
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent effects of N,N-dimethylformamide and methanol on mass spectrometry imaging by tapping-mode scanning probe electrospray ionization†
Yoichi
Otsuka
 *abc,
Nijiho
Ote
d,
Mengze
Sun
a,
Shuichi
Shimma
e,
Osamu
Urakawa
f,
Shinichi
Yamaguchi
g,
Tomoya
Kudo
g and
Michisato
Toyoda
ac
*abc,
Nijiho
Ote
d,
Mengze
Sun
a,
Shuichi
Shimma
e,
Osamu
Urakawa
f,
Shinichi
Yamaguchi
g,
Tomoya
Kudo
g and
Michisato
Toyoda
ac
aDepartment of Physics, Graduate School of Science, Osaka University, Japan. E-mail: otsuka@phys.sci.osaka-u.ac.jp
bJST, PREST, Japan
cForefront Research Center, Graduate School of Science, Osaka University, Japan
dDepartment of Biological Sciences, School of Science, Osaka Universit, Japan
eDepartment of Biotechnology, Graduate School of Engineering, Osaka University, Japan
fDepartment of Chemistry, Graduate School of Science, Osaka University, Japan
gShimadzu Corporation, Japan
First published on 20th February 2023
Abstract
Mass spectrometry imaging (MSI) is an effective technique for visualizing the distribution of lipids in tissues. The direct extraction-ionization methods using minute volumes of solvent for local components have the advantage of rapid measurement without any sample pretreatment. For effective MSI of tissues, it is necessary to understand the effect of solvent physicochemical properties on ion images. In this study, we report solvent effects on the lipid imaging of mouse brain tissue by tapping-mode scanning probe electrospray ionization (t-SPESI) which is capable of extraction-ionization using sub-pL solvents. To precisely measure lipid ions, we developed a measurement system incorporating a quadrupole-time-of-flight mass spectrometer. The differences in signal intensity and spatial resolution of lipid ion images were investigated using N,N-dimethylformamide (non-protic polar solvent), methanol (protic polar solvent) and their mixture. The mixed solvent was suitable for the protonation of lipids, and it provided high spatial resolution MSI. Results indicate that the mixed solvent improves the extractant transfer efficiency and minimizes charged droplets from an electrospray. The solvent selectivity study revealed the importance of solvent selection based on physicochemical properties for the advancement of MSI by t-SPESI.
1. Introduction
Cells are functional and structural units of life that are organized in organs that comprise our bodies, and the combinations of intracellular and extracellular chemical reactions maintain homeostasis. Analytical techniques that measure microscale changes in cellular metabolism in tissues are essential for understanding pathological mechanisms and identifying biomarkers.Lipids are an important component of the cell membrane, which play crucial roles such as energy storage and signal transduction. The relationship between lipid metabolism disorders and disease states has been clarified by lipidomic studies using mass spectrometry (MS).1–6 The comprehensive analysis using chromatography and MS can provide detailed structural information about lipids; however, determining the distribution of lipids inside tissues is difficult owing to the loss of spatial information during the chromatographic extraction process.
MS imaging (MSI) is a practical approach that enables the direct visualization of chemical distributions in tissues. By ionizing components in a local tissue region prior to MS, a mass spectrum associated with the position of the tissue is obtained. The distribution of the ionized species can be obtained by scanning the ionizing region over the tissue. Methods using ion or laser beams have been reported for the desorption–ionization of sample components. In secondary ion MS (SIMS), nanoscale MSI can be performed by irradiating a sample with focused ion beams; however, high-energy primary ion beams can cause fragmentation of biomolecules.7 Meanwhile, matrix assisted laser desorption/ionization (MALDI) can desorb and ionize components of a tissue by irradiating pulsed laser beam after applying a matrix coating.8
Unlike SIMS and MALDI, some atmospheric pressure sampling and ionization methods9,10 have employed microextraction using a small volume of solvent in conjunction with electrospray ionization (ESI).11 These methods are rapid and require minimal sample preparation. In a desorption ESI (DESI),12 charged droplets generated by an electrospray are sprayed onto the surface of a sample using a high-speed gas flow. Other methods used for MSI of tissues include liquid extraction surface analysis,13 liquid micro-junction surface sampling probe,14,15 nanospray DESI,16 and single-probe,17 which utilize capillary probes that deliver directly the solvent to a sample surface.
We have previously reported the development of tapping-mode scanning probe ESI (t-SPESI) and the applications for MSI of tissues.18–22 t-SPESI allows the extraction and ionization of components in a local region of tissue with picoliters of solvent via an oscillating capillary probe. We developed the laser-based measurement technique of probe oscillation and the feedback control system to stabilize probe scanning over the non-flat sample surface. We reported lipid imaging of the mouse brain section with a spatial resolution of 6.5 μm using a mixture of N,N-dimethylformamide (DMF) and methanol (MeOH).22
DMF and MeOH are nonprotic and protic polar solvents, respectively, and their mixtures have been reported as non-destructive solvents in DESI for lipid imaging of tissues.23 Differences in mass spectra were reported from measurements with DMF, MeOH, and their mixtures. The effect of solvents on signal intensity and spatial resolution in lipid imaging by t-SPESI is still unclear. To advance the MSI of tissues by t-SPESI, it is necessary to understand the relationship between physicochemical properties of solvent and ion images.
Here, we developed a new t-SPESI system with improved mass resolution and accuracy for MSI of lipids. DMF, MeOH, and their mixtures were used to investigate the effects of solvents on the MSI of mouse brain sections.
