6 ± 1.4 × 10−6vs. 4.0 × 10−6 ± 2.2 × 10−6). Digesta from the digestion-chips were flowed through the gut-chip, demonstrating the capacity to study sample digestion and intestinal permeability in a single microfluidic platform holding great promise for use in pharmacokinetic studies.
:6 dilution was sufficient to mitigate the cytotoxicity of simulated digestion fluids. Caco-2/HT29-MTX co-cultures were grown on-chip under a continuous flow for 7 days to obtain a differentiated cell monolayer forming a 3D villi-like epithelium with clear tight junction formation, and with an apparent permeability (Papp) of Lucifer Yellow closely approximating values reported ex vivo (3.7 × 10−6 ± 1.4 × 10−6vs. 4.0 × 10−6 ± 2.2 × 10−6). Digesta from the digestion-chips were flowed through the gut-chip, demonstrating the capacity to study sample digestion and intestinal permeability in a single microfluidic platform holding great promise for use in pharmacokinetic studies.
1 Introduction
Oral administration remains the preferred route for the delivery of drugs or dietary supplements. Bioaccessibility, intestinal absorption, and bioavailability are three fundamental parameters that indicate respectively the fraction of an orally ingested compound that (a) is available for absorption following digestion, (b) is absorbed by the gut epithelium, and (c) reaches systemic circulation following intestinal absorption and first-pass metabolism in the liver.1 The capacity to predict these parameters in vitro is key to the successful development of novel foods and pharmaceuticals intended for oral administration. Indeed, the cost for the development of each new approved drug is currently estimated to surpass 1 billion USD and one of the main reasons is the poor predictive power of the existing models.2 In addition, degradation in the gastrointestinal tract (GIT) and toxicity towards cell-based models are two of a series of important steps recommended by the European Food Safety Agency (EFSA) for the risk assessment of nanomaterials (NMs) used in the food and feed chain.3Digestion in the GIT is a complex, multistage process and its study in vivo carries ethical, economic, and technical concerns,4 hence the importance of in vitro models. Static models are typically simple, low cost and use a single reaction vessel but do not fully recapitulate the human physiological digestive conditions. The current state-of-the-art is the benchtop protocol defined from a collaborative effort within the former INFOGEST COST action.5,6 Dynamic models, in their turn, use flow to allow time-resolved addition of simulant fluids and enzymes, pH monitoring and regulation, and include other dynamic features, such as peristalsis or gastric emptying. However, they are considerably more complex and less affordable, and the use of reusable vessels often leads to issues with sample contamination.7 Critically, both static and dynamic models are highly informative. However, there is a lack of models that offer the finesse required to study the bioaccessibility of valuable molecules in small volumes, as is the case of new NMs being researched. To date, a single example of a miniaturised digestion simulator has been reported.8 The device consisted of 3 microfluidic mixers connected by up to 6 metre long sections of tubing that served as reaction chambers. It used continuous flow and significantly reduced reaction volumes and was validated using reporter molecules and lactoferrin.
In vitro cell-based models to predict intestinal permeability allow reducing both the use of animal models and issues associated with interspecies variability, by using human cell lines, thus increasing the likelihood of new drugs reaching the market. Although static cell-based models play an important role in the prediction of intestinal permeability, they overlook the important aspects of gut physiology such as continuous flow in the intestinal lumen, peristalsis, and the capacity to support co-cultures with the gut microbiota. To address these limitations, microengineered organs-on-chip based on microfluidics have been developed to replicate a variety of tissues and organs.9 These typically consist of two or more channels separated by a semi-porous membrane or a scaffold based on extracellular matrix (ECM) proteins where cells are cultured to form an epithelium. Examples include, but are not limited to, the devices now commercialised by Emulate Inc. or MIMETAS. Studies within the last decade have demonstrated the capacity to (i) culture cells from different origins, such as epithelial cell lines,10 immune cells,10–12 and primary cells,13,14 (ii) include mechanical stimuli to mimic peristalsis,10 (iii) allow co-cultures with gut microbiota,10–12 and (iv) integrate a variety of sensors for real-time monitoring, including measurements of the trans-epithelial electrical resistance.15,16
Importantly, very few studies have combined in vitro digestion and intestinal absorption studies using cell-based models in a single platform.17,18 Adding to the complexity of combining two technologically challenging platforms, a major hurdle remains the cytotoxicity of the digestion fluids towards the cultured intestinal epithelium. Previous studies have exploited methods based on filtration, ultracentrifugation, heat inactivation, or protease inhibition to mitigate the cytotoxicity of the digestion fluids, but none could circumvent the necessity of significantly diluting the digested samples.17,19–22
Here, we describe a modular device based on microfluidics for complete assessment of the bioaccessibility and intestinal permeability of ingested bioactive compounds. The first module, the digestion-chip, allows in vitro digestion in continuous flow and was validated using a fluorescently labelled casein reporter molecule. This study defined the experimental conditions and highlighted the potential for future bioaccessibility assessments of bioactive compounds. The second module consisted of a cell-based gut-chip. To couple both modules, the cytotoxicity of the gastric and intestinal digestion fluids was tested using semi-confluent Caco-2 cell monolayers and differentiated co-cultures of Caco-2 and HT29-MTX. The effect of the digesta on the cell-based gut-chip was assessed by changes to the apparent permeability of Lucifer Yellow.
2 Experimental
2.1 Materials
Detailed information on material references and suppliers can be found in the ESI.†2.2 Device design and fabrication
Devices were designed using CleWin4 from WieWeb Software (Hengelo, the Netherlands). The designs can be found in the supporting dataset of this manuscript and are shown in Fig. 1. Both digestion and gut chips were fabricated by soft lithography from PDMS (Sylgard® 184) mixed in 1:10 and 1:15 ratios (w/w) respectively. The top PDMS slab of the digestion-chip (containing the staggered herringbone mixers – SHM) was cast against silicon master moulds fabricated using SU-8 2025 photoresist following a two-step UV photolithography process, as detailed in the ESI.† The mould for the bottom digestion-chip PDMS slab and both moulds of the gut-chip were fabricated from polymethyl methacrylate (PMMA) by CNC micro milling (FlexiCAM Viper CNC, FlexiCAM GmbH, Eibelstadt, Germany). To upscale production and prolong the lifetime of the original moulds, polyurethane counter-moulds were prepared from the first PDMS slabs according to Varadarajan et al.23 For this, the PDMS slabs were attached feature-side up to silicone baking moulds using double-sided tape. The assembly was degassed, while the two-component polyurethane (Smooth-Cast™ 310) solutions were each thoroughly shaken for 10 min and then combined in 1:1 ratio followed by further mixing for an additional 2 min. The mixture was then poured over the degassed PDMS device, further degassed for 10 min, and left overnight in a fume hood followed by hard baking at 65 °C for 4 h. Once cooled, the PDMS devices were removed leaving the polyurethane moulds ready to use.
