
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutodisplay of streptococcal protein G for construction of an orientation-controlled immunoaffinity layer
Seong Gi
Kim
abc,
JeeYoung
Kim
abc,
Mi Yeon
Kim
abc,
Jong-Min
Park
 abc,
Joachim
Jose
d and
Min
Park
*abc
abc,
Joachim
Jose
d and
Min
Park
*abc
aMajor in Materials Science and Engineering, Hallym University, 1 Hallymdaehak-gil, Chuncheon-si, Gangwon-do 24252, Republic of Korea. E-mail: minpark@hallym.ac.kr
bIntegrative Materials Research Institute, Hallym University, 1 Hallymdaehak-gil, Chuncheon-si, Gangwon-do 24252, Republic of Korea
cInterdisciplinary Program of Nano-Medical Device Engineering, Hallym University, 1 Hallymdaehak-gil, Chuncheon-si, Gangwon-do 24252, Republic of Korea
dInstitute of Pharmaceutical and Medicinal Chemistry, PharmaCampus, Westfälische Wilhelms-Universität, 48 Corrensstraβe, Münster, 48149, Germany
First published on 17th January 2023
Abstract
An immunoaffinity layer with orientation-controlled antibodies was constructed to express streptococcal protein G in Escherichia coli cells using autodisplay technology. The sequence of protein G, a specific IgG-binding protein, was inserted into the autodisplay vector using recombinant technology and the constructed plasmid vector was transformed into E. coli cells. Protein G was confirmed to be autodisplayed with a high density of 2 × 105 copies per cell by SDS-PAGE analysis, and its IgG-binding affinity was confirmed by fluorescence microscopy. Autodisplayed protein G showed higher affinity than the IgG-binding Z-domain for goat IgG. Immunoassays based on E. coli cells were established to detect horseradish peroxidase (HRP) and C-reactive protein (CRP). Protein G autodisplaying E. coli cells were utilized as a solid support and immunoassays showed improved sensitivity by orientation control of autodisplayed protein G. The outer membrane (OM) of protein G autodisplaying E. coli was isolated and layered to construct an immunoaffinity layer. The OM was coated on a microplate to perform the immunoassays, which showed limits of detection of 5 and 0.2 ng mL−1 for HRP and CRP, respectively. An OM layer with autodisplayed protein G was applied as the immunoaffinity layer of a surface plasmon resonance (SPR) biosensor. After CRP detection, the SPR responses showed good linearity, with an R2 value of 0.99. The immunoaffinity layer with orientation control by autodisplayed protein G was confirmed to be applicable in immunoassays and immunosensors to improve sensitivity.
Introduction
Biosensors and bioassays are analytical devices and methods for determining the concentration of analytes in physiological samples, such as blood, serum, urine, and cerebrospinal fluid.1,2 Biosensors consist of three parts: a molecular recognition layer, transducer, and signal generator.3 Molecular recognition molecules are essential for the detection of target analytes in biological complexes, and these molecules form a molecular recognition layer.4 Among the various molecular recognition molecules, antibodies are one of the most frequently used because of their specificity, simplicity, cost effectiveness, and ease of immobilization.5–7 Because of these advantages, immunosensors have been widely used for the detection of various samples and have been applied in clinical diagnostics.8,9 There are numerous studies that showed that immunoaffinity layers have been fabricated based on the immobilization of antibodies on the transducer to form an antibody-based molecular recognition layer10–12 Similarly, biosensors require molecular recognition molecules, and antibodies are one of the most widely used for bioassays.13 Enzyme-linked immunosorbent assay (ELISA) is one of the most frequently used immunoassays for detecting target analytes in complex biological solutions.14 Similar to the immunoaffinity layer, captured antibodies are immobilized on a solid support to bind to the target analyte in sandwich ELISA. In both cases, immobilized antibodies are key components for target detection.15For detection by immunosensors or immunoassays, the analyte should be bound to the captured antibodies. Thus, the density of antigen-binding sites on the surface of a biosensor or solid support is directly related to sensitivity.16 To improve sensitivity, the exposure of as many antigen-binding sites as possible is required.17 Antibodies consist of 2 heavy chains and 2 light chains to form a ‘Y’ shaped structure.18 Among the three branches, two endpoints at which the heavy and light chains meet are the antigen-binding site called the Fab region, and the other endpoint at which two heavy chains meet is called the Fc region.19 In addition, antibodies are relatively large proteins (approximately 150 kDa), and antigen-binding sites are small and localized.20 Because of this asymmetric structure, when immobilized on a solid support, antibodies can have random molecular orientations, such as side-on, tail-on, head-on, or flat-on.21 When antibodies are randomly immobilized, such as physical adsorption, less than 20% of antibodies have been reported to have a suitable orientation.22 Therefore, to improve the sensitivity of immunosensors or immunoassays, orientation control of immobilized antibodies is important for efficient exposure of antigen-binding sites, as well as density control.