2. Materials and methods
2.1 MSI of mouse brain sections
Mouse brain (C57, Japan Bio Serum, Japan) was prepared into sections of 8 μm thickness using a cryomicrotome (CM1950, Leica, Germany), and the sections were placed on glass slides. Tissue sections were frozen at −80 °C in Falcon tubes containing silica gel. Before a measurement, the tube was returned to room temperature. DMF (high performance liquid chromatography (HPLC) grade, 13024-71) and MeOH (HPLC grade, 21929-81) from Nacalai Tesque, Japan, and their equivalent mixture by volume were used as solvents. Different mouse sections were used in the measurements with different solvents. Representative optical microscope images of mouse brain section samples are shown in Fig. S1.†Silica emitters with a 5 μm opening aperture (SilicaTip Emitters, FS360-20-5-N, New Objective, USA) were used as capillary probes. The solvent was supplied at a flow rate of 20 nL min−1 using a laboratory syringe pump. The resonant frequencies of the probe were 694.9, 694.5, and 684.1 Hz for experiments using DMF, mixed solvent, and MeOH, respectively. The measurement scheme was the same as in the previous publication.22 For data acquisition, the probe was scanned at a constant speed in the right direction from the upper left position of the measurement area (it corresponds to the ion image). The scanning speeds for measurements using DMF, mixed solvent, and MeOH solvent were 353, 370, and 373 μm s−1, respectively. When a single line scan was completed, the probe returned to the start position of the scan and moved down 65 μm for the next scan. At the beginning of each single-line scan, a trigger signal was input to the mass spectrometer to start the measurement. Mass spectra were acquired in positive ionization mode over the m/z range of 100–2000 with a 100 ms accumulation time. Thus, the pixel size of the ion images was 35–37 μm and 65 μm in horizontal and vertical axis, respectively. The signal intensity of a single pixel in an ion image corresponds to the sum of the ion signals measured in 68 or 69 extraction-ionization events on average. The voltage applied to the solvent through the stainless-steel union was +5 kV.
The ion inlet, which was used to guide the ions produced by t-SPESI into the desolvation line (DL) of the MS instrument (LCMS-9030, Shimadzu, Japan), was made with a stainless-steel tube (outer diameter = 1/8 in and tube thickness = 0.5 mm) and was heated at 300 °C and a voltage of 100 V was applied by an external power supply. The ions entering the DL were guided into the mass spectrometer. The DL and heat block were heated to 300 °C. The vacuum inside the developed interface unit was set to 90.6 kPa. Before any MSI measurement, mass calibration was performed by conducting ESI of a sodium iodide solution from the side probe connected to the interface unit. Compared to the previous measurement system,22 the mass resolving power of the mass spectrometer has been improved from 6000 to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 full width at half maximum.
000 full width at half maximum.
In the present measurements, the estimated average volume of solvent consumed in a single oscillation cycle (extraction–ionization) was about 480 fL by dividing the flow rate by the oscillation frequency of the probe.
2.2 Data analysis
Multiple raw data files in lcd format (Shimadzu) acquired for all probe scannings were converted to a single imaging data file in imdx format (Shimadzu) using an in-house data converter. Data analysis of the imaging data was carried out as follows using IMAGEREVERAL from Shimadzu. Data preprocessing was performed by normalizing signal intensities to the total ion count of a mass spectrum in each pixel. The region of interest (ROI) was set as the entire brain section, and its average mass spectrum was calculated. Peak detection was conducted in the m/z range of 600–1000. For locating monoisotopic masses, the set parameters of minimum number of isotopic cluster peaks and matching tolerance were 2 and 50 ppm, respectively. 93 ions of monoisotopic mass were selected for the level 2 putative annotation24 with results of Lipidmaps bulk structure searches.25 Adduct ions with proton ([M + H]+), sodium ion ([M + Na]+) and potassium ion ([M + K]+) were the targets, and the mass tolerance of ±0.005 m/z units was used.In the assignment of lipids, the abundant lipid with the smallest mass error from the theoretical value was selected if several candidates were found for the same ion peak. Based on a study on the quantification of lipids in mouse brain tissue using liquid chromatography (LC)-MS, the amount of lipids varies in the following order:26 phosphatidylcholines (PC) > phosphatidylethanolamine with alkyl ether substituent (PE-O) ≈ phosphatidylethanolamine (PE) > phosphatidylserine > hexosylceramides (HexCer) > phosphatidylcholines with alkyl ether substituent.
If several candidates for the m/z value of an ion peak with similar mass differences were found, the following additional rules were used for assignment. (1) The ion with several lipid suggestions was assigned as another cation adduct molecule of the assigned lipid when the image of an ion with several lipid suggestions was similar to that of an ion with a single lipid assignment. In addition, the difference between the m/z value of the ion with several lipid suggestions and the theoretical m/z value of a cation adduct for the ion with a single lipid assignment was <5 ppm. (2) It has been reported that the amounts of PE and PE-O increases and decreases, respectively in the hippocampus, motor cortex, olfactory bulb and thalamus;26 hence, if an unassigned ion was a candidate for both PE and PE-O, it was assigned as PE. (3) the abundant PCs previously reported in the MSI of mouse brain sections27–31 were used for the assignment.
Based on the above procedure, 25 ion images with a single lipid assignment were selected (Table S1†). The other 68 ion images and their m/z values with multiple candidates and a single candidate are summarized in the ESI (Fig. S2 and Table S2†). The intensity of an integrated signal within ±0.002 Da relative to the m/z of each ion peak was used for the ion images. The image contrast was automatically adjusted for each ion image using the linear rescaling function of IMAGEREVEAL. To compare the signal intensities of the lipid ions for each structure within the mouse brain, seven regions (i.e., fimbria (fi), corpus callosum (cc), thalamus (TH), hippocampal formation (HPF), hypothalamus (HY), cerebral cortex (CTX) and cortical subplate (CTXsp)) were assigned as ROIs (Fig. S3†), and the averaged signal intensities of the 25 ions with a single lipid assignment were calculated for all ROIs. We referred to the ROIs (mouse, P56, coronal, image number 72) from Allen Brain Atlas (https://atlas.brain-map.org/).
2.3 Physical properties of the solvents used
The surface tension and viscosity of DMF and MeOH mixtures were measured to examine the transfer process of the extracts in t-SPESI. The volume ratio of DMF in the mixtures were varied from 0% to 100%. Surface tension was measured using the Wilhelmy method. A dynamic contact angle analyzer (DCA-700, Kyowa Interface Science, Japan) was used to measure the force between platinum plate and solvent, and the obtained data were analyzed using a Kyowa Dynalyzer.Viscosity was measured using a openQCM Q-1 quartz crystal microbalance (Novaetch S.r.l., Italy). A quartz crystal with a base frequency of 10 MHz was used to measure the resonance frequency and half-width at half-maximum for air and solvent. The zero shear viscosity was determined according to Johannsmann's method32,33 The viscosities34 of DMF and MeOH were used to calibrate the instrument.