 | ||
| Fig. 1 (A) 2D design of the Digestion-Chip with an inset (B) showing the staggered herringbone mixers (SHM) introduced at each channel junction to promote mixing by chaotic advection. (C) SEM micrograph of a PDMS slab of the digestion-chip showing the SHM grooves. (D) Photograph of the digestion-chip with flowing food coloured solutions showing 4 inlets, and thorough mixing at each junction. (E) 3D CAD design representation of the digestion-chip. (F) 2D design of the top and bottom channels of the gut-chip. (G) SEM micrograph of the micropillar arrays with geometry shown in (H) used to fabricate the semi-porous PDMS membranes by spin coating and shown in (I). (J) 3D CAD design representation of the gut-chip. | ||
:15 w/w) at 2400 rpm for 5 min on top of silicon micropillar arrays (8 µm diameter, 32 µm pitch, 40 µm height, and offset every 2 rows – Fig. 1G and H) passivated by vapour deposition of trichloro(perfluorooctyl)-silane. The pore size was defined at 8 µm to offer enough support for Caco-2/HT29-MTX co-cultures while still providing large pores for cell invasion studies (not contemplated in this case). The pore rows were offset and the pitch set at 32 µm to increase the structural integrity of the membrane allowing its handling during fabrication and avoiding its collapse over the bottom channel. The bottom + membrane assembly was bonded to the top PDMS slab (with inlets and outlets opened using 1.5 mm diameter biopsy punches) following alignment using an alignment rig built in-house (ESI Fig. 1†).
2.3 Mixing by chaotic advection
To validate the efficiency of mixing by chaotic advection promoted by the SHMs, 0 µM and 5 µM fluorescein sodium salt solutions in 80/20 (v/v) glycerol/water mixture were flowed filling opposite halves of the channel cross section. Mixing in the channel was imaged through a 10× objective using a laser scanning confocal microscope (LSCM) (Zeiss LSM780; Oberkochen, Germany) equipped with a 488 nm argon laser.2.4 In vitro digestion
In vitro and on-chip gastro-intestinal digestions were carried out following the standardised INFOGEST COST action protocol first published by Minekus et al.5 in 2014 and later updated in 2019.6 The composition of the simulated digestion fluids can be found in ESI Table 1.† The activity of pepsin, used in the gastric phase, and of trypsin present in pancreatin, used in the intestinal phase, were quantified for each batch according to the procedure detailed in Brodkorb et al.6 and detailed in the ESI.† The concentration of bile salts was tested using a commercial kit. The simulated fluids were prepared in advance and stored at −20 °C before use. The enzyme (pepsin and pancreatin) and bile salt solutions were prepared fresh before each experiment to preserve their activity and CaCl2 was added just before each experiment to avoid precipitation.Briefly, for digestions, samples were mixed with simulated salivary fluid (SSF) in equal volumes and incubated for 2 min at 37 °C. All incubations were carried out in a thermomixer (Eppendorf™, Hamburg, Germany) for static in vitro digestions, or inside an orbital incubator (VWR 444-7082) for on-chip digestions. The bolus was then mixed 1:1 with simulated gastric fluid (SGF) and pepsin (2000 U mL−1 in H2O), pH adjusted to 3.0 (using 1 M HCl), and the reaction allowed to occur for 2 h. Finally, the chyme was mixed 1:1 with simulated intestinal fluid (SIF), pancreatin (100 U mL−1 in SIF) and bile salts (10 mM in SIF), pH adjusted to 7.0 (using 1 M NaOH), and the reaction allowed to occur for further 2 h. Samples were collected when convenient and the volumes adjusted accordingly. On-chip incubation times were controlled by varying the infusion flow rates considering the volumes of the gastric (96 µL) and intestinal incubation chambers (192 µL).
As proof-of-concept and to allow direct comparison between the static in vitro and on-chip digestions, a quenched, fluorescently labelled casein derivative (EnzCheck™ Protease Assay Kit, ThermoFisher) was used as the test molecule. The extension of digestion was estimated by measuring the fluorescence intensity (λex = 589 nm, λem = 617 nm) using a BioTeK® Synergy H1 microplate reader (Winnoski, VT, USA) or directly on-chip using a Zeiss LSM780 confocal laser scanning microscope (Oberkochen, Germany), with images analysed by ImageJ.
2.5 Cell culture
Cells were kept in a humidified chamber at 37 °C and 5% CO2. The culture medium was changed every 2–3 days and cells were routinely sub-cultured until achieving a maximum confluence of 70–80%. To detach adherent cells, a trypsin–EDTA solution (0.5 g L−1 trypsin; 0.2 g L−1 EDTA) was used for 5–10 min. For co-cultures, Caco-2 and HT29-MTX cells were mixed in 9:1 ratio and complete MEM medium was used. Caco-2 and HT29-MTX were used to represent the two most abundant cell populations in the intestinal epithelium (enterocytes and goblet cells, respectively). The ratio of Caco-2 to HT29-MTX cells in co-cultures has been studied over the years and there is a generalised consensus that between 90:10 (9:1) and 75:25 ratios provide the best approximation to the in vivo environment, because of both the actual ratio found in vivo and the increased functionality of cell culture models.24–26
2.6 Cell metabolic activity
Cell metabolic activity was determined by the resazurin (RZ) reduction assay on semi-confluent Caco-2 cells after 24 h of culture in 96-well plates or on differentiated Caco-2/HT29-MTX co-cultures in both 96-well plates and in cell hanging inserts (Merck Millipore Millicell® 1 µm pore size, PET membrane). For co-cultures, cells were plated at 1 × 105 cells per cm2 and grown for 21 days with media changed every other day. At day 21, the test samples were added and incubated with cells for 4 h or 24 h, after which the medium was removed and replaced by a 10 µg mL−1 RZ sodium salt solution in complete MEM, and incubated for 3 h at 37 °C. Deionised H2O (diH2O; 10% v/v) and DMSO (40% v/v) in complete MEM were used as positive and negative controls, respectively. The cell metabolic activity was determined by measuring the fluorescence of the product of resazurin reduction, resofurin, (λex = 560 nm, λem = 590 nm) using a BioTeK® Synergy H1 (Winnoski, VT, USA) microplate reader. The results are expressed as a percentage of cell viability in relation to untreated cells.2.7 On-chip cell culture
Gut-chips were sterilised by incubation with 70% (v/v) ethanol for 30 min under UV exposure, followed by a wash with PBS. To activate the surface of the PDMS membrane, 0.5 mg mL−1 sulfo-SANPAH solution in diH2O was introduced in the upper channel, and the device photo-activated by UV radiation (λ = 365 nm; 6.8 W; distance: 5 cm) for 30 min, as described in Sontheimer-Phelps et al.27 The solution was removed, and the channel washed with diH2O. An extracellular matrix (ECM) solution containing rat type I collagen (30 µg mL−1) and Matrigel (100 µg mL−1) in serum-free DMEM was added to both channels and incubated overnight at 4 °C, followed by 1 h of incubation at 37 °C the next day. The ECM solution was removed, washed with serum-free media, and Caco-2/HT29-MTX cells were seeded in the upper channel at 1 × 105 cells per cm2. Cells in the devices were incubated overnight at 37 °C in a CO2 incubator to promote cell adhesion. The devices were inspected under the microscope, and then transferred onto an in-house built imaging platform and 8-channel syringe pump (ESI Fig. 1†), connected via 1.6 mm OD PTFE tubing through commercial bubble traps from Darwin Microfluidics (Paris, France) and continuously perfused with degassed supplemented media at 120 µL h−1 in both channels.2.8 Immunocytochemistry