Antibody-binding proteins, such as staphylococcal protein A or streptococcal protein G, are known to have Fc region-binding properties. Because of this antibody-binding property, they have been widely used in research as well as industries, including antibody purification.23,24 In addition, Fc bound antibodies realize orientation control by effective exposure of antibody-binding sites, so these proteins have also been frequently used in immunoassays and biosensors to improve sensitivity by orientation control.4,25,26 Recently, the Z-domain, an engineered peptide from the B-domain of protein G, was employed to control the orientation of antibodies in sensitive immunoassays and biosensors.27,28 In these studies, autodisplay technology was utilized for the effective immobilization of the Z-domain. Autodisplay is a surface display technology for Gram-negative bacteria that expresses target proteins using autotransporters.4,29,30 The autodisplayed target protein is connected to the β-barrel, a naturally existing protein on the outer membrane (OM) of bacterial cells, through an α-helical linker to anchor on the OM. These Z-domain autodisplaying Escherichia coli cells were used to improve the sensitivity of immunoassays and biosensors by controlling the orientation of the antibodies.27 Protein A, including the Z-domain, has specific binding affinity toward antibodies from various mammalian species, including humans, rats, guinea pigs, and rabbits with strong binding affinity.31,32 However, among the frequently used mammalian species, protein A has weak affinity for mouse immunoglobulin (Ig) G1 and goat IgG.24 On the other hand, protein G has strong binding affinity to mouse and goat IgGs with an affinity constant (equilibrium constant) of 14.3 × 10−9 M−1.33 Protein G also has a specific binding affinity toward the Fc region of antibodies, so it can be utilized for the orientation control of antibodies.34 In addition, in the case of human and rabbit IgG, protein G has a greater affinity constant than protein A33. Thus, autodisplay of protein G promises expansion of antibody usage in immunoassays or immunosensors and is expected to improve due to high binding affinity and orientation control.
In this study, an immunoaffinity layer with orientation-controlled antibodies was developed based on the autodisplay of protein G. Streptococcal protein G was expressed as a target of the autotransporter using autodisplay technology. A protein G autodisplaying vector was constructed using recombinant technology and transformed into E. coli cells. The autodisplay of protein G was confirmed by SDS-PAGE, and IgG-binding affinity was analyzed using fluorescence microscopy. The activity confirmed that protein G autodisplaying E. coli cells were utilized as a solid support to carry out immunoassays, and autodisplayed protein G was confirmed to improve sensitivity by orientation control of antibodies. Then, the OM of protein G autodisplaying E. coli cells was isolated as a liposome and layered on a microplate and surface of a surface plasmon resonance (SPR) biosensor to construct an immunoaffinity layer with orientation control of antibodies. OM-based immunoassay was performed using a protein G autodisplaying OM-coated microplate to test the applicability of the immunoaffinity layer. Then, the OM layer with autodisplayed protein G was introduced into the SPR biosensor and confirmed to be suitable as an immunoaffinity layer with orientation control for sensitive biosensors.
Materials and methods
Materials
Goat polyclonal antibodies against C-reactive protein (CRP), goat polyclonal antibodies against horseradish peroxidase (HRP), goat polyclonal antibodies against CRP labeled with HRP, and fluorescein-conjugated rabbit polyclonal antibodies against human immunoglobulin (Ig) G were obtained from Abcam (Cambridge, UK). Tris-HCl buffer (pH 8.0), Tris-glycine buffer, phosphate-buffered saline (PBS), Luria–Bertani (LB) broth, ampicillin, protein G, and sodium dodecyl sulfate (SDS)-loading buffer were purchased from LPS solution (Daejeon, Korea). 96-Well microplates were obtained from SPL Life Sciences (Pocheon, Korea). The 3,3′,5,5′-tetramethylbenzidine (TMB) substrate kit was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Triton X-100, isopropyl β-D-1-thiogalactopyranoside (IPTG), sucrose, lysozyme, HRP, CRP, and other analytical-grade chemicals were obtained from Merck (Darmstadt, Germany). The restriction enzymes, XhoI and KpnI, and ligase were purchased from New England Biolabs (Ipswich, MA, US).Construction of a protein G autodisplaying vector
Gene encoding streptococcal protein G (Uniprot: P06654) was amplified by polymerase chain reaction (PCR) with additional sequences coding for 5′ end XhoI and 3′ end KpnI restriction sites.35 The PCR product was excised using KpnI and XhoI and confirmed by agarose gel electrophoresis. Simultaneously, the inducible auto-displaying vector pET-SH7, which has both XhoI and KpnI restriction sites at the 5′ and 3′ ends of the autodisplaying target sequence, was excised by KpnI and XhoI.36 Both double-digested DNA fragments from the PCR product encoding protein G and the autodisplaying backbone were extracted from the gel extraction kit (Qiagen, Hilden, Germany) and then ligated by ligase to construct a protein G autodisplaying vector named pJP002 (Fig. 1a). | ||
| Fig. 1 Schematic diagram of (a) autodisplay of protein G and (b) construction of an immunoaffinity layer with orientation control of antibodies using the isolated OM with autodisplayed protein G. (SP: signal peptide, IM: inner membrane, PP: periplasm, and OM: outer membrane). | ||
Autodisplay of streptococcal protein G
After the construction of the protein G autodisplaying vector, the pJP002 plasmids were transformed into E. coli BL21(DE3) cells by heat shock. Transformed E. coli cells were grown overnight in LB broth media containing ampicillin at 37 °C with vigorous shaking. Overnight cultured cells were 100-fold diluted with fresh media until the optical density (OD) reached 0.5 (600 nm) at 37 °C with vigorous shaking. Next, 1 mM IPTG was added to E. coli cells for the induction of protein G with an autotransporter, and the E. coli cells were incubated at 30 °C for 2 h. After incubation, the cells were harvested and the OD600 value was adjusted to 1.0.Outer membrane (OM) preparation and analysis of autodisplayed protein G