3. Results and discussion
3.1 t-SPESI system development
To improve the mass resolution of MSI, we developed a new measurement system that incorporates a quadrupole-time-of-flight (Q-TOF) MS instrument, and its block diagram is shown in Fig. 1a. The t-SPESI unit consists of a probe oscillation control unit and sample stage. A capillary probe made of fused silica was fixed to a probe holder attached to a piezoelectric actuator, and it is oscillated vertically at its resonant frequency by sine wave voltage. The angle of probe from the sample surface was 45°. A solvent was delivered to the probe apex using a syringe pump through polyether ether ketone (PEEK) tubing, stainless-steel union and precolumn filter made of PEEK (Fig. 1b). To charge the solvent, the union was connected to a high-voltage power supply. | ||
| Fig. 1 (a) Block diagram of the developed t-SPESI system. (b) Schematic illustration of the tubing used to supply a charged solvent to the capillary probe. | ||
When the sample and probe-end come into contact, a liquid bridge of solvent was formed between them, causing the extraction of the sample components into the solvent. The extractant was transferred by the upward motion of the probe, and the extractant on the probe tip mixed with the solvent flowing inside the probe. The solution was converted to charged droplets by ESI due to the electrical potential difference between the solution on the probe tip and the opposing ion inlet.19
For probe oscillation amplitude measurement, the side of the probe was irradiated by a laser beam, and the probe's shadow and the laser beam were guided to the two-segment photodiode. The signal difference (VA–B) of each segment denoted by A and B was measured by the differential amplifier. To reduce floor noise found in a previous system,22 an optical bandpass filter was placed in front of the photodiodes that detect the laser beam. Moreover, the electrical circuit was redesigned using low-noise amplifiers and power supply.
The probe oscillation amplitude was measured by inputting the measured value of VA–B and sine wave voltage for the probe oscillation to the signal and reference terminals of the lock-in amplifier, respectively. The amplitude signal was inputted into a proportional-integral feedback control unit, which outputs a feedback signal for a target amplitude value. The feedback signal was inputted to a piezo Z stage via a high-voltage amplifier, and the height of the sample stage was adjusted to maintain a constant oscillation amplitude while the probe was scanning over the sample.
We have newly designed and fabricated an interface unit (IFU) to introduce the ions generated by t-SPESI into the mass spectrometer. The IFU is an aluminum enclosure to which a 270 mm long stainless-steel ion inlet and ESI source (sub-probe) for calibration were connected. It was attached to the mass spectrometer by replacing a commercial ESI unit. The ion inlet, which extends outside the IFU, was heated and electrically biased using a heating block and external power supply, respectively. A spacing of ∼1 mm was maintained between ion inlet and DL that served as the ion entrance of the mass spectrometer. The IFU was evacuated using a diaphragm pump, and the flow rate (0.6 L min−1) was adjusted using a mass flow controller. The instruments used in this study is summarized in Table S3.†
3.2 Solvent effects on the MSI of mouse brain sections
To examine changes in the signal intensity of ions from different solvents, the signal intensity ratio of the mixed solvent and a pure solvent for 93 ions were calculated, and the results for the ratios with DMF and MeOH are shown in Fig. S5.† An intensity ratio of >1 means that the signal intensity in a pure solvent is greater than the mixed solvent. The signal intensities of 53% and 32% of the total ions were greater with DMF and MeOH, respectively, compared with the mixed solvent.
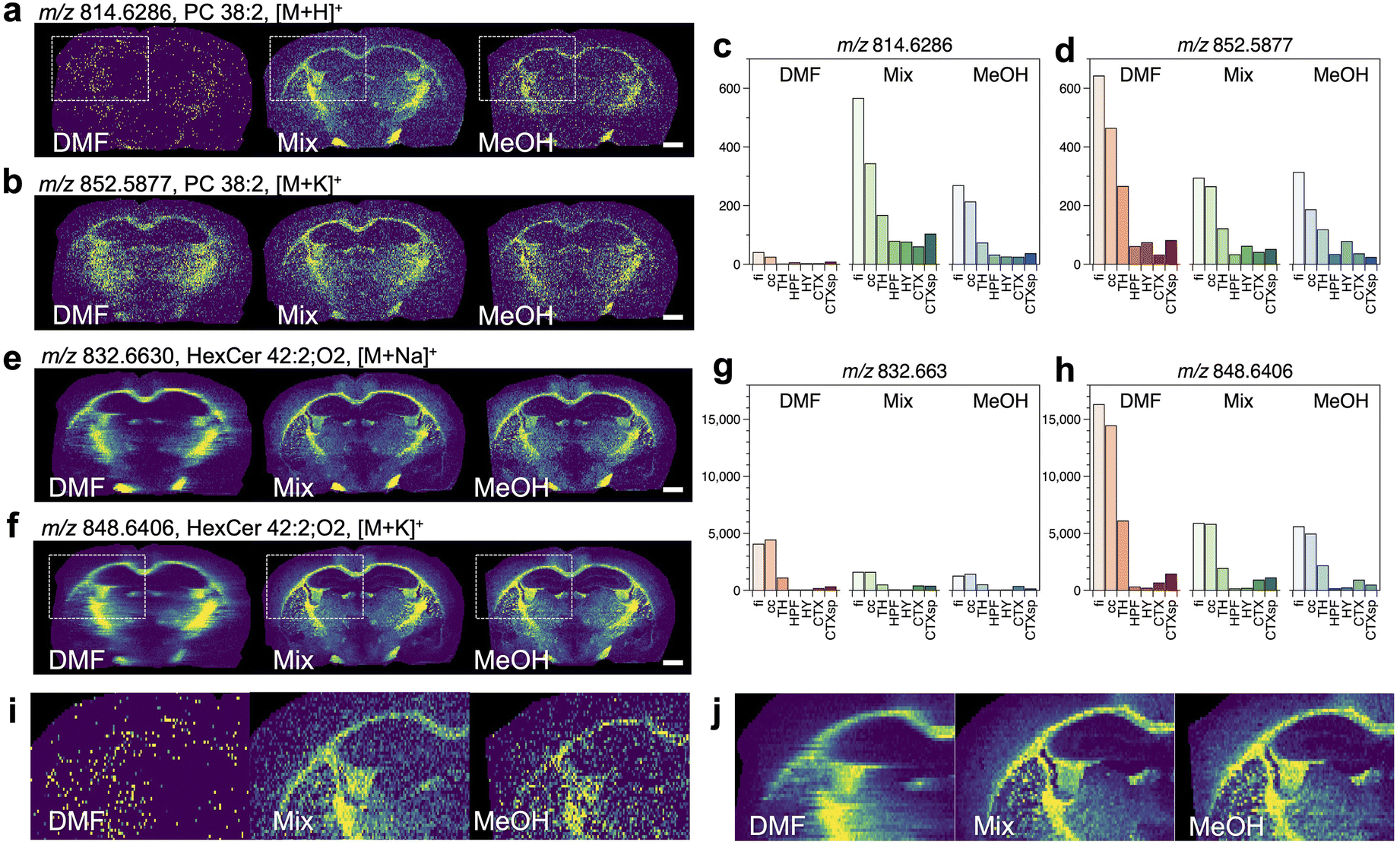 | ||