Cells were washed with PBS, fixed in 3.7% PFA in PBS (15 min at RT) and permeabilised by incubation with 0.2% Triton-X-100 solution for 10 min at 4 °C. Non-specific antibody bonding was then blocked by incubation with 2% (w/v) BSA solution followed by immunostaining with 5 µg mL−1 Alexa Fluor® 488-conjugated occludin monoclonal antibody (OC-3F10) and 0.1 µg mL−1 phalloidin-TRITC overnight at 4 °C. Finally, the cells were washed, the nuclei counterstained with 50 µg mL−1 DAPI for 5 min and preserved in Fluoroshield mounting media. Images were taken using a Zeiss LSM780 confocal laser scanning microscope (Oberkochen, Germany) equipped with 405 nm and 561 nm diode lasers and a 488 nm argon laser, and analysed using the Zen 2010 software and ImageJ.2.9 Intestinal permeability
Cells cultured on hanging cell culture inserts (for 7 or 21 days) or on chip (7 days) were used for intestinal permeability studies. Previous studies showed that cell differentiation on-chip is accelerated by the shear stress exerted by continuous flow.28 We tested the ideal culture time in our gut-chips using Caco-2/HT29-MTX co-cultures and determined that 7 days of culture reproducibly led to the formation of a robust epithelial barrier (ESI Fig. 2†).Gut-chips were connected to syringes pre-filled with HBSS or HBSS supplemented with the test samples. The epithelial barrier integrity was assessed by measuring the apparent permeability of 50 µM Lucifer Yellow from the apical to the basolateral channel. During the assay the flow rate was reduced to 20 µL h−1 and 20 µL of outflow was collected every hour from pipette tips inserted at the channel outlets. The apparent permeability coefficient of Lucifer Yellow (Papp, cm s−1) was calculated according to the eqn (1):
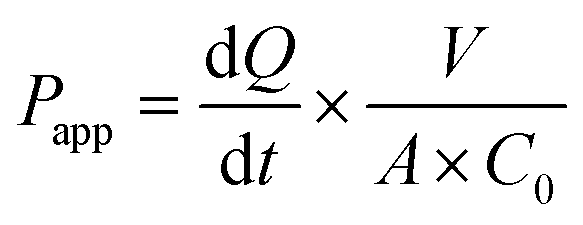 | (1) |
2.10 Statistical analysis
Results are displayed as Mean ± SD unless otherwise stated using GraphPad Prism 9.4.1 (San Diego, CA, USA). Data distributions were tested for normality using the Shapiro–Wilk test. The statistical significance was tested using the Kruskal–Wallis test followed by the Mann–Whitney U test.3 Results and discussion
3.1 Mixing by chaotic advection
Flow in the digestion-chip was in the Stokes or laminar flow regimes with the Reynolds number (ESI Table 2†) ranging from 4 × 10−2 to 4.25 at the range of flow rates used (0.024–2.3 mL h−1). In this regime, mixing can be problematic when relying solely on diffusion. Thus, a staggered herringbone mixer (SHM – first introduced by Stroock et al.29) was included following each junction of 2 or more channels to promote mixing by chaotic advection. Fig. 2 shows that complete mixing of 0 and 5 µM sodium fluorescein salt solutions in a glycerol/water (80/20 v/v) mixture was achieved after just 7.5 cycles of the SHM when flowed with the same mixture without fluorescein at 100 μL h−1. We also tested mixing in water-based solutions, which should provide a closer representation of the samples and digestive fluids, and at faster flow rates (2.3 mL h−1) at which less time would be available for diffusion to occur. In those conditions, complete mixing was also achieved before the end of the SHM (ESI Fig. 3†), which indicates that complete mixing between sample and digestion fluids was expected at all experimental conditions of this study. | ||
| Fig. 2 Confocal microscopy images showing mixing by chaotic advection over a staggered herringbone of a 0 µM and a 5 µM fluorescein sodium salt solution in an 80/20 (V/V) glycerol/water mixture. Tile scan images were taken at an intermediate focal point for complete channel visualisation, and z-stacks (0.5 µm increments) were acquired after determined cycles of mixing as shown by the yellow arrows (cycles 0, 1, 2, 2.5, 3, 4, 5 and 7.5). The tile scan image and the orthogonal view images were processed using 5-pixel and a 20-pixel median filter respectively. Fluorescence intensity profiles were extracted from ImageJ across a median line using the ‘plot profile’ function, and calculated as a percentage of the maximum intensity value across the same line. | ||
3.2 On-chip digestion
To validate the digestion-chip, we used a commercial casein derivative labelled with BODIPY® TR-X dye. The dye is quenched by proximity but becomes fluorescent upon hydrolysis, thus working as a digestion reporter molecule to determine protease activity. To study enzyme kinetics, casein was diluted in varying concentrations [S] according to the manufacturer's instructions, and mixed off-chip with SSF to represent the output of the oral digestion phase (amylase was not included due to the absence of starch in the test sample). Sample and SGF (2000 U mL−1 pepsin) were then flowed at 1152 µL h−1 through separate inlets leading to a SHM for mixing followed by a long serpentine channel for incubation. At ∼2.3 mL h−1 the total incubation time was 220 seconds. Note that the incubation time is a direct function of the channel dimensions (here fixed) and the flow rates used. The whole experiment was carried out under a confocal microscope equipped with an integrated incubator at 37 °C to allow determination of fluorescence at different digestion time points (every 12 s). There was an increase in fluorescence over time for every concentration tested (ESI Fig. 4A†). The increase was nearly negligible at the 2 and 5 µg mL−1 concentrations and became more evident from 10 µg mL−1 and above – in agreement with the results from Haan et al.8Fig. 3A shows that the digestion rates (calculated over the first 74 s of digestion – ESI Fig. 4B†) increased with concentration, appearing to plateau from 20 µg mL−1, fitting a typical Michaelis–Menten enzyme kinetics profile (R2 = 0.81). This indicated that digestion occurred in the digestion-Chip with active enzyme catalysis. Control experiments where no enzymes were added to the digestion fluids of both gastric and intestinal digestion phases did not lead to a fluorescence increase (data not shown – see full dataset https://doi.org/10.5281/zenodo.7324498). | ||
| Fig. 3 On-chip digestion of a casein reporter molecule labelled with BODIPY® TR-X. Fluorescence of hydrolysed substrate was measured using a microplate reader (λex = 589 nm; λem = 617 nm). (A) Enzyme kinetics studies where simulated gastric fluid containing a fixed concentration of pepsin [E] was introduced in one inlet and a sample containing a varying concentration of casein [S] was introduced in the other inlet. Data show the initial reaction rates (over 74 seconds) plotted vs. [S]. (B) Digestion following the static INFOGEST protocol of casein at 1 and 10 µg mL−1 and its comparison with the end-point of a full digestion using the digestion-chip (green triangles). (C) Time-resolved digestion simulations of the gastric and intestinal phases in the digestion-chip. Time-resolution was achieved by varying the flow rates (12–1152 µL h−1) of the sample and simulated digestion fluids (SGFs) thus attaining different incubation times. | ||