The OM of the E. coli cells was isolated as previously described.28 Briefly, a rigid peptidoglycan layer of E. coli cells was hydrolyzed by lysozyme treatment to form spheroplasts. An extraction buffer containing Triton X-100 was then added to spheroplasts for the formation of isolated OM particles. These particles were purified by sequential centrifugation. After OM isolation, autodisplayed protein G was analyzed by electrophoresis. The OM particles were denatured by mixing with sodium dodecyl sulfate containing sample buffer (Bio-Rad Laboratory, Hercules, CA, US) at 95 °C for 15 min and separated using polyacrylamide gel electrophoresis with a constant current of 30 mA. After separation, proteins in the gel were stained by treatment with Coomassie Blue and imaged using a ChemiDoc Gel Imaging System (Bio-Rad Laboratory, Hercules, CA, US).Immunoassay based on the OM layer
To form an immunoaffinity layer using isolated OM particles with autodisplayed protein G, 300 μg mL−1 of OM solution quantified using a BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, US) was added to a 96-well microplate for 2 h at room temperature and washed with 0.1% Tween-20 containing PBS for three times. After layer formation, the cells were treated with capture antibodies at a concentration of 2.3 μg mL−1 for 1 h to form an orientation-controlled immunoaffinity layer. The microplate was then rewashed, and analyte samples with various concentrations were added for 1 h. After washing, detection antibodies conjugated with HRP were added and incubated for 1 h. For quantification, TMB solution was added for 15 min, and the reaction was quenched with the addition of 2 M sulfuric acid. Then, the OD was measured at a wavelength of 450 nm with a reference wavelength of 650 nm using a microplate reader (Molecular Devices, San Jose, CA, US). All assays were performed in triplicate.SPR biosensor measurement
The OM layer was constructed on an unmodified bare gold chip installed on a commercial SPR biosensor system (Icluebio Co., Ltd, Seongnam, Korea) by incubation with 300 μg mL−1 isolated OM solution for 2 h. The flow rate of all samples and buffer injection was 0.3 mL min−1. After washing, 1 μg mL−1 capture antibodies and antibodies against CRP were bound to the OM layer with autodisplayed protein G. Then, the sensor was treated with CRP samples for 30 min. A 10 min washing step was performed between each process. The captured CRP was quantified by subtracting the SPR signal after CRP treatment from the signal before treatment. All treatment steps were measured under static conditions and performed in triplicate.Results and discussion
Autodisplay of streptococcal protein G with immunoglobulin G-binding affinity
The IgG-binding protein, streptococcal protein G, was expressed with an autotransporter to anchor on the OM of E. coli cells using autodisplay technology.6 Owing to its specific IgG-binding activity to various species, it has been applied to various industries and research studies, including immunoassays and biosensors.4 In particular, protein G binds to the Fc region of the antibody, realizing orientation control, thereby improving the sensitivity of immunoassays and immunosensors.37 As shown in Fig. 1a, the autotransporter was expressed from the constructed plasmid vector and passed through the inner membrane via a signal peptide.16 Then, the autotransporter folded to form a β-barrel to anchor to the OM of E. coli cells, and the target protein, IgG-binding protein G, was displayed through the linker toward the outside of the cells. After cultivation and induction of E. coli cells with a protein G autodisplaying vector, the OM of E. coli cells was isolated and analyzed by SDS-polyacrylamide gel electrophoresis (PAGE), as shown in Fig. 2. Outer membrane protein (Omp) F and OmpA, which have molecular weights of 37 kDa and 35 kDa, respectively, are the two most common porins and are also found in the clear band near 37 kDa in Fig. 2.38 This supports that the OM was successfully isolated and visualized by SDS-PAGE. Compared with the OM from intact E. coli cells without the autodisplaying vector (lane 1), the OM from protein G autodisplaying E. coli cells (lane 2) showed an additional distinctly thick band at 70 kDa. The size of this band was consistent with the molecular weight calculated from the amino acid sequence of the full autotransporter fusion protein without a signal peptide. This means that the transformation of the autodisplay vector produced an autotransporter containing protein G as a target, and the expressed protein was anchored to the OM of E. coli cells. OmpA is known to exist as 105 molecules per cell; therefore, it was used as an internal standard for calculating the expression level of the protein G autotransporter by densitometry.39 Using densitometry, protein G was determined to be 2 × 105 copies per cell. This number indicates that autodisplayed protein G was expressed and displayed on E. coli cells at a high density. In the case of the autodisplayed Z-domains, the expression density was reported to be 2.8 × 105 copies per cell.40 This value was comparable to or slightly higher than the autodisplay of protein G. Thus, the difference in signals was due to the binding affinity toward the antibodies from goat. | ||
| Fig. 2 Analysis of OM proteins. Lane M represents the molecular weight marker. Lane 1 and Lane 2 indicate OM proteins from intact BL21(DE3) cells without the autodisplay vector and from BL21(DE3) cells transformed with pJP002 for autodisplay of protein G, respectively. | ||
The IgG-binding property of autodisplayed protein G was tested by staining with fluorescein-labeled antibodies. Human IgG was added to protein G autodisplaying E. coli cells for 1 h and fluorescein-labeled secondary antibodies against human IgG were sequentially added for 1 h. Antibody-treated E. coli cells were then placed on glass slides and observed by fluorescence microscopy. As shown in Fig. 3, protein G autodisplaying E. coli cells showed intense fluorescence signals, indicating the binding of fluorescein-labeled antibodies. In this case, protein G autodisplaying E. coli cells were treated with a high concentration (100 μg mL−1) of human IgGs to bind all antibody binding sites. So all autodisplayed protein G were saturated with IgGs. In addition, even if a small amount of secondary antibodies binds with autodisplayed protein G, it also shows the antibody binding activity of autodisplayed protein G. In contrast, intact E. coli cells did not show a significant fluorescence signal after antibody staining. When the fluorescence image was overlaid on the phase image, all protein G autodisplaying E. coli cells exhibited fluorescence. These data indicate that protein G was autodisplayed in all E. coli cells, and autodisplayed protein G maintained specific antibody-binding activity. In addition, protein G was autodisplayed on the OM of E. coli cells because antibodies cannot pass through the membrane of E. coli. From these data, protein G was confirmed to be autodisplayed on the OM of E. coli cells at a high density and with IgG-binding activity, making it suitable for application in immunoassays and biosensors.