| Fig. 2 Comparison of ion images obtained for mouse brain sections classified as category 1, and the averaged signal intensities of the ROIs inside the brain (a–h). The m/z values of ions, assigned lipid species, and types of adduct ions are shown. The magnified ion images of (a) and (f) are shown in (i) and (j), respectively. Scale bar = 1 mm. | ||
Concerning the image of [M + H]+ for PC 38:2 (Fig. 2a), the signal intensities were higher in the fiber tracts region when the mixed solvent and MeOH were used. However, a clear distribution was not observed when DMF was used. Alternatively, the image of [M + K]+ for PC 38:2 showed signal distributions with higher intensities in the fiber tracts region for all the solvents (Fig. 2b). The signal intensities for [M + H]+ were higher in the fi and cc regions than other regions when the mixed solvent and MeOH were used, and they were substantially decreased when DMF was used (Fig. 2c). The signal intensities for [M + K]+ in the fi and cc regions were higher with DMF than the mixed solvents or MeOH (Fig. 2d). Similar results were found in the images of [M + H]+ and [M + K]+ for PC 38:1 (Fig. S7a–d†), which differs from PC 38:2 in the number of unsaturated bonds in the acyl chain.
Next, the ion images for HexCer (a group of sphingolipids) are compared. In the images of [M + Na]+ and [M + K]+ for HexCer 42:2; O2, a signal distribution was observed with all solvents (Fig. 2e and f). The signal intensities were higher in the fi and cc regions for all solvents, and lower signal intensities were obtained for [M + Na]+ than [M + K]+. Among the solvents studied, DMF showed the highest signal intensity for [M + K]+. Similar results were obtained for the ion images of [M + Na]+ and [M + K]+ for HexCer 40:2; O2 containing different acyl chain lengths (Fig. S7e–h†).
The magnified images of [M + H]+ for PC 38:2 and [M + K]+ for HexCer 42:2; O2 are shown in Fig. 2i and j, respectively. They correspond to the dashed regions in Fig. 2a and f, respectively. The boundaries between fi and HPF or CP (Fig. S8†) were recognized for the mixed solvent and MeOH. On the other hand, DMF caused a significant decrease of the signals in the image of [M + H]+ and the blurring of the image of [M + K]+, respectively. The comparison of ion image with different solvents indicated the signal difference between fi and adjacent VL, and the signal of localized lipids inside CP. On the other hand, it was difficult to visualize their spatial distribution when DMF was used (Fig. S9†).
 | ||
| Fig. 3 Comparison of ion images obtained for mouse brain sections classified as category 2, and the averaged signal intensities of the ROIs inside the brain (a–h). The m/z values of ions, assigned lipid species, and types of adduct ions are shown. The magnified ion images of (b) and (f) are shown in (i) and (j), respectively. Scale bar = 1 mm. | ||
For the image of [M + H]+ for PE 40:6, higher [M + H]+ signal intensities were observed with the mixed solvent and MeOH (Fig. 3a). In the image of [M + K]+ for PE 40:6, ion signals were obtained for all solvents (Fig. 3b). The signal intensities from the ROIs were compared. The signals of [M + H]+ from the fi and cc regions were lower in all the solvents, whereas the signal intensities from all ROIs were significantly lower when DMF was used (Fig. 3c). On the other hand, the signal intensity of [M + K]+ (Fig. 3d) was greater than that of [M + H]+ for all the solvents. Similar results were found in the images of [M + H]+ and [M + K]+ for PE 38:6 (Fig. S10a–d†), which differs in acyl chain length from PE 40:6.
The images of [M + K]+ obtained with all the solvents for PC 38:6 and PC 40:6 showed a higher signal intensity in TH that is in the central part of the brain compared with other regions (Fig. 3e and f). The signal intensities obtained for the ROIs were greater with DMF while the intensities with MeOH and the mixed solvent were comparable (Fig. 3g and h). In the images of [M + K]+ for PC 36:4 and PC 38:4, the signal intensities in HPF were significantly high for all the solvents (Fig. S10e and f†). The signal intensities obtained for the ROIs were greater with DMF while the intensities with MeOH and the mixed solvent were comparable (Fig. S10g and h†).
The magnified images of the dashed regions in Fig. 3b and f are shown in Fig. 3i and j, respectively. The boundaries between the fiber tracts region with lower signals and the regions with higher signals (i.e., HPF, CTX, and TH) were more apparent when the mixed solvent and MeOH were used while the boundaries were unclear when DMF was used.
 | ||
| Fig. 4 Comparison of ion images obtained for mouse brain sections classified as category 3, and the averaged signal intensities of the ROIs inside the brain (a–f). The m/z values of ions, assigned lipid species, and types of adduct ions are shown. Scale bar = 1 mm. | ||
Similar results were observed in the ion images of PC 34:1. Larger signal intensities were found from the fiber tracts and TH regions in the images of [M + H]+, [M + Na]+ and [M + K]+ when DMF was used. (Fig. S11a–f†). On the other hand, the ion images for each adduct ion were different when the mixed solvent and MeOH were used. The signal intensities from the internal capsule increased (Fig. S11a†) in the [M + H]+ images while the signal intensities from the fiber tracts, TH and HY decreased in the image of [M + Na]+ (Fig. S11b†). In the image of [M + K]+, the variation in signal intensities across the tissue was not significant, and the images showed a uniform contrast (Fig. S11c†). The signal intensities of [M + H]+ decreased in DMF (Fig. S11d†). The signal intensities of [M + Na]+ did not differ significantly among the solvents (Fig. S11e†). The signal intensities of [M + K]+ were strongest in DMF (Fig. S11f†).
In the images of [M + H]+, [M + Na]+ and [M + K]+ for SM 36:1; O2 with DMF, the signal intensities from fi, cc, and TH were greater compared with the other regions. On the other hand, the ion images obtained with the mixed solvent and MeOH showed a larger signal intensity distribution for HPF (Fig. S11g–i†), and the highest signal intensities for all the adduct ions were from HPF (Fig. S11j–l†).
A comparison of the ion images obtained for categories 1 (Fig. 2a, S7a†) and 2 (Fig. 3a, S10a†) revealed that the generation of [M + H]+ was suppressed by DMF. On the other hand, the generation was enhanced by the presence of MeOH, indicating the ability of MeOH as a proton donor. A mixture of DMF and MeOH has been reported to form hydrogen bonds and generate dipole–dipole interactions between the carbonyl oxygen of DMF and the hydroxyl group of MeOH.37–41 MS and theoretical analysis of DMF and MeOH cluster ions have verified the formation of the hydrogen bonds.42 These suggest the occurrence of energy stabilization of DMF and MeOH in the mixed solvent, which may reduce the proton-donation capability of MeOH for lipids. However, the signal intensities of [M + H]+ from a sample in the mixed solvent were higher compared with MeOH. This could be attributed to the higher solubility of lipids in the mixed solvent which increased the number of lipids available for ionization, as discussed in the following section 3.3.1.
Interestingly, in category 3, low signal intensities of [M + H]+ for PC 32:0, PC 34:1 and SM 36:1; O2 were observed from the fiber tracts, cc, and TH regions with DMF. Although the reason is unclear, the protonation of PCs and SM may be due to the presence of a proton source in these regions. The use of the mixed solvent and MeOH will induce the protonation of lipids in different regions, resulting in ion images that differ from DMF.
Next, regarding the MSI with the mixed solvent, the signal intensities for [M + K]+ were lower than those of [M + H]+ in categories 1 and 3. In contrast, the signal intensities for [M + K]+ were higher than that for [M + H]+ in category 2. Previous reports on lipid imaging of brain tissues have shown variations in the intensity of an adduct ion signal due to the presence of local cations in the tissues.43–46 The presented results indicate changes in the ratios of lipids to cations or protons in brain regions, resulting in differences in the signal intensities of the adduct ions. The results for category 1 suggest the abundance of potassium rather than sodium ions in white matter regions (Fig. 2e–h, S7e–h†). The higher signal intensities for [M + K]+ than that for [M + Na]+ in category 3 (PC 32:0 and PC 34:1) were consistent with a previous report.46
To suppress the changes in signal intensities due to an internal cation, potentially helpful approaches include the addition of an internal standard lipids46 and desalting of tissue sections.47–49 Furthermore, the addition of sodium iodide,50 ammonium fluoride35 or silver ion51 that utilize actively cation addition reactions may be practical for selective lipid ionization.
In order to check the reproducibility of the solvent effect on the ion images, we conducted additional measurements using different probes, brain sections, and solvents. The experimental conditions are shown in Table S4.† The averaged mass spectra (Fig. S12†) were varied with different solvents, and they were similar to the mass spectra shown in Fig. S4.† Several representative ion images (Fig. S13†) also showed similar results to those shown in Fig. 2–4 and S7, S10–11.† Meanwhile, the inter-experimental small variations of the mass spectra were confirmed. Possible factors affecting the ion signal intensities would be the flow rate of the solvent, the vibration frequency of the probe, and the relative position of the inlet and the probe apex. Further instrument development is needed to adjust these experimental parameters precisely in the future.
3.3 Effect of solvent physicochemical properties on extraction–ionization
In t-SPESI, a liquid bridge is formed when the probe tip contacts the sample surface, and molecules in the sample are extracted. The probe is then moved away from the sample, and the extractant is transferred to the probe tip. Finally, the extractant is mixed with the solvent supplied inside the probe before performing ESI. A dynamic change of the solvent on the probe tip affects likely the signal intensity and spatial resolution in MSI. In this section, we discuss the effect of the solvent on the extraction, transfer, and ionization processes of t-SPESI.The three Hansen parameters: the parameters of London dispersion force (δD), dipole-to-dipole force (δP) and hydrogen bonding force (δH) are used to describe the molecular interaction between solute and solvent. To evaluate the solubility of a solute in a solvent, Ra which represents the distance between the three solubility parameters for each solute and the solvent in Hansen space52 is given by eqn (1).
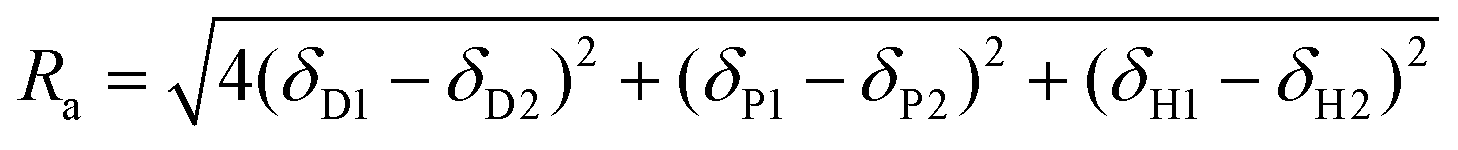 | (1) |
| RED = Ra/R0 | (2) |
We used the solubility parameters of DMF, MeOH and the mixed solvent58,59 (Table S5†), as well as the three lipids including PC 34:1, PE 34:2 and SM 34:1 (Fig. S14 and Table S6†)60 to calculate the values of Ra (Table S7†) and RED (Table 1). For DMF and the mixed solvent, the calculated RED values were ≤1 for all lipids, and the values were smaller for DMF than the mixed solvent. Meanwhile, the values were >1 for MeOH. These results suggest that the extraction efficiency of the studied solvents for the three lipids decreased in the order of DMF > the mixed solvent > MeOH.
| Solvent | PC (34:1) | PE (34:2) | SM (34:1) |
|---|---|---|---|
| MeOH | 1.5 | 1.4 | 1.1 |
| Mixed solvent | 1.0 | 0.9 | 0.6 |
| DMF | 0.8 | 0.7 | 0.5 |
| Ca = Uμ/γ | (3) |
To estimate the Ca for all solvents used in this study, the surface tension and viscosity of the mixed solvent were measured as a function of the mixing ratio, and the results are shown in Fig. 5. Linear increases in surface tension and viscosity were observed with increasing DMF, and these were consistent with previous results.34 The density of a solvent34,64 was used to calculate the values of Ca summarized in Table 2. The estimated stretch speed of a liquid bridge was in the range of 0.46–0.70 m s−1 (section S1†). The values of Ca decreased in the order: DMF, the mixed solvent and MeOH, suggesting that the transfer volume increased in the same order.
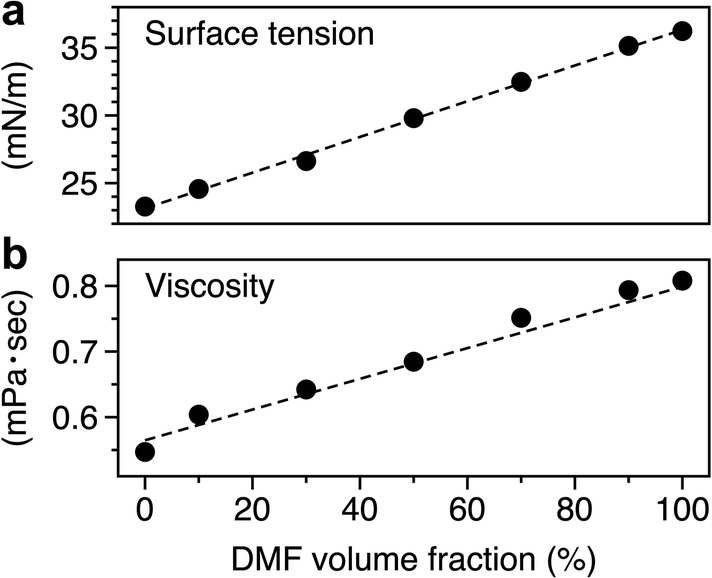 | ||
| Fig. 5 Relationship between the volume ratio of DMF in mixed solvents and the (a) surface tension or (b) viscosity. | ||
| Solvent | ρ (kg m−3) | μ (10−4 Pa s) | γ (10−2 N m−1) | U (m s−1) | Ca (10−2) |
|---|---|---|---|---|---|
| DMF | 944 | 8.08 | 3.62 | 0.70 | 1.6 |
| Mixed solvent | 896 | 6.84 | 2.98 | 0.64 | 1.5 |
| MeOH | 786 | 5.47 | 2.33 | 0.46 | 1.1 |
| VON ≈ (rcγcosθ/2ε0)1/2ln(4d/rc) | (4) |
In the ionization process of t-SPESI, ESI occurs after the probe is separated from the sample. The previous report suggested that the time from the application of voltage to stabilize ESI increased with increasing surface tension.66 It is conceivable that the time to start ESI in t-SPESI decreases according to the magnitude of surface tension in the order DMF > mixed solvent > methanol. The shorter the charging time to start ESI, the faster the extracted solution can be ionized (consumed).
The smaller transfer volume with DMF could lead to an increase in the solvent volume remaining on the tissue surface compared to other solvents. In addition, the extracted solution with DMF at the probe tip would not be sufficiently consumed during a single extraction–ionization event. In that case, the residual solutions on the probe tip would be supplied to the sample surface in the next extraction process. The enlarged extraction area, thereby, decreasing the spatial resolution of the MSI. In addition, the enlarged extraction area caused likely an increase in the concentration of lipids and cations in the extracted solution; thus, increasing the intensity of the ionic signals.
Above all, we conducted experimental and theoretical studies on the effects of three solvents on MSI by t-SPESI. The results indicate the better suitability of the mixed solvent compared with the pure solvents. Lipid solubility was higher in DMF; however, it showed lower transport and ionization efficiency, leading to a decreased spatial resolution. As a nonprotic solvent, it also suppressed the protonation of lipids. On the contrary, MeOH indicated higher transport and smaller charged droplets generation, but lower lipid solubility that produced lower signal intensities than the mixed solvent. To optimize the solvent for lipid imaging by t-SPESI, we propose to select solvents with lower RED, Ca, and surface tension among those that can dissolve lipids without deformation of the tissue.
4. Conclusions
The effects of the physicochemical properties of solvents on the MSI of tissues by t-SPESI were studied. Lipid imaging of mouse brain tissue was performed using the developed t-SPESI system connected to a Q-TOF-MS instrument. Comparison of DMF, MeOH, and the mixed solvent revealed that the mixed solvent provided higher signal intensities of protonated molecules; hence, it is more suitable for high spatial resolution imaging.The physicochemical properties of the mixed solvent were important factors in the balance among the extraction, transfer, and ionization processes in t-SPESI. The manipulation of charged liquids by oscillating capillary probes is a promising technique for miniaturizing liquids on substrates. Simultaneous miniaturization of the extraction region and high-efficiency extraction-ionization will realize the MSI of a single cell. The precise control of low-volume liquid in the pL to fL range is crucial for the advancement of MSI, and it will contribute to new knowledge in analytical chemistry and fluid mechanics.
Author contributions
Yoichi Otsuka: Conceptualization, formal analysis, methodology, project administration, resources, software, visualization, verification, funding acquisition, supervision, writing – original draft. Nijiho Ote: Data curation, investigation, visualization, writing – original draft. Mengze Sun: Sample preparation, data analysis. Shuichi Shimma: Resources, writing – review & editing. Osamu Urakawa: Formal analysis, methodology, resources, software, writing – review & editing. Shinichi Yamaguchi: Software. Tomoya Kudo: Methodology. Michisato Toyoda: Supervision, resources, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank Dr. Arihiro Kanazawa and Dr. Sadahito Aoshima at Osaka University for their guidance in measuring surface tension. This work was supported by JSPS Grants-in-Aid for Scientific Research on Innovative Areas “Chemistry for Multimolecular Crowding Biosystems” (JSPS KAKENHI Grant No. JP20H04710), AMED under Grant Number JP20he0322005, JP20lm0203014, Naito Foundation, and JST, PRESTO Grant Number JPMJPR20E4.References
- M. P. Wymann and R. Schneiter, Nat. Rev. Mol. Cell Biol., 2008, 9, 162–176 CrossRef CAS.
- M. Li, L. Yang, Y. Bai and H. Liu, Anal. Chem., 2014, 86, 161–175 CrossRef CAS.
- L. Yang, M. Li, Y. Shan, S. Shen, Y. Bai and H. Liu, J. Sep. Sci., 2016, 39, 38–50 CrossRef CAS.
- D. J. Stephenson, L. A. Hoeferlin and C. E. Chalfant, Transl. Res., 2017, 189, 13–29 CrossRef CAS.
- T. Hu and J.-L. Zhang, J. Sep. Sci., 2018, 41, 351–372 CrossRef CAS.
- T. Xu, C. Hu, Q. Xuan and G. Xu, Anal. Chim. Acta, 2020, 1137, 156–169 CrossRef CAS.
- J. M. McMahon, N. N. Dookeran and P. J. Todd, J. Am. Soc. Mass Spectrom., 1995, 6, 1047–1058 CrossRef CAS PubMed.
- R. M. Caprioli, T. B. Farmer and J. Gile, Anal. Chem., 1997, 69, 4751–4760 CrossRef CAS.
- S. Rankin-Turner, J. C. Reynolds, M. A. Turner and L. M. Heaney, Anal. Sci. Adv., 2022, 3, 67–89 CrossRef CAS.
- Y. Otsuka, Mass Spectrom., 2021, 10, A0095–A0095 CrossRef CAS.
- J. Fenn, M. Mann, C. Meng, S. Wong and C. Whitehouse, Science, 1989, 246, 64–71 CrossRef CAS.
- Z. Takáts, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef.
- V. Kertesz and G. J. Van Berkel, J. Mass Spectrom., 2010, 45, 252–260 CrossRef CAS.
- T. Wachs and J. Henion, Anal. Chem., 2001, 73, 632–638 CrossRef CAS.
- G. J. Van Berkel, A. D. Sanchez and J. M. E. Quirke, Anal. Chem., 2002, 74, 6216–6223 CrossRef CAS.
- P. J. Roach, J. Laskin and A. Laskin, Analyst, 2010, 135, 2233–2236 RSC.
- W. Rao, N. Pan, X. Tian and Z. Yang, J. Am. Soc. Mass Spectrom., 2016, 27, 124–134 CrossRef CAS.
- Y. Otsuka, S. Shide, J. Naito, M. Kyogaku, H. Hashimoto and R. Arakawa, Rapid Commun. Mass Spectrom., 2012, 26, 2725–2732 CrossRef CAS.
- Y. Otsuka, J. Naito, S. Satoh, M. Kyogaku, H. Hashimoto and R. Arakawa, Analyst, 2014, 139, 2336–2341 RSC.
- T. Kohigashi, Y. Otsuka, R. Shimazu, T. Matsumoto, F. Iwata, H. Kawasaki and R. Arakawa, Mass Spectrom., 2016, 5, S0054–S0054 CrossRef.
- Y. Otsuka, S. Satoh, J. Naito, M. Kyogaku and H. Hashimoto, J. Mass Spectrom., 2015, 50, 1157–1162 CrossRef CAS.
- Y. Otsuka, B. Kamihoriuchi, A. Takeuchi, F. Iwata, S. Tortorella and T. Matsumoto, Anal. Chem., 2021, 93, 2263–2272 CrossRef CAS.
- L. S. Eberlin, C. R. Ferreira, A. L. Dill, D. R. Ifa, L. Cheng and R. G. Cooks, ChemBioChem, 2011, 12, 2129–2132 CrossRef CAS.
- L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W. M. Fan, O. Fiehn, R. Goodacre, J. L. Griffin, T. Hankemeier, N. Hardy, J. Harnly, R. Higashi, J. Kopka, A. N. Lane, J. C. Lindon, P. Marriott, A. W. Nicholls, M. D. Reily, J. J. Thaden and M. R. Viant, Metabolomics, 2007, 3, 211–221 CrossRef CAS.
- E. Fahy, S. Subramaniam, R. C. Murphy, M. Nishijima, C. R. H. Raetz, T. Shimizu, F. Spener, G. van Meer, M. J. O. Wakelam and E. A. Dennis, J. Lipid Res., 2009, 50, S9–S14 CrossRef.
- D. Fitzner, J. M. Bader, H. Penkert, C. G. Bergner, M. Su, M.-T. Weil, M. A. Surma, M. Mann, C. Klose and M. Simons, Cell Rep., 2020, 32, 108132 CrossRef CAS.
- D. Gode and D. A. Volmer, Analyst, 2013, 138, 1289 RSC.
- A. K. Jarmusch, C. M. Alfaro, V. Pirro, E. M. Hattab, A. A. Cohen-Gadol and R. G. Cooks, PLoS One, 2016, 11, e0163180 CrossRef.
- M. M. B. Nielsen, K. L. Lambertsen, B. H. Clausen, M. Meyer, D. R. Bhandari, S. T. Larsen, S. S. Poulsen, B. Spengler, C. Janfelt and H. S. Hansen, Sci. Rep., 2016, 6, 39571 CrossRef CAS.
- J. Martínez-Gardeazabal, E. González de San Román, M. Moreno-Rodríguez, A. Llorente-Ovejero, I. Manuel and R. Rodríguez-Puertas, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1548–1557 CrossRef.
- S. N. Nguyen, R. L. Sontag, J. P. Carson, R. A. Corley, C. Ansong and J. Laskin, J. Am. Soc. Mass Spectrom., 2018, 29, 316–322 CrossRef CAS.
- D. Johannsmann, Phys. Chem. Chem. Phys., 2008, 10, 4516 RSC.
- R. Yoshizawa, O. Urakawa and T. Inoue, Nihon Reoroji Gakkaishi, 2022, 50, 333–337 CrossRef CAS.
- C. Yang, Y. Sun, Y. He and P. Ma, J. Chem. Eng. Data, 2008, 53, 293–297 CrossRef CAS.
- C. Gonzalez-Riano, A. Gradillas and C. Barbas, J. Chromatogr. Open, 2021, 1, 100018 CrossRef.
- X. Han and R. W. Gross, Mass Spectrom. Rev., 2005, 24, 367–412 CrossRef CAS.
- C. M. Kinart, Phys. Chem. Liq., 1994, 27, 115–121 CrossRef CAS.
- A. M. Ezhil Raj, L. B. Resmi, V. B. Jothy, M. Jayachandran and C. Sanjeeviraja, Fluid Phase Equilib., 2009, 281, 78–86 CrossRef.
- M. M. H. Bhuiyan and M. H. Uddin, J. Mol. Liq., 2008, 138, 139–146 CrossRef CAS.
- Z. Trabelsi, M. Dallel, H. Salhi, D. Das, N. A. Al-Omair and N. Ouerfelli, Phys. Chem. Liq., 2015, 53, 529–552 CrossRef CAS.
- M. S. Bakshi and G. Kaur, J. Chem. Eng. Data, 1997, 42, 298–300 CrossRef CAS.
- B. Zhang, Y. Cai, X. Mou, N. Lou and X. Wang, Chem. Phys., 2002, 280, 229–238 CrossRef CAS.
- A. L. Dill, D. R. Ifa, N. E. Manicke, A. B. Costa, J. A. Ramos-Vara, D. W. Knapp and R. G. Cooks, Anal. Chem., 2009, 81, 8758–8764 CrossRef CAS.
- R. Fernández, P. González, S. Lage, J. Garate, A. Maqueda, I. Marcaida, M. Maguregui, B. Ochoa, F. J. Rodríguez and J. A. Fernández, Anal. Chem., 2017, 89, 8565–8573 CrossRef.
- H.-M. Bergman, L. Lindfors, F. Palm, J. Kihlberg and I. Lanekoff, Anal. Bioanal. Chem., 2019, 411, 2809–2816 CrossRef CAS.
- I. Lanekoff, S. L. Stevens, M. P. Stenzel-Poore and J. Laskin, Analyst, 2014, 139, 3528–3532 RSC.
- H. Y. J. Wang, C. B. Liu and H. W. Wu, J. Lipid Res., 2011, 52, 840–849 CrossRef CAS.
- H. Y. J. Wang, H. W. Wu, P. J. Tsai and C. B. Liu, Anal. Bioanal. Chem., 2012, 404, 113–124 CrossRef CAS.
- P. M. Angel, J. M. Spraggins, H. S. Baldwin and R. Caprioli, Anal. Chem., 2012, 84, 1557–1564 CrossRef CAS.
- J. Chen, K. B. Green and K. K. Nichols, Invest. Ophthalmol. Visual Sci., 2013, 54, 5730–5753 CrossRef CAS.
- K. D. Duncan, R. Fang, J. Yuan, R. K. Chu, S. K. Dey, K. E. Burnum-Johnson and I. Lanekoff, Anal. Chem., 2018, 90, 7246–7252 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters a User's Handbook Second Edition, 2007 Search PubMed.
- D. Mathieu, ACS Omega, 2018, 3, 17049–17056 CrossRef CAS.
- M. M. R. Devalapalli, H. S. Cheruvu, T. Yertha, V. B. Veeravalli, S. Sampathi and S. Shivakumar, J. Sep. Sci., 2017, 40, 3662–3674 CrossRef CAS.
- H. Brandt, T. Ehmann and M. Otto, Rapid Commun. Mass Spectrom., 2010, 24, 2439–2444 CrossRef CAS.
- M. Murase, Y. Yamada, Y. Goto and N. Mizoshita, Bull. Chem. Soc. Jpn., 2022, 95, 1068–1074 CrossRef CAS.
- J. Pape, K. L. Vikse, E. Janusson, N. Taylor and J. S. McIndoe, Int. J. Mass Spectrom., 2014, 373, 66–71 CrossRef CAS.
- L. Paseta, G. Potier, S. Abbott and J. Coronas, Org. Biomol. Chem., 2015, 13, 1724–1731 RSC.
- S. Zhang, C. Campagne and F. Salaün, Appl. Sci., 2019, 9, 402 CrossRef CAS.
- D. Nakamura, M. Hirano and R. Ohta, Chem. Commun., 2017, 53, 4096–4099 RSC.
- A. K. Sankaran and J. P. Rothstein, J. Nonnewton. Fluid Mech., 2012, 175–176, 64–75 CrossRef CAS.
- D. F. James and M. Pouran, Rheol. Acta, 2009, 48, 611–624 CrossRef CAS.
- H. Chen, T. Tang and A. Amirfazli, Langmuir, 2015, 31, 11470–11476 CrossRef CAS.
- B. Kamihoriuchi, Y. Otsuka, A. Takeuchi, F. Iwata and T. Matsumoto, Mass Spectrom., 2019, 7, S0078–S0078 CrossRef.
- Electrospray and MALDI Mass Spectrometry, ed. R. B. Cole, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010 Search PubMed.
- D. T. Usmanov, K. Hiraoka, S. Ninomiya, L. C. Chen, H. Wada, M. Matsumura, S. Sanada-Morimura, K. Nakata and H. Nonami, Anal. Methods, 2017, 9, 4958–4963 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an01953a |
| This journal is © The Royal Society of Chemistry 2023 |