Aiming to compare on-chip digestion with a well-established in vitro digestion model, the same casein reporter was digested following the standardised INFOGEST static digestion protocol.5,6Fig. 3B shows fluorescence data from periodic measurements of 100 µL samples taken from the reaction vessel every 30 min. At a substrate concentration of 1 µg mL−1, fluorescence reached a plateau in the gastric phase after 90 min of reaction suggesting that the substrate had been fully converted. There was a further increase following the addition of SIF (100 U mL−1 pancreatin), reaching a plateau after another 30 min, showing that casein could be further hydrolysed by pancreatin.30 At 10 µg mL−1, the fluorescence increased over the full 2 h period of digestion. There is again a steep increase following the addition of SIF, appearing to plateau from 210 min of digestion onwards. The approximately 10 fold difference in fluorescence (∼5000 vs. ∼50000) between the 1 and 10 µg mL−1 concentrations at the 240 min end-point demonstrates a direct proportion between fluorescence and the digestion of the casein reporter. Critically, full on-chip digestion yielded an average fluorescence of 17917 ± 3871 (Mean ± SD, N = 7; Fig. 3B, green triangles). While reproducible, this was considerably lower than the extension of digestion obtained by the static INFOGEST method. To ascertain whether reaction in the device stopped before all the substrate had been converted, further SGF or SIF with freshly prepared pepsin and pancreatin were added to the output of the digestion-chip and allowed to react at 37 °C in a reaction plate for another 60 min. This did not lead to a further increase in fluorescence (data not shown – see full dataset https://doi.org/10.5281/zenodo.7324498) indicating that the lower extent of digestion observed on-chip was not due to enzyme inactivation. We also verified that pH had not changed inside the devices. The pH measurements at the outlets of the gastric and intestinal phases were not significantly different from that of the initial digestion solutions (3.2 vs. 3.0 and 7.2 vs. 7.0, respectively). Finally, due to PDMS’ reputation as a solid solvent with issues related to the absorption and adsorption of molecules frequently reported,31–34 we checked whether digested casein was being retained by the polymer matrix. For this, casein was digested off-chip and then flowed through the digestion-chip for 300 min. The fluorescence was measured before and after flowing through the device and a reduction of 13% in relative fluorescence intensity was observed (ESI Fig. 5A†). The presence of casein aggregates was also observed on-chip by confocal microscopy (ESI Fig. 5B and C†). While this may contribute to a lower extension of on-chip digestion, it does not fully account for the disparity compared to the INFOGEST protocol. However, it is reasonable to assume that just like digested casein, undigested substrate could be retained in the device and thus become unavailable for digestion. We also hypothesise that lack of constant mixing during the long incubation steps in both gastric and intestinal phases could contribute to different digestion kinetics and lower overall extension. We are thus currently working on a new device iteration using a different material for fabrication and where mixing is achieved throughout the incubation phases in large reaction chambers by the action of a small magnet operated by magnetic stirrers built in house.
Finally, time-resolved on-chip digestion was obtained by varying the flow rates (from 12 up to 1152 µL h−1) of the sample (casein at 10 µg mL−1) and simulated digestion fluids leading to decreasing incubation times. For time-resolved intestinal digestion, the gastric phase was first processed off-chip and then fed to the device as chyme. Fig. 3C shows a digestion kinetics profile within the first 60 min identical to the one obtained in the static INFOGEST method. The fluorescence values seem to plateau after 60 min, or even to slightly decrease (ESI Fig. 4C†), suggesting a potential deterioration or absorption/adsorption by PDMS of the hydrolysed substrate.
The results shown herein demonstrate the versatility of our digestion-chip to perform both full gastrointestinal digestions comprising the oral, gastric and intestinal phases, and time-resolved digestions at either the gastric or intestinal phase. This yields significant potential for bioaccessibility studies of valuable samples. Nevertheless, the use of alternative materials for device fabrication such as polymethyl methacrylate may be key to obtain improved on-chip digestion – one of our current research goals.
3.3 Cytotoxicity of simulating digestive fluids
Most orally administered drugs and nutrients do not exert their effects locally in the gut but rather target a systemic effect or an effect at a distant organ. Thus, while studying the bioaccessibility of these compounds provides valuable information, the study is not complete without determining the fraction that reaches systemic circulation – i.e. the compound bioavailability. However, the bioavailability of an orally ingested compound depends not only on its digestion and on the fraction absorbed by the intestinal epithelium, but also on the fraction that escapes metabolism in the gut and liver as it is carried through the portal vein – the so-called first pass effect. This phenomenon is particularly difficult to study in vitro and thus most in vitro studies do not progress beyond intestinal absorption studies using intestinal epithelial models typically based on hanging inserts. Interestingly, even studies of digested compounds on in vitro intestinal epithelial models have proved challenging owing to the cytotoxicity of the enzymes present in the digestive fluids,17,19–22 with studies showing dilutions superior to 200-fold being needed to avoid toxicity.21 Here, we investigated how combining dilution with a commercial protease inhibitor (Pefabloc® SC PLUS) could be used to mitigate the toxicity of the digestive fluids.The use of serine protease inhibitors has been linked to a reduction of the TEER in Caco-2 cultures due to inhibition of the membrane-anchored matriptase, which co-localises with E-cadherin in apical junctional complexes.35 However, in Pefabloc® SC PLUS, the PLUS additive prevents covalent attachment to proteins when used in extended incubation times at an alkaline pH, thus avoiding cell toxicity. In addition, to assess the Papp of LY, Pefabloc® SC was used in cell cultures only following addition to the digesta and dilution in HBSS. Given that Pefabloc® SC binds covalently (and irreversibly) to the serine at the active centre of serine proteases, and given the high concentrations of proteases in the digesta, the actual concentration of Pefabloc® SC exposed to the cell cultures is drastically reduced. Indeed, we monitored the TEER in cell culture inserts following exposure to digesta inhibited with Pefabloc® SC PLUS and there was no reduction over the 4 hours of the experiments (ESI Table 3†).