 | ||
| Fig. 3 The microscopic images of protein G autodisplaying E. coli cells after the treatment of fluorescein labeled antibodies. | ||
Immunoassays based on protein G autodisplaying E. coli cells
IgG-binding protein G autodisplaying E. coli was applied to the immunoassay and utilized as a solid support for antibody immobilization. As mentioned previously, staphylococcal protein A, a widely used protein owing to its specific binding affinity to IgG from various species, is known to have a weak bond with goat IgG, whereas streptococcal protein G has a strong bond.16,41 Because goat IgG is one of the most widely used antibodies in rabbits, mice, and humans, the weak binding affinity of protein A to goat IgG restricts its use for antibody immobilization in diagnostic tests. Thus, we compared the IgG-binding affinity of autodisplayed protein G with an autodisplayed Z-domain, an engineered protein from the B-domain of protein A.27 HRP was selected as the model analyte because peroxidase can be directly quantified in the presence of peroxide by various substrates such as TMB using colorimetry, fluorescence, or luminescence without treatment with detection antibodies.42E. coli cells with autodisplayed affinity proteins were prepared and resuspended in various concentrations of the capture antibody and goat polyclonal anti-HRP for 1 h. After washing, 1 ng mL−1 analyte HRP was added and treated for 1 h. HRPs bound to E. coli cells were quantified using a TMB chromogenic reaction, and OD values were measured at a wavelength of 450 nm, as described in Fig. 4a. Both protein G and Z-domain autodisplaying E. coli showed increasing OD450nm values with an increase in anti-HRP concentrations. This indicates that both autodisplayed affinity proteins have binding affinity with goat polyclonal antibodies. However, protein G autodisplaying E. coli (■) showed higher OD450nm values than Z-domain (●) at all anti-HRP concentrations. In addition, the minimum antibody concentrations required for the detection of constant HRP were calculated to be 150 and 500 ng mL−1 for autodisplayed protein G and Z-domain, respectively. These data indicate that autodisplayed protein G has higher affinity than the Z-domain for goat antibodies, and this phenomenon is consistent with the previously known binding affinity of protein A and protein G to goat IgG. Signal differences between the two autodisplayed affinity proteins increased until the concentration of antibodies reached 2.3 μg mL−1 and saturated after that concentration (Fig. 4a, inset). From these results, the optimal concentration of the capture antibody for immunoassays and biosensor applications was 2.3 μg mL−1. | ||
| Fig. 4 Immunoassays based on protein G autodisplaying E. coli cells. (a) IgG-binding affinity test by treatment of various concentrations of goat polyclonal antibodies to protein G and Z-domain autodisplaying E. coli cells. Inset: signal difference between assays using protein G and Z-domain autodisplaying E. coli cells. (b) HRP immunoassay and (c) CRP immunoassay using protein G autodisplaying E. coli cells. For comparison, Z-domain autodisplaying E. coli cells, intact E. coli cells without the autodisplay vector, and randomly immobilized antibodies by physical adsorption were used. | ||
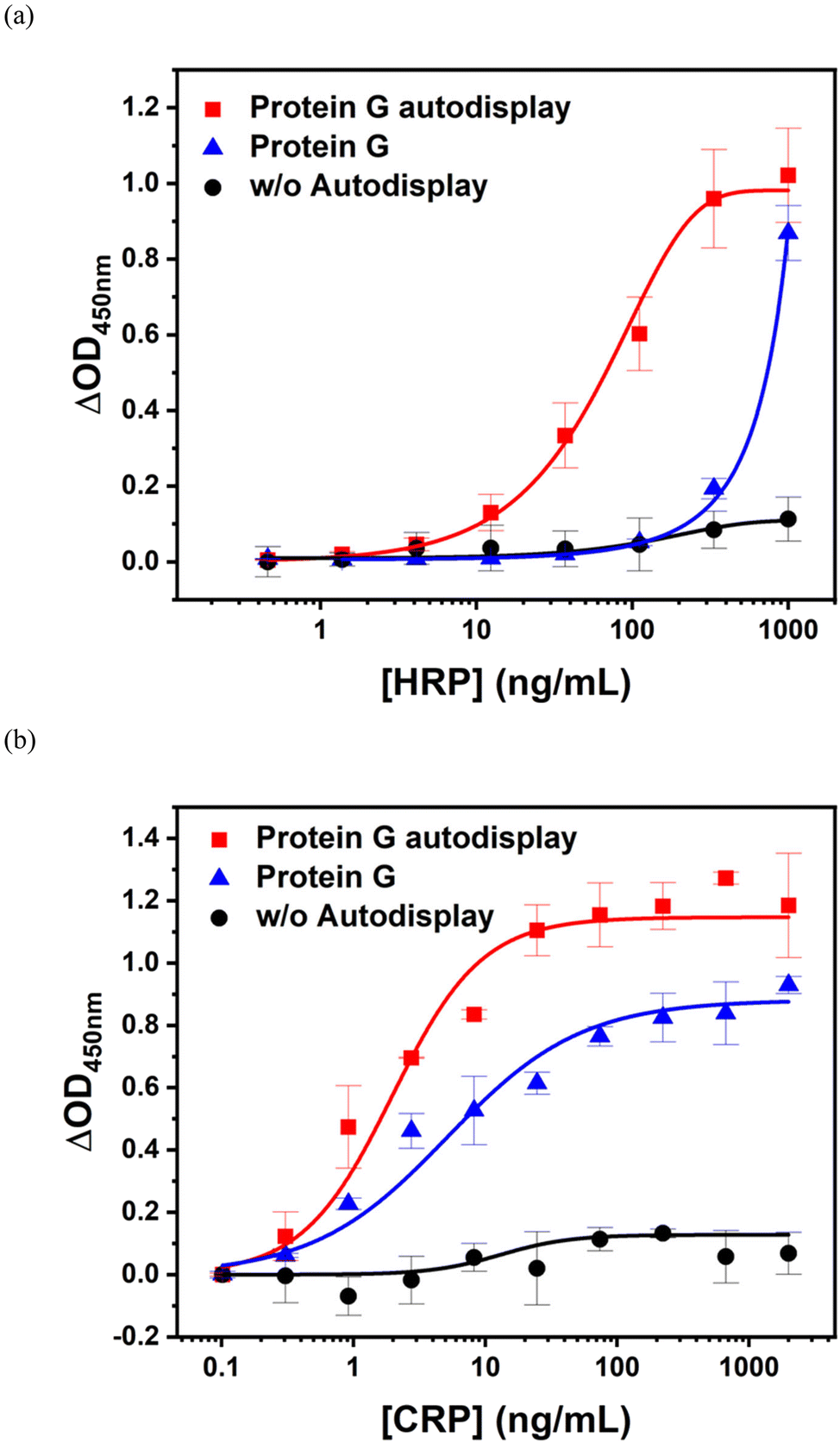
HRP and CRP immunoassays were performed using protein G autodisplaying E. coli cells. As shown in Fig. 4b, an E. coli cell-based immunoassay was performed in the HRP concentration range of 0.1–300 ng mL−1. The assay curve of protein G autodisplaying E. coli (■) showed a sigmoidal growth curve and reached saturation with 100 ng mL−1 HRP. The limit of detection (LOD) was evaluated using three Sigma and calculated to be approximately 1 ng mL−1, with a dynamic range of 1–100 ng mL−1. For comparison of IgG-binding affinity with protein G, Z-domain autodisplaying E. coli cells (▲) were tested as solid supports and showed an LOD of 20 ng mL−1. The assay curve did not saturate at an HRP concentration of 300 ng mL−1. These results suggest that protein G has a much higher binding affinity to goat IgG than protein A, even after autodisplay of these affinity proteins. In addition, to confirm the orientation effect of antibodies, they were immobilized on a microplate by simple physical adsorption with a random orientation (▼) to be carried out by the HRP immunoassay. The OD450nm values from the physical adsorption assay were lower than those from protein G autodisplaying E. coli at all HRP concentrations, and the LOD of this assay was estimated to be 20 ng mL−1. These results indicate that the sensitivity of protein G autodisplaying E. coli cells was improved by orientation control of the antibodies. In contrast, the assay using intact E. coli cells without the autodisplay vector (●) showed no significant increase up to 100 ng mL−1 of HRP. This implies that intact E. coli cells can reduce non-specific binding without any blocking step.
Based on the confirmation of applicability to immunoassays, CRP was detected using protein G autodisplaying E. coli cells in the concentration range of 0.25–1000 ng mL−1 (Fig. 4c). CRP is a serum protein synthesized by the liver and is a widely used biomarker for the diagnosis of various inflammatory diseases, such as infection, autoimmune disorders, rheumatoid arthritis, nephrotic syndrome, and chronic renal failure.43 Similar to the HRP immunoassay, the assay curve for protein G (■) showed a sigmoidal growth curve. In this case, the curve was saturated at a CRP concentration of 300 ng mL−1. The LOD of the CRP immunoassay based on protein G autodisplaying E. coli cells was estimated to be 2 ng mL−1; therefore, the dynamic range of the CRP immunoassay was confirmed to range from 2 ng mL−1 to 300 ng mL−1. Z-domain autodisplaying E. coli cells (▲) showed lower OD450nm values than protein G (■) at all CRP concentrations, and the LOD was calculated to be 4 ng mL−1, with a dynamic range of 4–200 ng mL−1. At saturation, protein G showed a 3-fold higher OD450nm value than the Z-domain. This indicates that the binding capacity of protein G is higher than that of the Z-domain because of the difference in binding affinity to goat IgG. In the case of physical adsorption (▼), similar to the Z-domain, the OD450nm values were lower than those of protein G at all CRP concentrations. The LOD of this assay was calculated to be 6 ng mL−1. The protein G autodisplaying E. coli cell-based immunoassay showed an LOD that was three times higher than that based on Z-domains or physical adsorption. As protein G has stronger binding affinity than the Z-domain, the LOD difference is due to the differences in the number of antibodies bound to the autodisplayed E. coli cells. Compared to physical adsorption with a random orientation, sensitivity is improved by orientation control of autodisplayed protein G. From these data, protein G autodisplaying E. coli was confirmed to have stronger binding affinity against goat IgG than the Z-domain and is suitable for application in immunoassays as a solid support for antibody immobilization to improve sensitivity by orientation control.
Construction of an immunoaffinity layer with orientation control of antibodies using the OM of protein G autodisplaying E. coli and its application to SPR biosensors
E. coli cells with autodisplayed affinity proteins can act as an antibody immobilizing substrate and can be washed by simple centrifugation steps using a table-top centrifuge. However, these buffer changes by sequential centrifugation may cause cell loss, and the centrifugation process is difficult to automate. In addition, for the construction of an immunoaffinity layer on the transducer of biosensors, additional immobilization steps of E. coli cells are required, and these may decrease the IgG-binding efficiency. In addition, the size of E. coli cells on the micrometer scale hinders the detection of biosensors that can only detect near the surface of the transducer, such as electrochemical biosensors, or evanescence field-based biosensors such as SPR biosensors.16 To increase the expandability of protein G autodisplay technology to construct an orientation-controlled immunoaffinity layer, the OMs of E. coli cells were isolated as liposomes and layered on the surface of a two-dimensional substrate via hydrophobic interactions, as shown in Fig. 1b.HRP was selected as the model analyte for the OM-based immunoassay. After layering the OM with autodisplayed protein G on a 96-well microplate, capture antibodies and anti-HRP antibodies were used to form an immunoaffinity layer. Then, HRP solution with a concentration range of 0.5 to 1000 ng mL−1 was placed for 1 h at room temperature. After washing, the TMB reaction was quantified, and the chromogenic reaction was measured by optical density at a wavelength of 450 nm using a microplate reader. As shown in Fig. 5a, the OM layer with autodisplayed protein G (■) showed increasing OD450nm values with an increase in HRP concentration. This implies that the layered OM of autodisplayed E. coli is able to form an immunoaffinity layer with orientation-controlled antibodies. The assay curve for the OM with autodisplayed protein G showed a sigmoidal growth curve and reached saturation at 300 ng L−1. The LOD was calculated as 5 ng mL−1; therefore, the dynamic range of the OM-based HRP immunoassay was estimated as 5–300 ng mL−1. In addition, the signals from the OM layer were much higher than those from physical adsorption (Fig. 4b ▼). Then, to test the density control of autodisplayed protein G, commercial protein G was immobilized on a microplate (▲) and the HRP immunoassay was carried out. The OD450nm values from the protein G-based assay were also lower than those from protein G autodisplaying E. coli at all HRP concentrations (LOD = 100 ng mL−1). These results indicate that the autodisplay technology was suitable for the improvement of the sensitivity through the density control as well as orientation control of the antibodies. Layered OMs from intact E. coli cells without the autodisplay vector (●) were utilized as a comparison group and showed no significant increase in OD450nm values over the whole range. This indicated that the OM layer was compactly coated on the surface of the substrate and prevented the non-specific binding of both antibodies and antigens, even without a blocking step.
 | ||
| Fig. 5 (a) HRP and (b) CRP immunoassays based on the OM with autodisplayed protein G. | ||
Based on the applicability of the OM layer to immunoassays, a CRP immunoassay was performed at concentrations of 0.1 to 2000 ng mL−1. Similar to the HRP immunoassay, OMs with autodisplayed protein G were isolated and layered on a microplate, and antibodies against CRP were immobilized with orientation control. CRP samples were then treated and HRP-conjugated anti-CRP antibodies were added to quantify bound CRP. After the measurement of the TMB chromogenic reaction, the OD450nm values were obtained as shown in Fig. 5b. Protein G autodisplaying the OM layer (■) showed an increasing sigmoidal curve with increased CRP concentrations. The assay curve was saturated at a CRP concentration of 100 ng ml−1. The estimated LOD of the CRP immunoassay was 0.2 ng mL−1 so the dynamic range of the OM-based CRP immunoassay was calculated to be 0.2 to 100 ng mL−1. These data imply that the immunoaffinity layer based on the OM with autodisplayed protein G is suitable to be applied to immunoassays of biomarkers, including CRP. In addition, the dynamic ranges and LODs of the OM-based immunoassays shown in Fig. 5 are comparable to those of the E. coli cell-based immunoassays shown in Fig. 4 and the signals from the OM layer were also higher than those from physical adsorption (Fig. 4c ▼) and the commercial protein G immobilized microplate (Fig. 5b ▲). These results imply that the OM layer with autodisplayed protein G is successfully introduced to construct a sensitive immunoaffinity layer by the density and orientation control of the antibodies. The OM layer without autodisplayed protein (●) also showed no significant increase in signals over the whole range, so it was again confirmed to have very low non-specific binding by the CRP immunoassay. From these results, the OM layer with autodisplayed protein G was confirmed to have antibody-binding activity comparable to that of protein G on E. coli cells, and the OM coating on the microplate was applicable to immunoassays.
The constructed and confirmed immunoaffinity layer with orientation control based on the OM with autodisplayed protein G was applied to an SPR biosensor, which is an optical biosensor based on an evanescence field that can be detected within a few hundred nanometers of the surface of a sensor chip.44 In this case, the OM layer was suitable for application because the E. coli cells were larger than the detection length. Gold was selected as the transducer surface owing to its biologically novel and hydrophobic properties. Similar to the microplate, the Au chip was coated with isolated OM particles by hydrophobic interactions and the layering process was monitored with the SPR biosensor, as shown in Fig. 6. When OM particles were introduced, the SPR response increased significantly during incubation. After the washing step, the SPR response increased by 40 A.U. owing to the formation of the OM layer with autodisplayed protein G (Fig. 6a). Next, the capture antibodies and anti-CRP antibodies were injected. Bound antibodies against autodisplayed protein G on the OM layer generated an increased SPR response by 2 A.U. This increased SPR response indicated that the immunoaffinity layer was generated by sequential OM layering and antibody immobilization. After the formation of the immunoaffinity layer, CRP samples with concentrations of 1, 10, 100, and 1000 ng mL−1 were sequentially injected. As shown in Fig. 6a, the SPR responses increased when the CRP samples were treated. This illustrates that CRP binds to the immunoaffinity layer based on autodisplayed protein G, and the binding process was available to be monitored using an SPR biosensor. The increased SPR responses were estimated to be 0.9, 2.4, 3.5, and 5.0 A.U. for 1, 10, 100, and 1000 ng mL−1 CRP, respectively (inset of Fig. 6a). These signals showed an increase with an increase in concentration. When linear fitting was performed based on CRP concentrations on a log scale, it showed good linearity with R2 values greater than 0.99. These results indicate that accurate measurement of CRP concentrations in the range of 1–1000 ng mL−1 was realized by the application of the OM layer with autodisplayed protein G to the immunoaffinity layer of the SPR biosensor.
 | ||
| Fig. 6 SPR response using an orientation-controlled immunoaffinity layer based on (a) OM with autodisplayed protein G and (b) a protein G layer. (c) SPR response using a physically adsorbed antibody layer. Insets: relative SPR responses according to the CRP concentration. (d) SPR response based on the OM from intact E. coli without autodisplay. | ||
For comparison with another orientation controlled immunoaffinity layer, commercial protein G was applied to the SPR biosensor. As shown in Fig. 6b, antibodies were immobilized on the protein G layer after blocking. The increased SPR responses were estimated to be 0.1, 0.1, 0.8, and 2.6 A.U. for 1, 10, 100, and 1000 ng mL−1 CRP, respectively (inset of Fig. 6b). These data support that layering of the OM with autodisplayed targets on the surface of the transducer has high immobilizing efficiency and the OM layer with autodisplayed protein G is suitable for the density control of antibodies. In the case of the immunoaffinity layer by physical adsorption (Fig. 6c), the increased SPR responses were 0.1, 0.7, 1.7, and 3.1 A.U. for 1, 10, 100, and 1000 ng mL−1 CRP, respectively (inset of Fig. 6c). This result indicates that the orientation control by autodisplayed protein G improves the sensitivity of the SPR biosensor. In the comparison group, an OM layer without an autodisplay was generated on the Au chip of the SPR biosensor, as shown in Fig. 6d. Even in this case, the SPR response was increased by 40 A.U. by the formation of an OM layer. However, when the capture antibodies were injected, there was no significant increase in the SPR response. In addition, when CRP samples with the same concentrations were used, there was no significant increase in the SPR response. This implies that the antibodies were not bound to the surface of the SPR biosensor. This supports the idea that the layered OM blocks the immobilization of antibodies, and antibody immobilization is only available when protein G is autodisplayed. These experiments confirmed that an immunoaffinity layer with orientation controlled by autodisplayed protein G was introduced into an SPR biosensor and was suitable for CRP measurements with high sensitivity and low non-specific binding.
Conclusions
The IgG-binding protein, streptococcal protein G, was expressed as a fusion protein on the OM of E. coli cells by autodisplay technology to generate an immunoaffinity layer for immunoassays and biosensors. The DNA sequence of protein G was inserted into the target sequence of the autodisplay vector using recombinant technology to construct the autodisplaying vector pJP002, and the constructed plasmid vector was transformed into BL21(DE3) E. coli cells. Autodisplay of protein G was analyzed by SDS-PAGE of OM proteins and confirmed to be expressed on the OM at 2 × 105 copies per cell by densitometry in comparison with OmpA. In addition, E. coli cells with autodisplayed protein G were confirmed to have IgG-binding affinity by fluorescence microscopy after the addition of fluorescein-labeled IgG. The IgG-binding affinity was compared with that of another IgG-binding protein, the Z-domain. When various concentrations of goat polyclonal antibodies were immobilized on E. coli cells with autodisplayed protein G and Z-domain, protein G showed higher affinity to goat IgG. After affinity confirmation, immunoassays based on protein G autodisplaying E. coli cells were performed to detect HRP and CRP. E. coli cells were used as a solid support for the immobilization of the capture antibodies. HRP immunoassay showed an LOD of 1 ng mL−1 with a dynamic range of 0.1–100 ng mL−1. In the CRP immunoassay, CRP was detected using a sandwich-type immunoassay and the assay showed an LOD of 2 ng mL−1 with a dynamic range of 2–300 ng mL−1. In both immunoassays, autodisplayed protein G showed better sensitivity than autodisplayed Z-domains and physical adsorption. This means that autodisplayed protein G has a high affinity for goat IgG and has the potential to improve the sensitivity of immunoassays by controlling the orientation of antibodies. After the establishment of the E. coli cell-based immunoassay, the OMs of protein G autodisplaying E. coli cells were isolated and layered on a microplate and Au chip to construct an immunoaffinity layer for immunoassays and SPR biosensors. OM-based immunoassays to detect HRP and CRP showed LODs of 5 and 0.2 ng mL−1 with dynamic ranges of 5–300 and 0.2–100 ng nL−1 for HRP and CRP, respectively. When the OM layer was applied to the SPR biosensor for CRP detection, the SPR responses showed good linearity in the CRP concentration range of 1–1000 ng mL−1 with an R2 value of 0.99. These results indicated that protein G was successfully autodisplayed on the OM of E. coli cells with IgG-binding affinity, and autodisplayed protein G was suitable for use as a solid support for antibodies for immunoassays. In addition, the OM with autodisplayed protein G could be extended to construct an immunoaffinity layer with orientation control by layering on a 2D substrate, such as a microplate or surface of the transducer of biosensors. Thus, the autodisplay of protein G is potentially applicable in various immunoassays and immunosensor formats to improve sensitivity by orientation control and density control.Author contributions
S. G. K.: data curation, formal analysis, writing – original draft, and visualization. J. K.: formal analysis and visualization. M. Y. K.: data curation and formal analysis. J. P.: conceptualization and data curation. J. J.: conceptualization and validation. M. P.: supervision, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the Hallym University Research Fund, 2022 (HRF-202201-008).References
- P. Thakur and A. Ward, Health Phys., 2019, 116, 694–714 CrossRef CAS PubMed.
- S.-M. Kang, BioChip J., 2021, 1–14 Search PubMed.
- X. Ma, W. Ding, C. Wang, H. Wu, X. Tian, M. Lyu and S. Wang, Sens. Actuators, B, 2021, 331, 129422 CrossRef CAS.
- M. Park, BioChip J., 2019, 13, 82–94 CrossRef CAS.
- C. Gouvêa, Biosensors for health, environment and biosecurity, 2011, pp. 71–86 Search PubMed.
- M. Park, Sensors, 2020, 20, 2775 CrossRef CAS PubMed.
- M. Aydin, E. B. Aydin and M. K. Sezgintürk, Adv. Clin. Chem., 2021, 102, 1–62 CrossRef PubMed.
- K. Mahato, S. Kumar, A. Srivastava, P. K. Maurya, R. Singh and P. Chandra, Handbook of immunoassay technologies, Elsevier, 2018, pp. 359–414 Search PubMed.
- Y. Kanai, Y. Ohmuro-Matsuyama, M. Tanioku, S. Ushiba, T. Ono, K. Inoue, T. Kitaguchi, M. Kimura, H. Ueda and K. Matsumoto, ACS Sens., 2020, 5, 24–28 CrossRef CAS PubMed.
- J. Kang and M.-G. Kim, BioChip J., 2020, 14, 18–31 CrossRef CAS.
- X. Huang, S. Sang, Z. Yuan, Q. Duan, X. Guo, H. Zhang and C. Zhao, ACS Sens., 2021, 6, 3933–3939 CrossRef CAS PubMed.
- X. Sun, Y. Ye, S. He, Z. Wu, J. Yue, H. Sun and X. Cao, Biosens. Bioelectron., 2019, 143, 111607 CrossRef CAS PubMed.
- A. C. Register, S. S. Tarighat and H. Y. Lee, Int. J. Mol. Sci., 2021, 22, 5350 CrossRef CAS PubMed.
- R. M. Lequin, Clin. Chem., 2005, 51, 2415–2418 CrossRef CAS PubMed.
- A. Makaraviciute and A. Ramanaviciene, Biosens. Bioelectron., 2013, 50, 460–471 CrossRef CAS PubMed.
- M. Park, J. C. Pyun and J. Jose, J. Pharm. Biomed. Anal., 2018, 147, 174–184 CrossRef CAS PubMed.
- S. Buus, A. Sette, S. M. Colon and H. M. Grey, Science, 1988, 242, 1045–1047 CrossRef CAS PubMed.
- J. Zorea, R. P. Shukla, M. Elkabets and H. Ben-Yoav, Anal. Bioanal. Chem., 2020, 412, 1709–1717 CrossRef CAS PubMed.
- D. Boyd, A. Ebrahimi, S. Ronan, B. Mickus, M. Schenauer, J. Wang, D. Brown and A. Ambrogelly, mAbs, 2018, 10, 346–353 CrossRef CAS PubMed.
- P. S. Norman, J. Allergy Clin. Immunol., 1995, 96, 274 CrossRef.
- A. K. Trilling, J. Beekwilder and H. Zuilhof, Analyst, 2013, 138, 1619–1627 RSC.
- B. Lu, M. R. Smyth and R. O'Kennedy, Analyst, 1996, 121, 29R–32R RSC.
- S. Hober, K. Nord and M. Linhult, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 848, 40–47 CrossRef CAS PubMed.
- J. B. Fishman and E. A. Berg, Cold Spring Harbor Protocols, 2019, 2019, pdb. prot099143 Search PubMed.
- D. Lin, R. G. Pillai, W. E. Lee and A. B. Jemere, Microchim. Acta, 2019, 186, 1–9 CrossRef CAS PubMed.
- Q. Li, X. Dou, L. Zhang, X. Zhao, J. Luo and M. Yang, Anal. Bioanal. Chem., 2019, 411, 6057–6066 CrossRef CAS PubMed.
- J.-M. Park, M. Y. Kim, J. Jose and M. Park, Int. J. Mol. Sci., 2022, 23, 459 CrossRef CAS PubMed.
- M. Park, G. Yoo, J. H. Bong, J. Jose, M. J. Kang and J. C. Pyun, Biochim. Biophys. Acta, 2015, 1848, 842–847 CrossRef CAS PubMed.
- J. Jose and T. F. Meyer, Microbiol. Mol. Biol. Rev., 2007, 71, 600–619 CrossRef CAS PubMed.
- J. Jose, Appl. Microbiol. Biotechnol., 2006, 69, 607–614 CrossRef CAS PubMed.
- G. Kronvall, H. M. Grey and R. C. Williams, J. Immunol., 1970, 105, 1116–1123 CrossRef CAS.
- W. Choe, T. A. Durgannavar and S. J. Chung, Materials, 2016, 9, 994 CrossRef PubMed.
- B. Akerström and L. Björck, J. Biol. Chem., 1986, 261, 10240–10247 CrossRef.
- A. E. Sauer-Eriksson, G. J. Kleywegt, M. Uhlén and T. A. Jones, Structure, 1995, 3, 265–278 CrossRef CAS PubMed.
- A. M. Gronenborn, D. R. Filpula, N. Z. Essig, A. Achari, M. Whitlow, P. T. Wingfield and G. M. Clore, Science, 1991, 253, 657–661 CrossRef CAS PubMed.
- S. D. Schumacher and J. Jose, J. Biotechnol., 2012, 161, 113–120 CrossRef CAS PubMed.
- H. Yamazoe, Mater. Sci. Eng., C, 2019, 100, 209–214 CrossRef CAS PubMed.
- X. Wang, D. Teng, Q. Guan, R. Mao, Y. Hao, X. Wang, J. Yao and J. Wang, AMB Express, 2017, 7, 1–12 CrossRef PubMed.
- R. Koebnik, K. P. Locher and P. Van Gelder, Mol. Microbiol., 2000, 37, 239–253 CrossRef CAS PubMed.
- J. Jose, J. W. Chung, B. J. Jeon, R. M. Maas, C. H. Nam and J. C. Pyun, Biosens. Bioelectron., 2009, 24, 1324–1329 CrossRef CAS PubMed.
- M. Bendayan and S. Garzon, J. Histochem. Cytochem., 1988, 36, 597–607 CrossRef CAS PubMed.
- J. Gibbs and M. Kennebunk, Corning Life Sciences ELISA Technical Bulletin, 2001, vol. 14 Search PubMed.
- C. Fava and M. Montagnana, Front. Pharmacol., 2018, 9, 55 CrossRef PubMed.
- Y. Yanase, H. Suzuki, T. Tsutsui, T. Hiragun, Y. Kameyoshi and M. Hide, Biosens. Bioelectron., 2007, 22, 1081–1086 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |