
Top-down mass spectrometry and assigning internal fragments for determining disulfide bond positions in proteins†
Benqian
Wei
 a,
Muhammad A.
Zenaidee
ac,
Carter
Lantz
a,
Brad J.
Williams
d,
Sarah
Totten
d,
Rachel R.
Ogorzalek Loo
a and
Joseph A.
Loo
*ab
a,
Muhammad A.
Zenaidee
ac,
Carter
Lantz
a,
Brad J.
Williams
d,
Sarah
Totten
d,
Rachel R.
Ogorzalek Loo
a and
Joseph A.
Loo
*ab
aDepartment of Chemistry and Biochemistry, University of California Los Angeles, Los Angeles, CA, USA. E-mail: jloo@chem.ucla.edu
bDepartment of Biological Chemistry, University of California Los Angeles, Los Angeles, CA, USA
cAustralian Proteome Analysis Facility, Macquarie University, Macquarie Park, NSW, Australia
dWaters Corporation, Milford, MA, USA
First published on 14th November 2022
Abstract
Disulfide bonds in proteins have a substantial impact on protein structure, stability, and biological activity. Localizing disulfide bonds is critical for understanding protein folding and higher-order structure. Conventional top-down mass spectrometry (TD-MS), where only terminal fragments are assigned for disulfide-intact proteins, can access disulfide information, but suffers from low fragmentation efficiency, thereby limiting sequence coverage. Here, we show that assigning internal fragments generated from TD-MS enhances the sequence coverage of disulfide-intact proteins by 20–60% by returning information from the interior of the protein sequence, which cannot be obtained by terminal fragments alone. The inclusion of internal fragments can extend the sequence information of disulfide-intact proteins to near complete sequence coverage. Importantly, the enhanced sequence information that arise from the assignment of internal fragments can be used to determine the relative position of disulfide bonds and the exact disulfide connectivity between cysteines. The data presented here demonstrates the benefits of incorporating internal fragment analysis into the TD-MS workflow for analyzing disulfide-intact proteins, which would be valuable for characterizing biotherapeutic proteins such as monoclonal antibodies and antibody–drug conjugates.
Introduction
Disulfide bonds are among the most important posttranslational modifications (PTMs) in proteins, as they have a substantial impact on protein structure, stability, and biological activity.1–4 Determining disulfide bonding patterns is critical for understanding protein folding and higher-order structure as non-native disulfide bridges and aggregates can have detrimental effects on a protein's three-dimensional structure and consequently their function.5,6 The advancement of biotherapeutics such as monoclonal antibodies and antibody–drug conjugates have further driven the development of more efficient and accurate experimental strategies including mass spectrometry (MS) and ion mobility-MS to characterize disulfide bond linkages,7–13 as disulfide connectivity, which ensures its proper folding and consequently biological function and immunogenicity, is considered as a critical quality attribute during antibody manufacturing.14,15 Mass spectrometry has established itself as a frontrunner for these characterizations owing to its exceptional sensitivity, low sample requirements, and the ability to be coupled with chromatographic separations to generate and detect diagnostic fragment ions possessing various disulfide connectivities,16–19 which cannot be achieved easily by conventional methods such as nuclear magnetic resonance (NMR) and X-ray crystallography.20,21Conventional “bottom-up” MS approaches employ chemical reduction and alkylation to cleave disulfide bonds and cap the free cysteines, followed by enzymatic digestion of the protein prior to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.22,23 Although protein sequence usually can be unambiguously determined using this approach, information on disulfide bond locations and connectivities can be lost.18,19 To compensate for this limitation, alternative strategies including proteolysis without prior reduction or with partial reduction have been utilized to generate disulfide-linked peptides for LC-MS/MS measurements.24–29 This allows for the elucidation of disulfide bonding patterns by comparing the peptides resulting from the reduced regions with the peptides from constrained regions to identify disulfide-linked peptides. However, it is difficult to control the amount of disulfide reduction using this approach, which results in complex mixtures of peptides with differing amounts of capped cysteines, making data analysis challenging.30 Moreover, with limited disulfide reduction, protein sequence coverage may not be sufficient to capture all disulfide linkage information. This problem will be exacerbated with increasing protein size and/or proteins that contain a large number of disulfide bonds.31
Top-down mass spectrometry (TD-MS), where direct mass measurement and subsequent fragmentation of intact gas-phase protein ions in the mass spectrometer to obtain the primary sequence information, has gained in popularity in recent years for interrogating proteins with various PTMs, including but not limited to disulfide bonds.32–36 TD-MS bypasses the time-consuming digestion and separation steps, allowing for all disulfide information to be preserved. By comparing the accurate measured mass with the theoretical sequence mass of disulfide intact proteins, the number of disulfide bonds can be readily determined. The modification sites can be further identified by subsequent fragmentation of the intact protein ions with high sequence coverage. However, challenges still remain. Accessing disulfide bond information usually requires concurrent fragmentation of the protein backbone and disulfide bonds to gain extensive sequence coverage, which is important for localizing disulfide bridges, whereas TD-MS suffers from low relative fragmentation efficiency, limiting sequence coverage.37–39 To increase sequence coverage, various fragmentation methods (alternative to the traditionally employed collision-based techniques) have been employed to characterize disulfide-intact peptides and proteins including electron-based dissociation (ExD),9,31,40–43 photon-based dissociation (PD),19,44–48 and their hybrid methods with varying success.12,30,49,50 An additional approach to increase TD-MS sequencing efficiency is to incorporate the assignment of internal fragments,51,52 generated by multiple gas-phase cleavages of the polypeptide backbone, into the data analysis workflow.53
While the analysis of internal fragment ions has been largely ignored by the TD-MS community due to the general lack of software tools to accurately and reliably assign them, the concept of the formation of internal fragment ions in TD-MS spectra is not novel. Previous studies have shown that the inclusion of internal fragments results in much richer sequence information of small peptides,51,54–57 intact proteins,58–64 protein complexes,65,66 and aid the identification of ambiguous proteoforms in mammalian cell lysates by top-down ptoteomics.67 In addition, a recent study by Chin et al. demonstrated the utility of internal fragments to enhance sequence coverage and to decipher disulfide bonds of disulfide-rich peptides.68 Schmitt et al. also applied internal fragments to determine sequence motifs located within a disulfide constrained loop of SOD1 protein that could not be achieved by terminal fragments alone.63 The benefits of including internal fragments for characterizing disulfide-intact proteins are two-fold. Identifiable internal fragment ions within disulfide constrained regions can be generated without the need to cleave the disulfide bond,68 lowering the barrier to obtaining more sequence information. Second, by including internal fragments, the chance of identifying product ions that result from cleavage of disulfide bonds to access disulfide linkage information is higher than analyzing terminal fragments alone.
Here, we show that assigning internal fragments generated from collisionally activated dissociation (CAD) and ExD can increase the sequence coverage of disulfide-intact proteins by accessing the interior of the protein sequence constrained by multiple disulfide bonds. Importantly, by correlating the number of disulfide bonds cleaved by internal fragments to their sequence positions, the relative locations of disulfide bonds can be determined. By specifically analyzing internal fragments with disulfide bonds remaining intact, disulfide connectivity can be determined. This study demonstrates the benefits of considering internal fragments when analyzing these heavily constrained proteins, which would be valuable for characterizing biotherapeutic proteins that contain a large number of disulfide bonds.
Experimental
Materials and sample preparation
The proteins β-lactoglobulin from bovine milk, ribonuclease A from bovine pancreas, α-lactalbumin from bovine milk, trypsin inhibitor from glycine max, and m-nitrobenzyl alcohol (mNBA) were purchased from Sigma-Aldrich (St Louis, MO, USA). Lysozyme from chicken white egg was acquired from EMD Millipore (Darmstadt, Germany). LC/MS-grade water, methanol and formic acid were obtained from Fisher Chemical (Hampton, NH). All proteins were used without further purification. Protein samples were prepared in 49.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49.5:1 water/methanol/formic acid to a final concentration of 10 or 20 μM. Supercharging agent mNBA was added to the ribonuclease A and α-lactalbumin solutions at a 0.25% (v/v) concentration.
:49.5:1 water/methanol/formic acid to a final concentration of 10 or 20 μM. Supercharging agent mNBA was added to the ribonuclease A and α-lactalbumin solutions at a 0.25% (v/v) concentration.
Mass spectrometry
All samples were measured with a 15-Tesla solariX Fourier transform ion cyclotron resonance (FTICR)-MS instrument equipped with an infinity ICR cell (Bruker Daltonics, Billerica, MA, USA). The protein solutions were loaded into in-house pulled capillaries coated with gold, and electrosprayed by applying a voltage between 0.7 and 1.5 kV on the electrospray ionization capillary. Individual charge states of each multiply-protonated protein (11+ to 15+ for β-lactoglobulin, 8+ to 12+ for lysozyme, 8+ to 14+ for ribonuclease A, and 11+ to 14+ for α-lactalbumin) were isolated in the quadrupole, with an isolation window of 10 m/z before fragmentation. Three fragmentation methods including CAD, electron capture dissociation (ECD), and electron induced dissociation (EID) were applied to each isolated ion. For CAD fragmentation, collision energies were adjusted to achieve the same lab-frame energy for different charge states of each protein. The lab-frame energy is defined as the multiplication product of charge state and collision energy. The lab-frame energies used for each protein are: β-lactoglobulin, 336 V; lysozyme, 438 V; ribonuclease A, 330 V; α-lactalbumin, 286 V to achieve optimal fragmentation. For ECD fragmentation, the pulse length was set at 0.02 s, with a lens voltage at 50 V and bias voltage at 2 V. For EID fragmentation, the pulse length was set at 0.02 s, with a lens voltage at 50 V and bias voltage ranging from 26 to 30 V.CAD-MS/MS of trypsin inhibitor (TI) was done by isolating [TI + 17H]17+ with an isolation window of 10 m/z. The CAD energy was set at 20 V, which reduced the precursor ion signal to ∼40% of the mass spectral level.
ECD-MS/MS of β-lactoglobulin and lysozyme were also performed on a Waters SELECT SERIES™ Cyclic IMS Q-ToF mass spectrometer (Waters, Milford, MA, USA) with an electromagnetostatic ExD cell (e-MSion Inc., Corvallis, OR) mounted before the cyclic ion mobility cell to allow for pre-IMS ECD fragmentation. All ECD parameters were optimized to achieve the best fragmentation.
Data analysis
Results and discussion
Internal fragments can access the interior protein sequence constrained by multiple disulfide bonds
To demonstrate that internal fragments can enhance sequence information of disulfide intact proteins, three fragmentation methods were applied, CAD, ECD, and EID on various isolated precursor charge states of four disulfide-intact proteins, including β-lactoglobulin (2 disulfide bonds), lysozyme (4 disulfide bonds), ribonuclease A (4 disulfide bonds), and α-lactalbumin (4 disulfide bonds). The disulfide connectivity of these proteins is shown in Scheme 1. EID fragmentation of β-lactoglobulin, [B-lac + 14H]14+ generated rich mass spectra filled with informative peaks (Fig. 1A). Many of the peaks in the spectra that were not assigned as terminal fragments can be assigned as internal fragments (Fig. 1A inset), demonstrating that more information can be extracted from a single MS/MS spectrum when considering internal fragments. Importantly, the location of all the assigned fragments for B-lac demonstrates that internal fragments span much of the interior sequence enclosed by multiple disulfide bonds, providing complementary sequence information to terminal fragments (Fig. 1B). Similar results were also observed for EID of lysozyme, [Lys + 10H]10+ (Fig. 1C and D). In both cases, the extent of information extracted from a single mass spectrum can be enhanced significantly when including internal fragments. Further, ECD and CAD of the same isolated precursor ions show similar fragmentation patterns, although ECD is less energetic than EID and CAD, and generated significantly fewer internal fragments (Fig. S1 and S2†).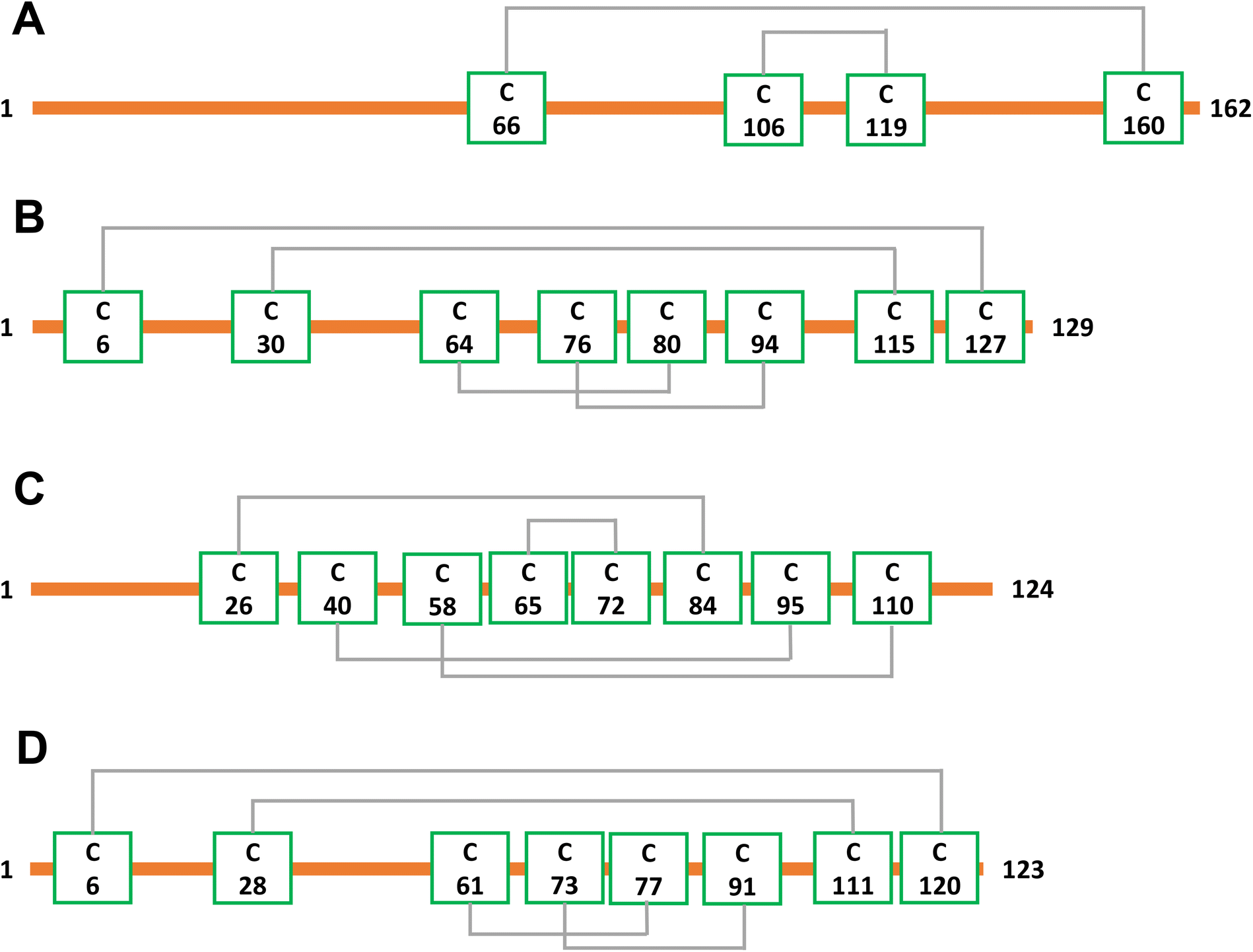 | ||
| Scheme 1 Disulfide bond connectivities of the four proteins examined, (A) β-lactoglobulin (2 disulfide bonds), (B) lysozyme (4 disulfide bonds), (C) ribonuclease A (4 disulfide bonds), (D) α-lactalbumin (4 disulfide bonds). | ||
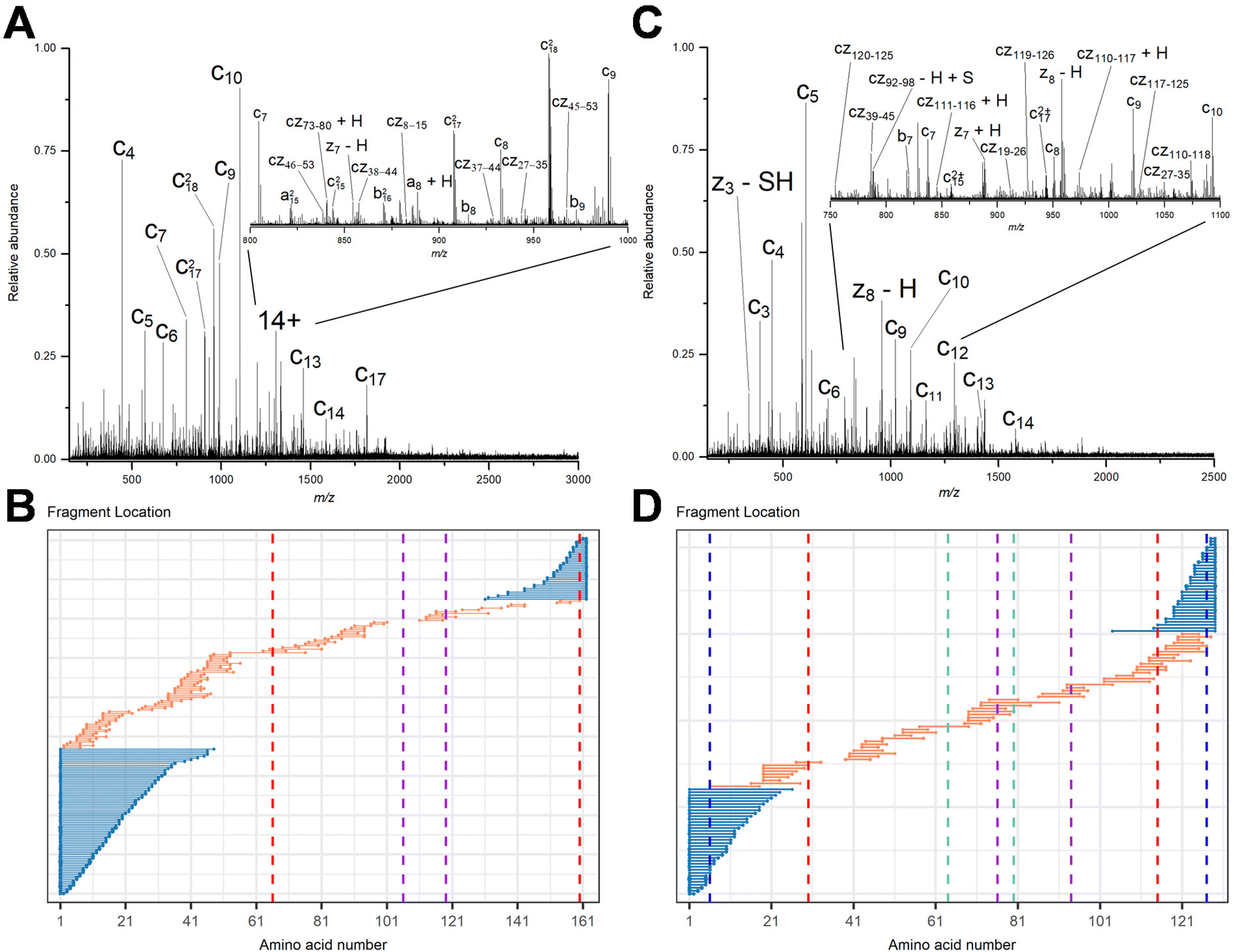 | ||
| Fig. 1 Representative EID MS/MS spectra of (A) β-lactoglobulin, [B-lac + 14H]14+ and (C) lysozyme, [Lys + 10H]10+. Fragment location maps indicating the region of the protein sequence covered by terminal fragments (blue) and internal fragments (orange) for (B) EID of β-lactoglobulin, [B-lac + 14H]14+ (spectrum in A) and (D) EID of lysozyme, [Lys + 10H]10+ (spectrum in C). Vertical dashed lines in panels B and D represent cysteines positions, with the same color indicating a disulfide bond is formed between those two cysteines. | ||
To compare sequence information obtained from terminal fragments with internal fragments, all assigned unique fragments generated from every charge state for each protein were integrated. Assigning internal fragments generated from CAD, ECD, and EID increases the sequence coverage by 20–60% for all proteins examined. For example, sequence coverage increases from 43% to 83% for EID of β-lactoglobulin (Fig. 2D), 37% to 84% for EID of lysozyme (Fig. 3F), 40% to 87% for EID of ribonuclease A (Fig. S3F†), and 36% to 90% for EID of α-lactalbumin (Fig. S4F†) after including internal fragments. Incorporating internal fragments can cover almost every single inter-residue site to achieve near complete sequence coverage (99%) for CAD of lysozyme (Fig. 3F), with CAD of α-lactalbumin also close to 100% sequence coverage (96%, Fig. S4F†). This is primarily due to the fact that the generation of terminal fragments beyond regions enclosed by disulfide bonds is difficult (vide infra); most often, an S–S bond would need to be cleaved in order to release the terminal fragment. This is further discussed below.
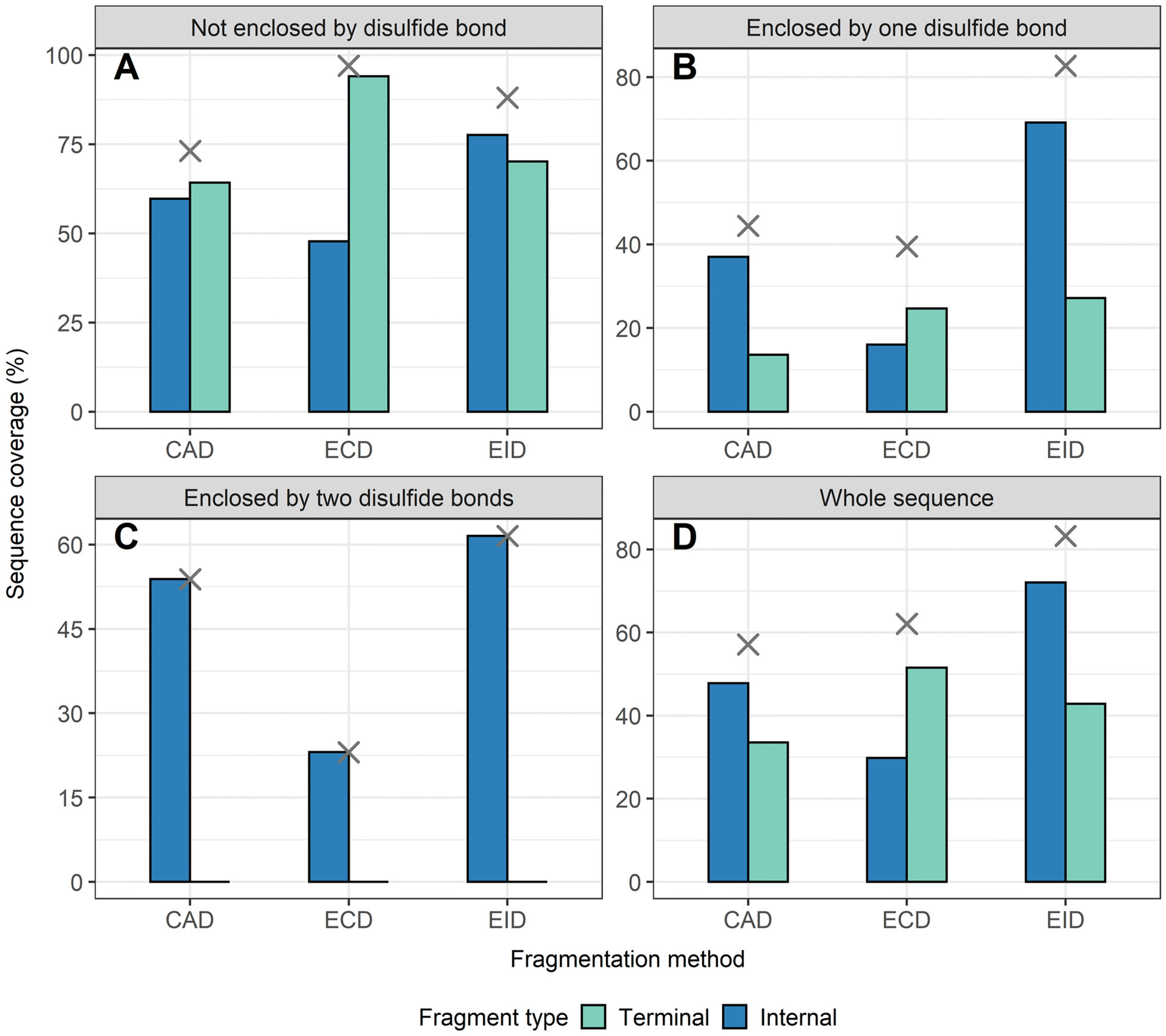 | ||
| Fig. 2 The extent of sequence information obtained by terminal and internal fragments for β-lactoglobulin at different sequence regions after integrating data from all five charge states (11+ to 15+) and for all three fragmentation methods (CAD, ECD, and EID) examined, (A) sequence not enclosed by disulfide bond, (B) sequence enclosed by one disulfide bond, (C) sequence enclosed by two disulfide bonds, (D) whole sequence. Cross marks in each panel indicate the sequence coverage after combing terminal and internal fragments. | ||
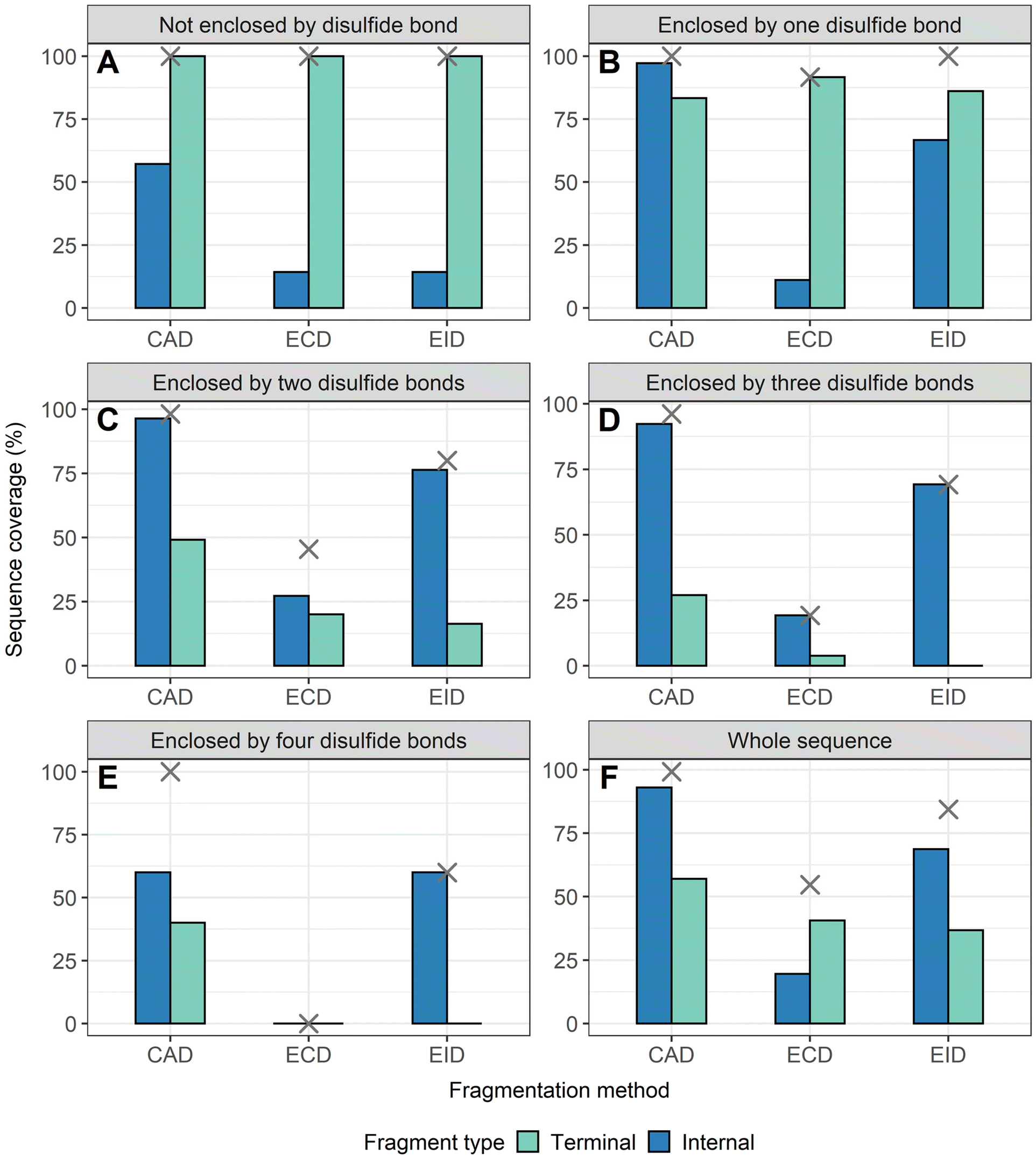 | ||
| Fig. 3 The extent of sequence information obtained by terminal and internal fragments for lysozyme at different sequence regions after combining data from all five charge states (8+ to 12+) and for all three fragmentation methods (CAD, ECD, and EID) examined, (A) sequence not enclosed by disulfide bond, (B) sequence enclosed by one disulfide bond, (C) sequence enclosed by two disulfide bonds, (D) sequence enclosed by three disulfide bonds. (E) Sequence enclosed by four disulfide bonds. (F) Whole sequence. Cross marks in each panel indicate the sequence coverage after combing terminal and internal fragments. | ||
The sequence of these proteins can be classified into different regions depending on the number of disulfide bonds enclosed. For example, β-lactoglobulin has two disulfide bonds with a connectivity of Cys66–Cys160 and Cys106–Cys119 (Scheme 1A), thus the β-lactoglobulin sequence can be classified into three regions: (i) sequence not enclosed by disulfide bond (residues 1–66, 160–162), (ii) sequence enclosed by one disulfide bond (residues 66–106, 119–160), and (iii) sequence enclosed by two disulfide bonds (residues 106–119). Similarly, the sequence of lysozyme, which possesses four disulfide bonds (Scheme 1B) can be classified into five regions including sequence not enclosed by a disulfide bond (residues 1–6, 127–129), sequence enclosed by one disulfide bond (residues 6–30, 115–127), sequence enclosed by two disulfide bonds (residues 30–64, 94–115), sequence enclosed by three disulfide bonds (residues 64–76, 80–94), and sequence enclosed by four disulfide bonds (residues 76–80). For the other two proteins with four disulfide bonds, the primary protein sequence can also be separated into specific regions (ribonuclease A, Scheme 1C, and α-lactalbumin, Scheme 1D). To investigate the utility of internal fragments for accessing highly disulfide constrained regions, the extent of sequence information obtained from terminal and internal fragments at different sequence regions were compared and a clear trend can be observed. Generally, most internal fragments originate from the interior of the sequence within disulfide bonded regions, while terminal fragments originate from the outermost sequence. For example, for CAD of β-lactoglobulin, terminal fragments cover more sequence not enclosed by disulfide bond than internal fragments (64% vs. 60%, Fig. 2A), corresponding to a change of +4%, while no terminal fragments and only internal fragments cover the sequence enclosed by two disulfide bonds (0% vs. 54%, Fig. 2C), corresponding to a change of −54%. Similarly, for CAD of lysozyme with four disulfide bonds and five distinct sequence regions, the sequence coverage change when comparing terminal vs. internal fragments are +43%, −14%, −47%, −65%, and −20%, respectively, when going deeper into the middle of the sequence (Fig. 3A–E). This data clearly demonstrates that internal fragments significantly enhances sequence information of the regions constrained by multiple disulfide bonds. A similar trend was observed for ECD and EID of these two proteins (Fig. 2 and 3) and the other two proteins possessing four disulfide bonds (ribonuclease A, Fig. S3, and α-lactalbumin, Fig. S4†), with the relative sequence coverage decreasing for terminal fragments while increasing for internal fragments when reaching the interior protein sequence (Fig. 2A–C, 3A–E, S3A–E, and S4A–E†). Notably, some specific sequence regions can only be accessed by internal fragments, such as the sequence enclosed by two disulfide bonds of β-lactoglobulin (Fig. 2C) and sequence enclosed by four disulfide bonds of ribonuclease A (Fig. S3E†) and α-lactalbumin (Fig. S4E†), highlighting the ability of internal fragments to cover regions that cannot be reached by terminal fragments. The data shown here shows promise for the inclusion of internal fragments in obtaining more comprehensive sequence information for disulfide-intact proteins.
Internal fragments can determine the relative position of disulfide bonds
To determine the position of disulfide bonds for these proteins, the number of disulfide bond cleavages were analyzed. We show here that terminal fragments result from cleavage of disulfide bonds located on the exterior of the protein, while internal fragments can result from cleavage of disulfide bonds within the interior of the protein. For example, terminal fragments generated by EID of β-lactoglobulin (2 disulfide bonds) resulted from more cleavages at the outermost disulfide bond (Cys66–Cys160) than internal fragments (38 vs. 11, Fig. 4A), while only internal fragments originated from the cleavage of the interior disulfide bond (9 times at the Cys106–Cys119 bond, Fig. 4B). This trend is more pronounced for proteins with a greater number of disulfide bonds. For example, EID of lysozyme (4 disulfide bonds) showed that the Cys6–Cys127 bond was cleaved 62 times by terminal fragments but only 6 times by internal fragments (Fig. 5A). For the Cys30–Cys115 bond, located more interior of the protein sequence, the difference between disulfide cleavages from terminal and internal fragments was reversed, 10 vs. 16, respectively (Fig. 5B). For the Cys64–Cys80 bond and the Cys76–Cys94 bond, the disulfide cleavages comparison is 0 vs. 17 and 0 vs. 19 (terminal vs. internal, Fig. 5C and D). This trend was also observed for CAD and ECD of β-lactoglobulin and lysozyme, and the other two disulfide bonded proteins (ribonuclease A, Fig. S5, and α-lactalbumin, Fig. S6†). Surprisingly, for disulfide bonds buried within the protein, their cleavages were only explained by internal fragments (Fig. 4B, 5C and D, S5D, S6C and D†), highlighting the use of internal fragments to access disulfide bond information that cannot be obtained by terminal fragments.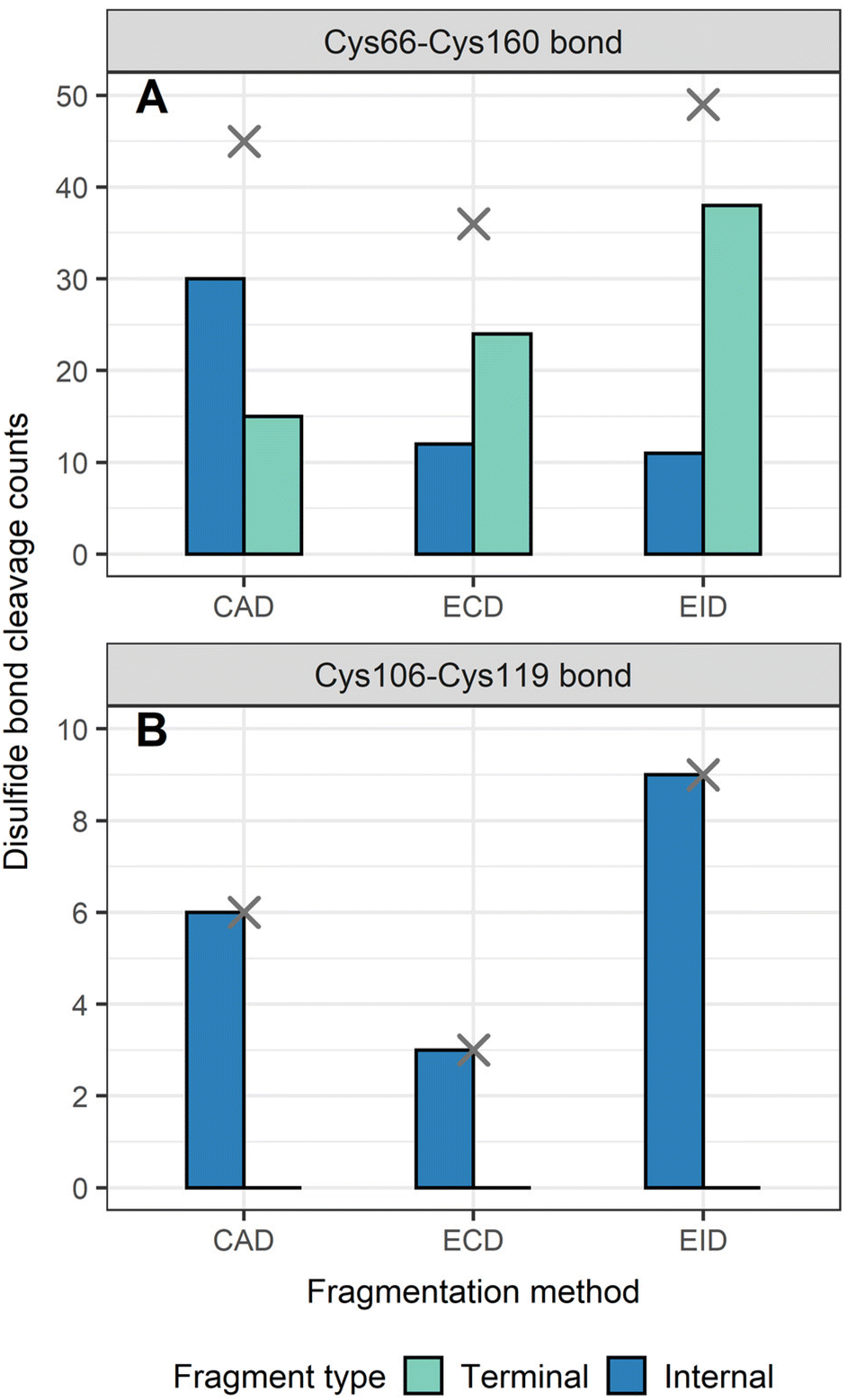 | ||
| Fig. 4 Number of disulfide bonds cleaved by terminal and internal fragments for β-lactoglobulin after integrating data from all five charge states (11+ to 15+) and for all three fragmentation methods (CAD, ECD, and EID) examined, (A) Cys66–Cys160 bond, (B) Cys106–Cys119 bond. Cross marks in each panel indicate the disulfide bond cleavage counts after combing terminal and internal fragments. | ||
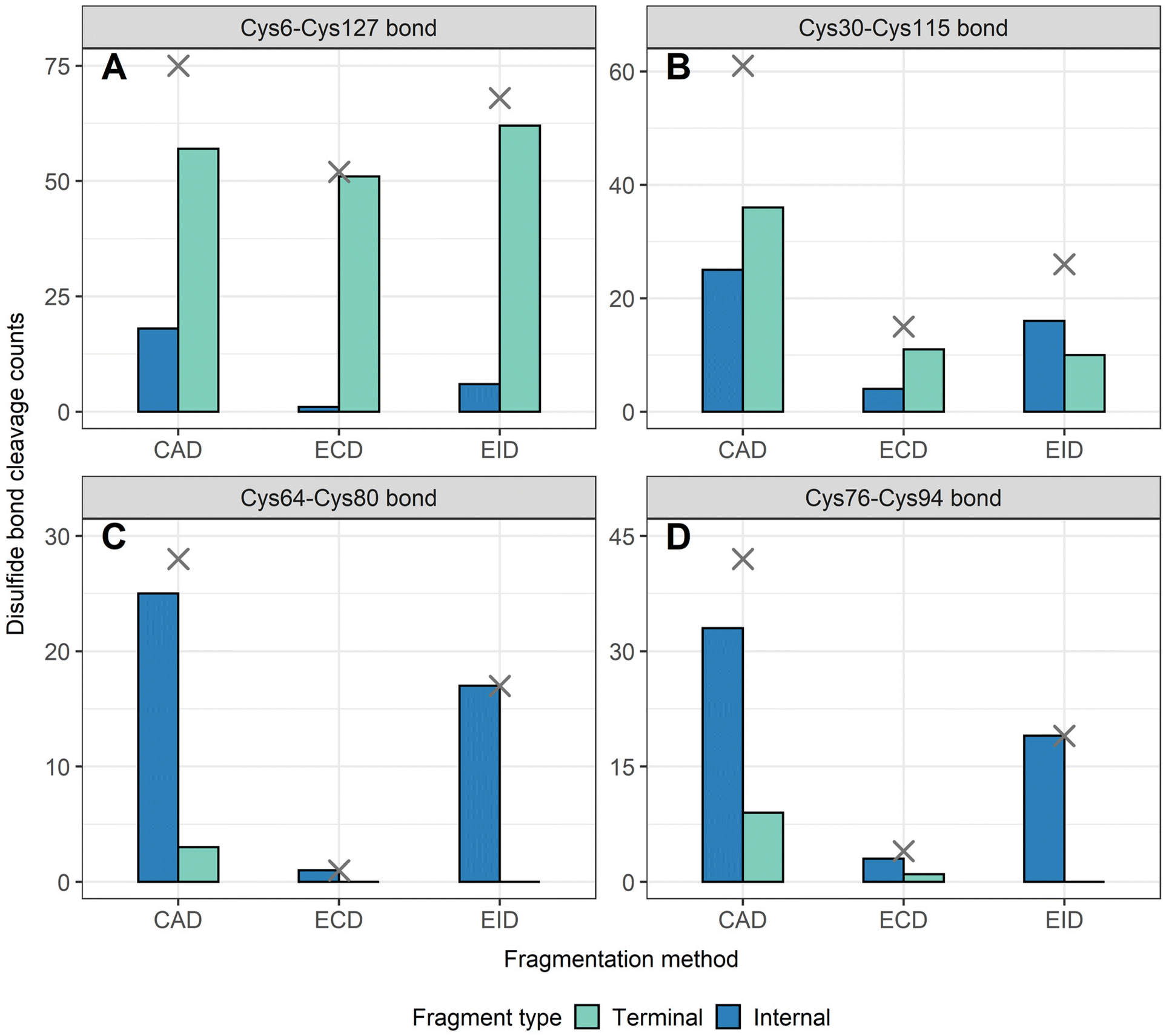 | ||
| Fig. 5 Number of disulfide bonds cleaved by terminal and internal fragments for lysozyme after combining data from all five charge states (11+ to 15+) for all three fragmentation methods (CAD, ECD, and EID) examined, (A) Cys6–Cys127 bond, (B) Cys30–Cys115 bond, (C) Cys64–Cys80 bond, (D) Cys76–Cys94 bond. Cross marks in each panel indicate the disulfide bond cleavage counts after combing terminal and internal fragments. | ||
These data indicate that by correlating the relative number of disulfide cleavages resulting from internal fragments to their sequence positions, the relative locations of disulfide bonds can be determined. The outermost disulfide bonds are explained more by terminal fragments, as their formation usually only require one backbone cleavage in addition to one disulfide bond cleavage. In contrast, in order for internal fragments to explain these outermost disulfide bond cleavages, simultaneous cleavages of one disulfide bond and multiple protein backbone bonds are required, raising the energy barrier compared to terminal fragments. When going deeper into the protein sequence, more internal fragments result from cleavage of innermost disulfide bonds. In these highly constrained regions, simultaneous cleavages of multiple disulfide bonds and one protein backbone bond are needed to generate terminal fragments, while the formation of internal fragments still only require one disulfide bond cleavage in addition to multiple protein backbone cleavages. These results can be rationalized by considering the relative energies required to cleave the protein backbone (∼10–15 kcal mol−1) compared to the disulfide bond (∼45–60 kcal mol−1).38,69 Because the energy barrier of cleaving a disulfide bond is higher than cleaving a protein backbone bond, the energy requirement of forming internal fragments in the interior protein sequence could be lower than for terminal fragments, and thus internal fragments could more easily result from cleavage of disulfide bonds buried within the protein. To support our data, ECD of β-lactoglobulin and lysozyme were conducted using a different mass spectrometry system (Waters Select Series Cyclic IMS Q-TOF). Similar trends for both sequence coverages and disulfide bond cleavages were observed (Fig. S7 and S8†), further demonstrating the utility of internal fragments to cover the interior protein sequence and determine the relative positions of disulfide bonds.
Internal fragments retaining intact disulfide bonds can determine disulfide connectivity
To determine the disulfide connectivity between cysteines, we focus on fragments that only result from protein backbone cleavages and retain the intact disulfide bonds. Fragments that arise from these types of cleavages can be divided into type I fragments and type II fragments (Scheme 2). Type I fragments correspond to fragments (terminal and internal) that traverse an even number of dehydrocysteine residues (e.g., 2, 4, 6) and contain mass shifts associated with the multiplication product of the number of disulfide bonds and dehydrocysteines (no. of disulfide bonds × −2 Da, Scheme 2). Type II fragments correspond to internal fragments formed between adjacent cysteine residues; thus no disulfide bonds are involved (Scheme 2). Type I fragments suggest that intact disulfide bonds are maintained within the cysteines involved, while type II fragments suggest that those two adjacent cysteines are highly unlikely to be connected.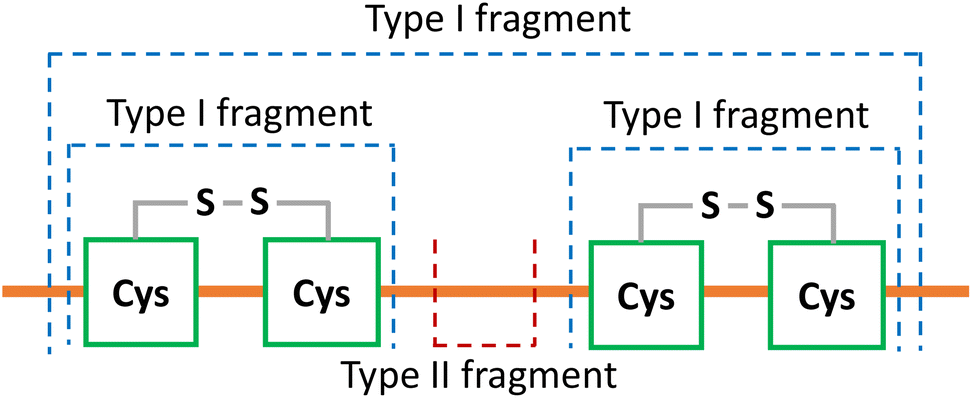 | ||
| Scheme 2 The two types of fragments retaining intact disulfide bonds to determine disulfide connectivity. A hydrogen loss (−1 Da) was applied on every cysteine residue to suggest the integrity of disulfide bonds involved. Type I fragment traverses an even number of dehydrocysteines (2, 4, 6 etc.), suggesting that intact disulfide bonds are formed within the cysteines involved. Type II fragment is generated between adjacent cysteines with no disulfide bonds involved, suggesting that those two adjacent cysteines are highly unlikely to be connected. | ||
To determine disulfide connectivity using type I and type II fragments, CAD fragmentation of trypsin inhibitor (181 residues, 20.1 kDa, 2 disulfide bonds, Fig. S9A†), [TI + 17H]17+ (Fig. 6A) was investigated, as the non-overlapping feature of the two disulfide bonds of trypsin inhibitor makes it a good test example. Type I fragments can be used to determine the disulfide connectivity of the two disulfide bonds of trypsin inhibitor. For example, the two dehydrocysteines (Cys39 and Cys86) located close to the N-terminus were traversed by 9 type I terminal fragments and 70 type I internal fragments, and the two dehydrocysteines (Cys136 and Cys145) located closer to the C-terminus were traversed by 8 type I terminal fragments and 7 type I internal fragments, which strongly suggests that the connectivity between these cysteines should be “Cys39–Cys86” and “Cys136–Cys145” for these two disulfide bonds (Fig. 6A). Four examples of type I internal fragments traversing these two disulfide bonds are shown (Fig. S9†). It should be noted that fragments traversing an even number of dehydrocysteines do not guarantee the integrity of disulfide bonds involved; however, the likelihood of them being cleaved is much lower. For example, only one internal fragment (by42–137) traversed the middle two dehydrocysteines (Cys86 and Cys136), whereas the formation of 3 type II fragments between Cys86 and Cys136 (by96–115, by100–115, by125–132) indicates that these two cysteines are not likely to be connected.
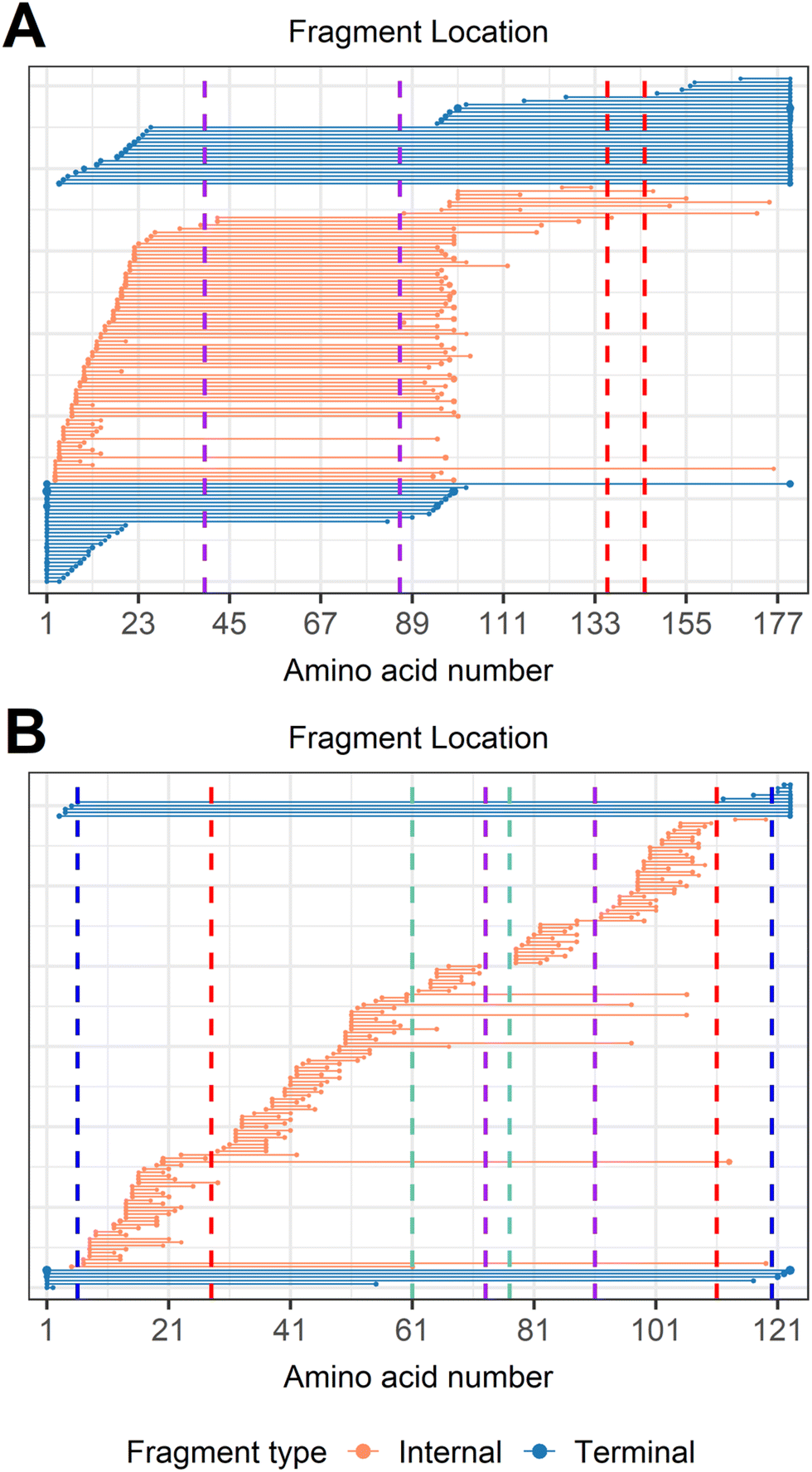 | ||
| Fig. 6 Fragment location maps after importing a hydrogen loss localized modification on every cysteine. (A) CAD of trypsin inhibitor, [TI + 17H]17+, and (B) CAD of α-lactalbumin after integrating data from all four charge states examined (11+ to 14+). Vertical dashed lines represent cysteines positions, with the same color indicating a disulfide bond is formed between those two cysteines. Internal fragments traversing an even number of dehydrocysteines (type I fragments) suggest that intact disulfide bonds are formed within those cysteines, while internal fragments formed between adjacent cysteines (type II fragments) suggest that those two cysteines are not likely to relate to each other. | ||
Similar results could also be gleaned when α-lactalbumin (123 residues, 14.2 kDa, 4 disulfide bonds, Scheme 1D), which possesses a more complicated disulfide linkage, was analyzed (Fig. 6B). Disulfide connectivity of α-lactalbumin was determined by interrogating the innermost disulfide bonds, and expanding to the outermost disulfide bonds. The middle four dehydrocysteines (Cys61, Cys73, Cys77, Cys91) were traversed by 4 type I internal fragments (by50–97, by51–106, by53–97, by60–106, Fig. S10†), indicating that two disulfide bonds are formed within these four cysteines (Fig. 6B). Type II internal fragments were then used to aid the assignment of the exact connectivity within these four cysteines. The formation of 8 type II internal fragments between Cys61 and Cys73, and 12 type II internal fragments between Cys77 and Cys91 strongly suggests that the connectivity of “Cys61–Cys73” and “Cys77–Cys91” is not likely. In addition, the lack of type I internal fragments traversing the middle two dehydrocysteines (Cys73 and Cys77) indicates that the “Cys73–Cys77” connectivity is not likely either. Should Cys73 and Cys77 be connected, type I internal fragments traversing the dehydro form of these two cysteines would have been generated, as demonstrated by CAD of trypsin inhibitor (Fig. 6A). Therefore, the only possible connectivity of these four cysteines is “Cys61–Cys77” and “Cys73–Cys91”. Expanding to the outermost cysteines, the formation of 1 type I internal fragment traversing the middle six dehydrocysteines (by20–113), and 8 type I terminal fragments traversing all eight dehydrocysteines indicates that two more disulfide bonds are formed between the four cysteines located on the exterior protein sequence. The presence of type I internal fragment by20–113 determines the connectivity of “Cys28–Cys111”, provided that the middle four cysteines are associated with each other. This is further supported by the fact that 28 type II internal fragments are formed between Cys6 and Cys28, 40 type II internal fragments are formed between Cys28 and Cys61, 28 type II internal fragments are formed between Cys91 and Cys111, and 1 type II internal fragment is formed between Cys111 and Cys120. These type II internal fragments rule out the possibility of “Cys6–Cys28”, “Cys28–Cys61”, “Cys91–Cys111”, and “Cys111–Cys120” connectivities. Therefore, the two outermost disulfide bond connectivities can be determined as “Cys28–Cys111” and “Cys6–Cys120”. It is noteworthy that only internal fragments can access the middle four cysteines, demonstrating again the value of analyzing internal fragments to obtain comprehensive disulfide bond information. The disulfide connectivity of lysozyme can be elucidated and determined in a similar way using these two types of fragments (Fig. S11†).
Conclusions
Here we report the utility of internal fragments to enhance information obtained from disulfide-intact proteins. We demonstrate that internal fragments can access the interior protein sequence constrained by multiple disulfide bonds that cannot be reached by terminal fragments, resulting in a sequence coverage increase of 20–60% to cover nearly the complete sequence of disulfide-intact proteins. We show that terminal fragments result from cleavage of disulfide bonds located on the exterior of the protein while internal fragments represent cleavage of more disulfide bonds buried within the interior of the protein. By correlating the relative number of internal fragments that result in disulfide cleavages to their sequence positions, the relative positions of disulfide bonds can be determined. Lastly, we show that internal fragments retaining intact disulfide bonds, which are traditionally overlooked, can be used to determine the disulfide connectivity. By analyzing internal fragments, it is possible to gain more sequence information and elucidate disulfide linkage patterns for proteins with unknown disulfide connectivities, which would be valuable for characterizing biotherapeutic proteins that contain many disulfide bonds.Data availability
All experimental supporting data associated with this article are available in the main manuscript and in the ESI.†Author contributions
Benqian Wei: investigation, formal analysis, writing – original draft, writing – review & editing. Muhammad A. Zenaidee: investigation, supervision, writing – review & editing. Carter Lantz: investigation, writing – review & editing. Brad J. Williams and Sarah Totten: investigation, writing – review & editing. Rachel R. Ogorzalek Loo and Joseph A. Loo: supervision, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Support from the US National Institutes of Health (R01GM103479, R35GM145286, S10RR028893), the US National Science Foundation (NSF) (CHE1808492), and the US Department of Energy (DEFC02-02ER63421) are gratefully acknowledged. C. L. acknowledges support from the Ruth L. Kirschstein National Research Service Award Program (GM007185).References
- M. Matsumura, G. Signor and B. W. Matthews, Nature, 1989, 342, 291–293 CrossRef CAS.
- W. J. Wedemeyer, E. Welker, M. Narayan and H. A. Scheraga, Biochemistry, 2000, 39, 4207–4216 CrossRef CAS PubMed.
- P. J. Hogg, Trends Biochem. Sci., 2003, 28, 210–214 CrossRef CAS.
- M. V. Trivedi, J. S. Laurence and T. J. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614–625 CrossRef CAS.
- M. O. Glocker, B. Arbogast and M. L. Deinzer, J. Am. Soc. Mass Spectrom., 1995, 6, 638–643 CrossRef CAS.
- M. J. Feige, I. Braakman and L. M. Hendershot, in Oxidative Folding of Proteins: Basic Principles, Cellular Regulation and Engineering, The Royal Society of Chemistry, 2018, pp. 1–33, 10.1039/9781788013253-00001.
- W. Zhang, L. A. Marzilli, J. C. Rouse and M. J. Czupryn, Anal. Biochem., 2002, 311, 1–9 CrossRef CAS.
- J. Wypych, M. Li, A. Guo, Z. Zhang, T. Martinez, M. J. Allen, S. Fodor, D. N. Kelner, G. C. Flynn, Y. D. Liu, P. V. Bondarenko, M. S. Ricci, T. M. Dillon and A. Balland, J. Biol. Chem., 2008, 283, 16194–16205 CrossRef CAS.
- S.-L. Wu, H. Jiang, Q. Lu, S. Dai, W. S. Hancock and B. L. Karger, Anal. Chem., 2009, 81, 112–122 CrossRef CAS.
- D. Bagal, J. F. Valliere-Douglass, A. Balland and P. D. Schnier, Anal. Chem., 2010, 82, 6751–6755 CrossRef CAS.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- J. B. Shaw, W. Liu, Y. V. Vasil’ev, C. C. Bracken, N. Malhan, A. Guthals, J. S. Beckman and V. G. Voinov, Anal. Chem., 2020, 92, 766–773 CrossRef CAS PubMed.
- E. Deslignière, T. Botzanowski, H. Diemer, D. A. Cooper-Shepherd, E. Wagner-Rousset, O. Colas, G. Béchade, K. Giles, O. Hernandez-Alba, A. Beck and S. Cianférani, J. Am. Soc. Mass Spectrom., 2021, 32, 2505–2512 CrossRef.
- L. Zhang, C. P. Chou and M. Moo-Young, Biotechnol. Adv., 2011, 29, 923–929 CrossRef CAS PubMed.
- M. Góngora-Benítez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef.
- J. J. Gorman, T. P. Wallis and J. J. Pitt, Mass Spectrom. Rev., 2002, 21, 183–216 CrossRef CAS.
- P. L. Tsai, S.-F. Chen and S. Y. Huang, Rev. Anal. Chem., 2013, 32, 257–268 CAS.
- J. Wiesner, A. Resemann, C. Evans, D. Suckau and W. Jabs, Expert Rev. Proteomics, 2015, 12, 115–123 CrossRef CAS.
- M. M. Quick, C. M. Crittenden, J. A. Rosenberg and J. S. Brodbelt, Anal. Chem., 2018, 90, 8523–8530 CrossRef CAS PubMed.
- A. Walewska, J. J. Skalicky, D. R. Davis, M.-M. Zhang, E. Lopez-Vera, M. Watkins, T. S. Han, D. Yoshikami, B. M. Olivera and G. Bulaj, J. Am. Chem. Soc., 2008, 130, 14280–14286 CrossRef CAS PubMed.
- L. Poppe, J. O. Hui, J. Ligutti, J. K. Murray and P. D. Schnier, Anal. Chem., 2012, 84, 262–266 CrossRef CAS PubMed.
- A. Shevchenko, M. Wilm, O. Vorm and M. Mann, Anal. Chem., 1996, 68, 850–858 CrossRef CAS PubMed.
- S. E. Ong and M. Mann, Nat. Chem. Biol., 2005, 1, 252–262 CrossRef CAS.
- S. F. Foley, Y. Sun, T. S. Zheng and D. Wen, Anal. Biochem., 2008, 377, 95–104 CrossRef CAS PubMed.
- Y. Zhang, H. D. Dewald and H. Chen, J. Proteome Res., 2011, 10, 1293–1304 CrossRef CAS PubMed.
- D. S. Zhao, Z. R. Gregorich and Y. Ge, Proteomics, 2013, 13, 3256–3260 CrossRef CAS PubMed.
- S. Nicolardi, M. Giera, P. Kooijman, A. Kraj, J.-P. Chervet, A. M. Deelder and Y. E. M. van der Burgt, J. Am. Soc. Mass Spectrom., 2013, 24, 1980–1987 CrossRef CAS PubMed.
- C. N. Cramer, K. F. Haselmann, J. V. Olsen and P. K. Nielsen, Anal. Chem., 2016, 88, 1585–1592 CrossRef CAS PubMed.
- C. N. Cramer, C. D. Kelstrup, J. V. Olsen, K. F. Haselmann and P. K. Nielsen, Anal. Chem., 2017, 89, 5949–5957 CrossRef CAS.
- P. Wongkongkathep, H. Li, X. Zhang, R. R. Ogorzalek Loo, R. R. Julian and J. A. Loo, Int. J. Mass Spectrom., 2015, 390, 137–145 CrossRef CAS PubMed.
- J. Zhang, R. R. Ogorzalek Loo and J. A. Loo, Int. J. Mass Spectrom., 2015, 377, 546–556 CrossRef CAS.
- N. Siuti and N. L. Kelleher, Nat. Methods, 2007, 4, 817–821 CrossRef CAS.
- Y. Zhang, W. Cui, H. Zhang, H. D. Dewald and H. Chen, Anal. Chem., 2012, 84, 3838–3842 CrossRef CAS PubMed.
- A. D. Catherman, O. S. Skinner and N. L. Kelleher, Biochem. Biophys. Res. Commun., 2014, 445, 683–693 CrossRef CAS PubMed.
- F. Lermyte, Y. O. Tsybin, P. B. O'Connor and J. A. Loo, J. Am. Soc. Mass Spectrom., 2019, 30, 1149–1157 CrossRef CAS PubMed.
- T. K. Toby, L. Fornelli and N. L. Kelleher, Annu. Rev. Anal. Chem., 2016, 9, 499–519 CrossRef CAS PubMed.
- A. J. Kleinnijenhuis, M. C. Duursma, E. Breukink, R. M. A. Heeren and A. J. R. Heck, Anal. Chem., 2003, 75, 3219–3225 CrossRef CAS.
- H. Lioe and R. A. J. OrsHair, J. Am. Soc. Mass Spectrom., 2007, 18, 1109–1123 CrossRef CAS PubMed.
- J. Chen, P. Shiyanov, L. Zhang, J. J. Schlager and K. B. Green-Church, Anal. Chem., 2010, 82, 6079–6089 CrossRef CAS PubMed.
- S. K. Gammelgaard, S. B. Petersen, K. F. Haselmann and P. K. Nielsen, J. Am. Soc. Mass Spectrom., 2021, 32, 1910–1918 CrossRef CAS PubMed.
- N. A. K. Roman, A. Zubarev, E. K. Fridriksson, M. A. Lewis, D. M. Horn, B. K. Carpenter and F. W. McLafferty, J. Am. Chem. Soc., 1999, 121, 2857–2862 CrossRef.
- B. Ganisl and K. Breuker, ChemistryOpen, 2012, 1, 260–268 CrossRef CAS PubMed.
- S. R. Cole, X. Ma, X. Zhang and Y. Xia, J. Am. Soc. Mass Spectrom., 2012, 23, 310–320 CrossRef CAS.
- D. Wen, Y. Xiao, M. M. Vecchi, B. J. Gong, J. Dolnikova and R. B. Pepinsky, Anal. Chem., 2017, 89, 4021–4030 CrossRef CAS PubMed.
- Y. M. E. Fung, F. Kjeldsen, O. A. Silivra, T. W. D. Chan and R. A. Zubarev, Angew. Chem., Int. Ed., 2005, 44, 6399–6403 CrossRef CAS PubMed.
- A. Agarwal, J. K. Diedrich and R. R. Julian, Anal. Chem., 2011, 83, 6455–6458 CrossRef CAS.
- J. Bonner, L. E. Talbert, N. Akkawi and R. R. Julian, Analyst, 2018, 143, 5176–5184 RSC.
- L. E. Talbert and R. R. Julian, J. Am. Soc. Mass Spectrom., 2018, 29, 1760–1767 CrossRef CAS PubMed.
- S. K. Gammelgaard, S. B. Petersen, K. F. Haselmann and P. K. Nielsen, ACS Omega, 2020, 5, 7962–7968 CrossRef CAS PubMed.
- M. J. P. Rush, N. M. Riley, M. S. Westphall and J. J. Coon, Anal. Chem., 2018, 90, 8946–8953 CrossRef CAS.
- K. D. Ballard and S. J. Gaskell, Int. J. Mass Spectrom. Ion Processes, 1991, 111, 173–189 CrossRef CAS.
- V. H. Wysocki, K. A. Resing, Q. Zhang and G. Cheng, Methods, 2005, 35, 211–222 CrossRef CAS PubMed.
- C. Lantz, M. A. Zenaidee, B. Wei, Z. Hemminger, R. R. Ogorzalek Loo and J. A. Loo, J. Proteome Res., 2021, 20, 1928–1935 CrossRef CAS PubMed.
- P. E. Barran, N. C. Polfer, D. J. Campopiano, D. J. Clarke, P. R. R. Langridge-Smith, R. J. Langley, J. R. W. Govan, A. Maxwell, J. R. Dorin, R. P. Millar and M. T. Bowers, Int. J. Mass Spectrom., 2005, 240, 273–284 CrossRef CAS.
- A. Michalski, N. Neuhauser, J. Cox and M. Mann, J. Proteome Res., 2012, 11, 5479–5491 CrossRef CAS.
- Y. A. Lyon, D. Riggs, L. Fornelli, P. D. Compton and R. R. Julian, J. Am. Soc. Mass Spectrom., 2018, 29, 150–157 CrossRef CAS PubMed.
- B. Wei, M. A. Zenaidee, C. Lantz, R. R. Ogorzalek Loo and J. A. Loo, Anal. Chim. Acta, 2022, 1194, 339400 CrossRef CAS PubMed.
- J. S. Cobb, M. L. Easterling and J. N. Agar, J. Am. Soc. Mass Spectrom., 2010, 21, 949–959 CrossRef CAS PubMed.
- K. R. Durbin, O. S. Skinner, R. T. Fellers and N. L. Kelleher, J. Am. Soc. Mass Spectrom., 2015, 26, 782–787 CrossRef CAS PubMed.
- J. Chen, P. Shiyanov and K. B. Green, J. Mass Spectrom., 2019, 54, 527–539 CrossRef CAS.
- F. Griaud, B. Denefeld, C.-Y. Kao-Scharf, J. Dayer, M. Lang, J.-Y. Chen and M. Berg, Anal. Chem., 2019, 91, 8845–8852 CrossRef CAS PubMed.
- M. A. Zenaidee, C. Lantz, T. Perkins, W. Jung, R. R. O. Loo and J. A. Loo, J. Am. Soc. Mass Spectrom., 2020, 31, 1896–1902 CrossRef CAS PubMed.
- N. D. Schmitt, J. M. Berger, J. B. Conway and J. N. Agar, Anal. Chem., 2021, 93, 6355–6362 CrossRef CAS PubMed.
- M. A. Zenaidee, B. Wei, C. Lantz, H. T. Wu, T. R. Lambeth, J. K. Diedrich, R. R. Ogorzalek Loo, R. R. Julian and J. A. Loo, J. Am. Soc. Mass Spectrom., 2021, 32, 1752–1758 CrossRef CAS PubMed.
- H. Li, Y. Sheng, W. Mcgee, M. Cammarata, D. Holden and J. A. Loo, Anal. Chem., 2017, 89, 2731–2738 CrossRef CAS PubMed.
- H. Li, H. H. Nguyen, R. R. Ogorzalek Loo, I. D. G. Campuzano and J. A. Loo, Nat. Chem., 2018, 10, 139–148 CrossRef CAS PubMed.
- Z. Rolfs and L. M. Smith, J. Proteome Res., 2021, 20, 5412–5418 CrossRef CAS PubMed.
- S. Chin, T. Chen, R. N. Hannoush and C. M. Crittenden, J. Pharm. Biomed. Anal., 2021, 195, 113893 CrossRef CAS PubMed.
- B. Paizs and S. Suhai, J. Am. Soc. Mass Spectrom., 2004, 15, 103–113 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2an01517j |
| This journal is © The Royal Society of Chemistry 2023 |