Fig. 4A shows that there was no cytotoxicity (p > 0.05 vs. control) of the digestive fluids from the gastric phase towards semi-confluent Caco-2 cells even at the lowest dilution tested (1:4). This is due to pepsin becoming inactive at pH above 6.8 and irreversibly denatured at pH 7.5 and above.36 Note that the large buffering capacity of complete MEM and HBSS brings the pH of the final test solutions to the pH range 6.97 to 7.60 (ESI Table 4†). Conversely, when incubated with the intestinal digestive fluids, cells showed a significant decrease in metabolic activity (p < 0.001), which was more marked at the lower dilutions. This indicated that pancreatin and/or bile salts,20 which are not present in the gastric phase and are active at a neutral pH, are likely to be responsible for the reduction in metabolic activity observed.
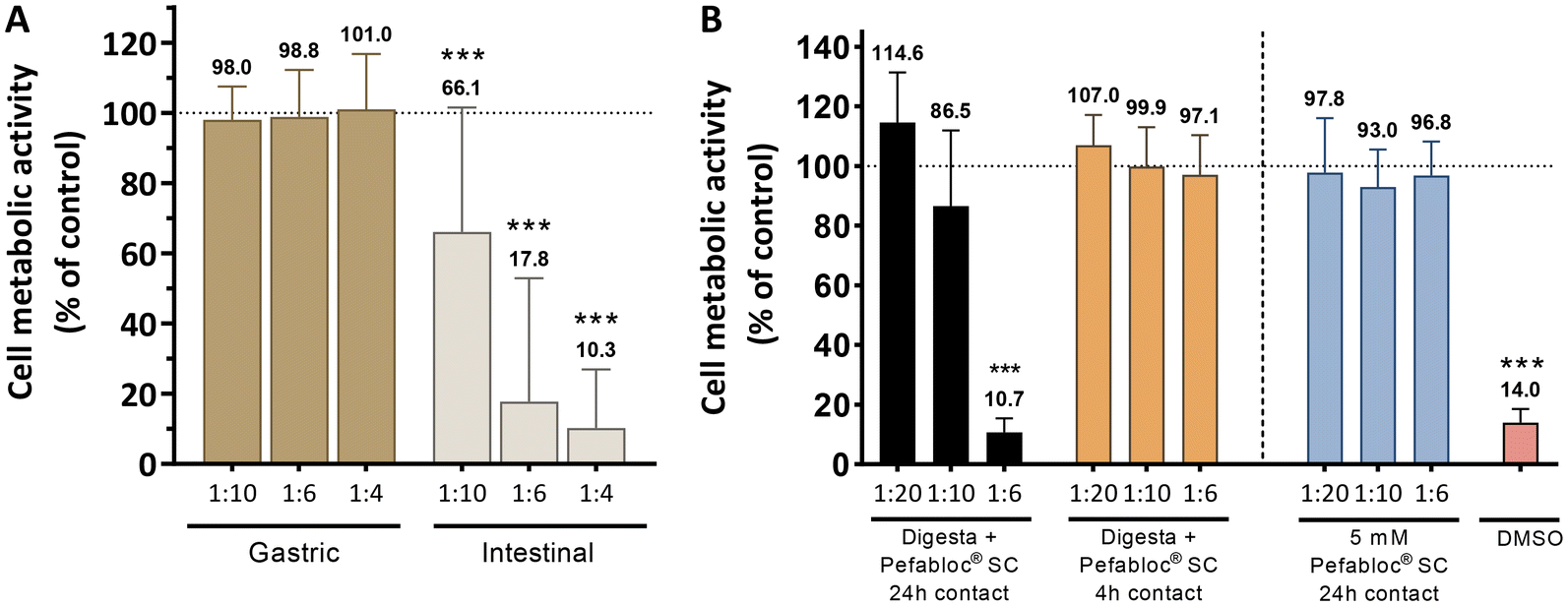 | ||
| Fig. 4 Cell metabolic activity of (A) semi-confluent Caco-2 cells following exposure for 24 h to simulated gastric and intestinal fluids of digestion diluted at 1:10, 1:6 and 1:4; and (B) Caco-2/HT29-MTX co-cultures following 4 h or 24 h exposure to digesta from complete simulated digestions treated with an irreversible protease inhibitor and diluted at 1:20, 1:10 and 1:6. 40% DMSO was used as a control. Values show Mean ± SD (N ≥ 3; ***p < 0.001 vs. control (HBSS – dotted line at 100%) with p-values obtained using the Kruskal–Wallis test followed by the Mann–Whitney U test). | ||
The use of the irreversible protease inhibitor Pefabloc® SC was not sufficient (Fig. 4B) to mitigate the cytotoxicity of the digesta resulting from a complete digestion simulation of the three main phases (oral, gastric and intestinal) when the contact time with differentiated Caco-2/HT29-MTX co-cultures was 24 h. However, when the contact time was limited to 4 h, which is more representative of a real scenario and the typical length of an intestinal permeability assay, cell metabolic activity was maintained even when a 1:6 dilution was used. These were the conditions used for subsequent permeability studies on the cell-based gut-chip. It should be noted that to the best of our knowledge this is the lowest dilution used in intestinal permeability studies with simulated digesta reported in the literature so far, with the previous best achievement set at 1:8 dilution and using simulated digestion fluids with lower total enzymatic activity.17,21
3.4 Gut-chip and intestinal permeability
To assess whether digesta from the digestion-chip could be used in intestinal permeability studies in hanging cell culture inserts or in a fully microfluidic-based platform, Caco-2/HT29-MTX co-cultures were grown on-chip under continuous flow for 7 days or in hanging inserts for 7 and 21 days. Firstly, the morphology of the epithelium was compared to static cultures on hanging inserts. Fig. 5A shows that cells grown on-chip formed an epithelium with a 3-dimensional structure resembling intestinal villi, in contrast with a flat, planar epithelium when cells were grown in static conditions even after 21 days. Immunocytochemistry of occludin and f-actin also showed that the cells formed an apical brush border with well-defined tight junctions (Fig. 5A, B and ESI Video 1†). This was in keeping with seminal studies on gut-chip technology that showed that flow-induced shear stress (here approximately 0.0025 dyn cm−2) accelerates cell differentiation and promotes spontaneous villi morphogenesis.37,38 The same authors showed that mechanical cyclic strain does not produce a significant additive effect.37 | ||
| Fig. 5 (A) Confocal microscopy images of occludin (green), f-actin (red) and nuclei (blue) of Caco-2/HT29-MTX co-cultures grown on the gut-chip and orthogonal views on the gut-chip and on day-7 and day-21 hanging inserts. Scale bar: 100 µm. (B) Confocal microscopy images of occludin (green), f-actin (red) and nuclei (blue) of Caco-2/HT29-MTX co-cultures grown on the gut-chip and challenged with 2 mM EDTA causing a disruption of cellular tight junctions. (C) Apparent permeability of Lucifer Yellow across Caco-2/HT29-MTX co-cultures grown on-chip and on hanging inserts and exposed to simulated digesta. Values show Mean ± SD (N ≥ 3; ns: not significant; *p < 0.05; **p < 0.01; ***p < 0.001 with p-values obtained using a non-parametric Mann–Whitney U test). | ||
Note that for the two devices to be used in a single platform for complete bioaccessibility and intestinal permeability studies, the flow rate of the digestion chip (outflow at 96 µL h−1) needs to be matched to that of the gut chip (inflow at 20 µL h−1) while considering the dilution factor determined in section 3.3. While we foresee continuous operation of the two devices, in this work the devices were operated in a semi-batched approach, and the combination in a single platform is the focus of one of our current research goals.
Intestinal permeability studies were run using Lucifer Yellow (LY), which is a small (521.8 Da), hydrophilic fluorescent molecule that is commonly used to determine paracellular transport and thus serves as an indicator of epithelial barrier integrity.39 Critically, when the permeability assay was run in the presence of EDTA, a divalent ion chelator that causes partial cell dissociation by disturbing the calcium-dependent adherens junctions (Fig. 5B), the apparent permeability of LY increased significantly when compared to the control condition (HBSS) in both the gut-chip and hanging inserts (Fig. 5C, p < 0.05). The apparent permeability of the EDTA-challenged epithelium was nearly identical in the 3 conditions (7-day and 21-day inserts, and 7-day gut chip). However, when the assay was run in HBSS (and thus the epithelial integrity was not compromised) there was a significant difference between the apparent permeability of LY in the hanging inserts when compared to the gut-chip (1.8 × 10−7 ± 2.5 × 10−7vs. 3.7 × 10−6 ± 1.4 × 10−6, p < 0.001). Although a higher Papp in the gut-chip could be perceived as a negative result, in fact it has been reported that in vitro intestinal epithelial models grown using Caco-2 cells are selective but are less permeable when compared to the in vivo scenario.40,41 A study by Rozehnal et al. using an ex vivo model of human small intestinal and colonic tissue mounted on using chambers determined the apparent permeability of LY ex vivo at 4.0 × 10−6 ± 2.2 × 10−6 (n = 11) and 4.4 × 10−6 ± 2.7 × 10−6 (n = 16) in the small intestine and in the colon respectively, which is a very close approximation to the Papp obtained in our Gut-Chip.
Finally, the Papp of LY was assessed on both gut-chips and 21-day inserts following exposure to simulated digesta. This study reiterated that in static conditions in hanging inserts, a 1:6 dilution appears to be not sufficient to mitigate the cytotoxicity of the digesta towards differentiated Caco-2/HT29-MTX co-cultures, which led to a dramatic increase in the Papp of LY of nearly 1000-fold. In contrast, when the digesta were diluted and protease activity was inhibited using Pefabloc® SC, there were no significant differences between the Papp of LY in the challenged epithelium and the control (HBSS), validating the data shown in section 3.3. In the gut-chip, 1:6 dilution was sufficient to maintain the Papp of LY similar to that of the control at 2.5 × 10−6 ± 6.0 × 10−7vs. 3.7 × 10−6 ± 1.4 × 10−6 (not significant). This was in agreement with the results from de Haan et al., where 1:8 dilution was used without any further treatment to the digesta in flow-through hanging inserts,17 and indicates that the intestinal epithelium grown under continuous flow may be more robust than in static conditions. Interestingly, when Pefabloc® SC was added there was a reduction in the Papp of LY to 0.8 × 10−6 ± 1.4 × 10−7. This suggests that there might be a competitive effect between LY and Pefabloc® SC, which is also a hydrophilic compound (4-benzenesulfonyl fluoride hydrochloride – AEBSF) and of similar molecular weight (239.5 Da). Transcellular passage of LY can also occur via organic anion transporters.39 AEBSF has been previously indicated as an inhibitor of organic anion transporters, such as MOAT-B (MRP4).42,43 It is thus important to use adequate controls in apparent permeability studies combining LY and AEBSF.
4 Conclusion
This work showed the development and validation of a modular microfluidic-based platform with the capacity to mimic the human gastro-intestinal tract for bioaccessibility and intestinal permeability studies. Although digestion occurred to a lesser extent on-chip when compared to the gold-standard static INFOGEST protocol, the results were consistent and reproducible, and a time-resolved digestion profile could be obtained on-chip in a fully ‘sample-in-answer-out’ fashion. This device could thus prove to be invaluable for bioaccessibility studies of minute samples, such as nanoformulated drugs or food ingredients, where conventional static protocols and/or large dynamic enzymatic reactors are not suitable. Nevertheless, we identified possible refinements to the current device, which are the focus of our current studies. These include (i) the use of a different material (PMMA) to avoid absorption/adsorption of small molecules by PDMS; (ii) automated pH and temperature control with closed loop feedback systems; (iii) constant mixing in 2 separate (gastric and intestinal) incubation chambers; and (iv) gastric emptying from the gastric to the intestinal phase. The cytotoxicity of the digestion fluids was mitigated using a combination of dilution (1:6) and protease inhibition using a commercial inhibitor – Pefabloc® SC. Critically, intestinal permeability assays using Lucifer Yellow as a paracellular transport reporter could be run on a cell-based gut-chip device in simultaneous contact with digesta from the digestion-chip. While the dilution factor (1:6) is, to the best of our knowledge, the lowest reported thus far, it is important to note that it is still only an approximation to the in vivo scenario and that there is room for improvement and for the motivation to search for better solutions to mitigate the toxicity of simulated digesta. In sum, the microfluidic platform shown here offers great potential to be used in robust in vitro bioaccessibility and intestinal permeability studies of new compounds intended for oral administration.
Data availability
The complete dataset supporting this article is available from https://doi.org/10.5281/zenodo.7324498.Author contributions
M. Xavier: conceptualisation, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, verification, visualisation, writing – original draft preparation, and writing – review and editing. P. Rodrigues: data curation, formal analysis, investigation, methodology, visualisation, and writing – review and editing. M. Neto: data curation, formal analysis, investigation, methodology, visualisation, and writing – review and editing. M. I. Guedes: data curation, formal analysis, investigation, methodology, visualisation, and writing – review and editing. V. Calero: data curation, formal analysis, investigation, methodology, verification, visualisation, and writing – review and editing. C. Gonçalves: conceptualisation, data curation, funding acquisition, investigation, methodology, project administration, supervision, verification, and writing – review and editing. L. Pastrana: conceptualisation, funding acquisition, project administration, supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the SbDtoolbox project (grant agreement NORTE-01-0145-FEDER-000047) funded by NORTE2020 and PORTUGAL2020 partnership agreement through the European Regional Development Fund, and by the European Union's Horizon 2020 research and innovation programme through the project GASTRIC, under the Marie Sklodowska-Curie grant agreement no. 101003440. The authors would like to thank the INL cleanroom micro and nano-fabrication staff for the fabrication of the silicon micropillar arrays and the photolithography hard masks used within this work, and the INL Nanophotonics and BioImaging (NBI) facility.References
- A. Alegría, G. Garcia-Llatas and A. Cilla, Static Digestion Models: General Introduction, in The Impact of Food Bioactives on Health, ed. K. Verhoeckx, P. Cotter, I. López-Expósito, C. Kleiveland, T. Lea, A. Mackie, et al., In Vitro and Ex Vivo Models, Springer, 2015, pp. 3–12 Search PubMed.
- E. W. Esch, A. Bahinski and D. Huh, Organs-on-chips at the frontiers of drug discovery, Nat. Rev. Drug Discovery, 2015, 14(4), 248–260 CrossRef CAS PubMed.
- M. Xavier, I. A. Parente, P. M. Rodrigues, M. A. Cerqueira, L. Pastrana and C. Gonçalves, Safety and fate of nanomaterials in food: The role of in vitro tests, Trends Food Sci. Technol., 2021, 109, 593–607 CrossRef CAS.
- T. Bohn, F. Carriere, L. Day, A. Deglaire, L. Egger and D. Freitas, et al., Correlation between in vitro and in vivo data on food digestion. What can we predict with static in vitro digestion models?, Crit. Rev. Food Sci. Nutr., 2018, 58(13), 2239–2261 CrossRef CAS PubMed.
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn and C. Bourlieu, et al., A standardised static in vitro digestion method suitable for food - an international consensus, Food Funct., 2014, 5(6), 1113–1124 RSC.
- A. Brodkorb, L. Egger, M. Alminger, P. Alvito, R. Assunção and S. Ballance, et al., INFOGEST static in vitro simulation of gastrointestinal food digestion, Nat. Protoc., 2019, 14(4), 991–1014 CrossRef CAS PubMed.
- D. DuPont, M. Alric, S. Blanquet-Diot, G. Bornhorst, C. Cueva and A. Deglaire, et al., Can dynamic in vitro digestion systems mimic the physiological reality?, Crit. Rev. Food Sci. Nutr., 2019, 59(10), 1546–1562 CrossRef CAS PubMed.
- P. de Haan, M. A. Ianovska, K. Mathwig, G. A. A. van Lieshout, V. Triantis and H. Bouwmeester, et al., Digestion-on-a-chip: a continuous-flow modular microsystem recreating enzymatic digestion in the gastrointestinal tract, Lab Chip, 2019, 19(9), 1599–1609 RSC.
- K. Ronaldson-Bouchard and G. Vunjak-Novakovic, Organs-on-a-Chip: A Fast Track for Engineered Human Tissues in Drug Development, Cell Stem Cell, 2018, 22(3), 310–324 CrossRef CAS PubMed.
- H. J. Kim, H. Li, J. J. Collins and D. E. Ingber, Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(1), E7–E15 CAS.
- M. Maurer, M. S. Gresnigt, A. Last, T. Wollny, F. Berlinghof and R. Pospich, et al., A three-dimensional immunocompetent intestine-on-chip model as in vitro platform for functional and microbial interaction studies, Biomaterials, 2019, 220, 119396 CrossRef CAS PubMed.
- M. S. Jeon, Y. Y. Choi, S. J. Mo, J. H. Ha, Y. S. Lee and H. U. Lee, et al., Contributions of the microbiome to intestinal inflammation in a gut-on-a-chip, Nano Convergence, 2022, 9(1), 1–13 CrossRef PubMed.
- S. Rajasekar, D. S. Lin, L. Abdul, A. Liu, A. Sotra and F. Zhang, et al., IFlowPlate—A Customized 384-Well Plate for the Culture of Perfusable Vascularized Colon Organoids, Adv. Mater., 2020, 32(46), 2002974 CrossRef CAS PubMed.
- M. Kasendra, A. Tovaglieri, A. Sontheimer-Phelps, S. Jalili-Firoozinezhad, A. Bein and A. Chalkiadaki, et al., Development of a primary human Small Intestine-on-a-Chip using biopsy-derived organoids, Sci. Rep., 2018, 8(1), 2871 CrossRef PubMed.
- R. S. Al-Lamki, J. R. Bradley and J. S. Pober, Human Organ Culture: Updating the Approach to Bridge the Gap from In Vitro to In Vivo in Inflammation, Cancer, and Stem Cell Biology, Front. Med., 2017, 4(SEP), 148 CrossRef PubMed.
- M. W. van der Helm, O. Y. F. Henry, A. Bein, T. Hamkins-Indik, M. J. Cronce and W. D. Leineweber, et al., Non-invasive sensing of transepithelial barrier function and tissue differentiation in organs-on-chips using impedance spectroscopy, Lab Chip, 2019, 19(3), 452–463 RSC.
- P. De Haan, M. J. C. Santbergen, M. Van Der Zande, H. Bouwmeester, M. W. F. Nielen and E. Verpoorte, A versatile, compartmentalised gut-on-a-chip system for pharmacological and toxicological analyses, Sci. Rep., 2021, 11, 4920 CrossRef CAS PubMed.
- Y. Imura, E. Yoshimura and K. Sato, Micro Total Bioassay System for Oral Drugs: Evaluation of Gastrointestinal Degradation, Intestinal Absorption, Hepatic Metabolism, and Bioactivity, Anal. Sci., 2012, 28, 197–199 CrossRef CAS PubMed.
- A. Bettencourt, L. M. Gonçalves, A. C. Gramacho, A. Vieira, D. Rolo and C. Martins, et al., Analysis of the characteristics and cytotoxicity of titanium dioxide nanomaterials following simulated in vitro digestion, Nanomaterials, 2020, 10(8), 1–18 CrossRef PubMed.
- J. Yi, F. Zhong, Y. Zhang, W. Yokoyama and L. Zhao, Effects of Lipids on in Vitro Release and Cellular Uptake of β-Carotene in Nanoemulsion-Based Delivery Systems, J. Agric. Food Chem., 2015, 63(50), 10831–10837 CrossRef CAS PubMed.
- X. Huang, Y. Liu, Y. Zou, X. Liang, Y. Peng and D. J. McClements, et al., Encapsulation of resveratrol in zein/pectin core-shell nanoparticles: Stability, bioaccessibility, and antioxidant capacity after simulated gastrointestinal digestion, Food Hydrocolloids, 2019, 93, 261–269 CrossRef CAS.
- E. Déat, S. Blanquet-Diot, J. F. Jarrige, S. Denis, E. Beyssac and M. Alric, Combining the dynamic TNO-gastrointestinal tract system with a Caco-2 cell culture model: Application to the assessment of lycopene and α-tocopherol bioavailability from a whole food, J. Agric. Food Chem., 2009, 57(23), 11314–11320 CrossRef PubMed.
- A. R. Varadarajan, R. N. Allan, J. D. P. Valentin, O. E. C. Ocampo, V. Somerville and F. Pietsch, et al., An integrated model system to gain mechanistic insights into biofilm-associated antimicrobial resistance in Pseudomonas aeruginosa MPAO1, npj Biofilms Microbiomes, 2020, 6, 46 CrossRef CAS PubMed.
- C. Hilgendorf, H. Spahn-Langguth, C. G. Regårdh, E. Lipka, G. L. Amidon and P. Langguth, Caco–2 versus Caco–2/HT29−MTX Co–cultured Cell Lines: Permeabilities Via Diffusion, Inside– and Outside–Directed Carrier–Mediated Transport, J. Pharm. Sci., 2000, 89(1), 63–75 CrossRef CAS PubMed.
- C. R. Kleiveland, Co-cultivation of Caco-2 and HT-29MTX, in The Impact of Food Bioactives on Health, ed. K. Verhoeckx, P. Cotter, I. López-Expósito, C. Kleiveland, T. Lea, A. Mackie, et al., In Vitro and Ex Vivo Models, Springer, 2015, pp. 135–144 Search PubMed.
- A. Béduneau, C. Tempesta, S. Fimbel, Y. Pellequer, V. Jannin and F. Demarne, et al., A tunable Caco-2/HT29-MTX co-culture model mimicking variable permeabilities of the human intestine obtained by an original seeding procedure, Eur. J. Pharm. Biopharm., 2014, 87(2), 290–298 CrossRef PubMed.
- A. Sontheimer-Phelps, D. B. Chou, A. Tovaglieri, T. C. Ferrante, T. Duckworth and C. Fadel, et al., Human Colon-on-a-Chip Enables Continuous In Vitro Analysis of Colon Mucus Layer Accumulation and Physiology, Cell. Mol. Gastroenterol. Hepatol., 2020, 9(3), 507–526 CrossRef PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow, Lab Chip, 2012, 12(12), 2165–2174 RSC.
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezic, H. A. Stone and G. M. Whitesides, Chaotic mixer for microchannels, Science, 2002, 295(5555), 647–651 CrossRef CAS PubMed.
- W. R. Akeson and M. A. Stahmann, A Pepsin Pancreatin Digest Index of Protein Quality Evaluation, J. Nutr., 1964, 83(3), 257–261 CrossRef CAS PubMed.
- E. Berthier, E. W. K. Young and D. Beebe, Engineers are from PDMS-land, Biologists are from Polystyrenia, Lab Chip, 2012, 12(7), 1224–1237 RSC.
- M. W. Toepke and D. J. Beebe, PDMS absorption of small molecules and consequences in microfluidic applications, Lab Chip, 2006, 6, 1484–1486 Search PubMed.
- R. Mukhopadhyay, When PDMS isn't the best, Anal. Chem., 2007, 79(9), 3249–3253 Search PubMed.
- P. M. Rodrigues, M. Xavier, V. Calero, L. Pastrana and C. Gonçalves, Partitioning of Small Hydrophobic Molecules into Polydimethylsiloxane in Microfluidic Analytical Devices, Micromachines, 2022, 13(5), 713 CrossRef PubMed.
- M. S. Buzza, S. Netzel-Arnett, T. Shea-Donohue, A. Zhao, C. Lin and K. List, et al., Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine, PNAS, 2010, 1079, 4200–4205 Search PubMed.
- E. Bathoorn, P. Daly, B. Gaiser, K. Sternad, C. Poland and W. MacNee, et al., Cytotoxicity and Induction of Inflammation by Pepsin in Acid in Bronchial Epithelial Cells, Int. J. Inflammation, 2011, 2011, 1–5 CrossRef PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow, Lab Chip, 2012, 12(12), 2165 RSC.
- H. J. Kim and D. E. Ingber, Gut-on-a-Chip microenvironment induces human intestinal cells to undergo villus differentiation, Integr. Biol., 2013, 5(9), 1130 CrossRef CAS PubMed.
- T. S. Frost, L. Jiang, R. M. Lynch and Y. Zohar, Permeability of Epithelial/Endothelial Barriers in Transwells and Microfluidic Bilayer Devices, Micromachines, 2019, 10(8), 533 CrossRef PubMed.
- B. Sarmento, F. Andrade, S. B. da Silva, F. Rodrigues, J. das Neves and D. Ferreira, Cell-based in vitro models for predicting drug permeability, Expert Opin. Drug Metab. Toxicol., 2012, 8(5), 607–621 CrossRef CAS PubMed.
- P. Artursson, K. Palm and K. Luthman, Caco-2 monolayers in experimental and theoretical predictions of drug transport, Adv. Drug Delivery Rev., 2001, 46(1–3), 27–43 CrossRef CAS PubMed.
- C. J. F. De Wolf, H. Yamaguchi, I. Van Der Heijden, P. R. Wielinga, S. L. Hundscheid and N. Ono, et al., GMP transport by vesicles from human, mouse erythrocytes, FEBS J., 2007, 274(2), 439–450 CrossRef CAS PubMed.
- A. A. K. El-Sheikh, Renal Transport and Drug Interactions of Immunosuppressants. [S.l.] : [S.n.]; 2008.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an02088b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |