
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomistic simulations on the carbidisation processes in Pd nanoparticles†
Apostolos Kordatos a,
Khaled Mohammeda,
Reza Vakilib,
Alexandre Goguetb,
Haresh Manyarb,
Emma Gibsonc,
Marina Carravettaa,
Peter Wellsa and
Chris - Kriton Skylaris*a
a,
Khaled Mohammeda,
Reza Vakilib,
Alexandre Goguetb,
Haresh Manyarb,
Emma Gibsonc,
Marina Carravettaa,
Peter Wellsa and
Chris - Kriton Skylaris*a
aSchool of Chemistry, University of Southampton, SO17 1BJ, UK. E-mail: C.Skylaris@soton.ac.uk
bSchool of Chemistry and Chemical Engineering Queen's University Belfast, BT7 1NN, UK
cSchool of Chemistry, University of Glasgow, G12 8QQ, UK
First published on 14th February 2023
Abstract
The formation of interstitial PdCx nanoparticles (NPs) is investigated through DFT calculations. Insights on the mechanisms of carbidisation are obtained whilst the material's behaviour under conditions of increasing C-concentration is examined. Incorporation of C atoms in the Pd octahedral interstitial sites is occurring through the [111] facet with an activation energy barrier of 19.3–35.7 kJ mol−1 whilst migration through the [100] facet corresponds to higher activation energy barriers of 124.5–127.4 kJ mol−1. Furthermore, interstitial-type diffusion shows that C will preferentially migrate and reside at the octahedral interstitial sites in the subsurface region with limited mobility towards the core of the NP. For low C-concentrations, migration from the surface into the interstitial sites of the NPs is thermodynamically favored, resulting in the formation of interstitial carbide. Carbidisation reaction energies are exothermic up to 11–14% of C-concentration and slightly vary depending on the shape of the structure. The reaction mechanisms turn to endothermic for higher concentration levels showing that C will preferentially reside on the surface making the interstitial carbide formation unfavorable. As experimentally observed, our simulations confirm that there is a maximum concentration of C in Pd carbide NPs opening the way for further computational investigations on the activity of Pd carbides in directed catalysis.
1. Introduction
Within heterogeneous catalysis, supported Pd nanoparticles (NPs) have been extensively investigated1–7 for their high activity towards many existing and emerging industrial applications;8,9 from the selective hydrogenation and oxidation of unsaturated hydrocarbons, to the conversion of biomass,10,11 and exhaust emission control technologies. One key example is the semi-hydrogenation of acetylene, which is a crucial purification step for ethylene feedstocks used in polymerization to produce polyethylene (∼50 megatons per year). Elsewhere, as the chemical industry pivots towards more sustainable feedstocks, Pd has proved very effective for the selective upgrading of waste biomass. In these example processes, the Pd structures are significantly altered by the catalytic conditions, forming interstitial carbide structures.Whether Pd forms interstitial structures of hydride, carbide, or nitride during catalysis, these changes are not innocent as they have a fundamental contribution to the catalytic process. For instance, in acetylene semi-hydrogenation, the formation of hydride phases (α-PdHx and β-PdHx)12–14 is considered a rate-limiting factor as full hydrogenation to ethane occurs through reversed hydrogen diffusion on the Pd surface. Carbidic Pd forms via incorporation of C atoms into the Pd lattice, suppressing the formation of hydride phases and thus increasing selectivity towards production of ethylene. Similarly, for the hydrogenation of furfural, PdCx affects the adsorption of species whilst reducing catalytic turnover. Understanding the formation and stability of carbidic Pd structures is crucial to determine their function in catalysis. Detailed information on the material's controlled synthesis (interactions on the surface and subsurface regions, interstitial C distribution) as well as insights on its behaviour in reaction conditions (deactivation) are still required. Since the adsorption type and microkinetics of hydrocarbons on these structures may vary, atomistic simulations through Density Functional Theory (DFT) and microkinetic modelling can provide advanced insights and complement with experimental methods such as X-ray absorption fine structure (XAFS) on the mechanistic investigation of carbidisation processes and reaction pathways of catalytic mechanisms.
In the study by Usoltsev et al.,15 the adsorption of ethylene (C2H4) on the Pd surface is followed by dehydrogenation to C2H3, C2H2 and C2H leading to the formation of the carbide phase. Experimental results also show the connection of the carbide formation with the Pd lattice expansion5,16 whilst Liu et al.17 highlights the improved catalytic behaviour of PdCx through the formation of more reactive sites. The influence of the support in the surface/subsurface chemistry has been also recently highlighted by Rötzer et al.18 showing different turnover frequencies for two amorphous SiO2 films. The catalytic activity between pristine Pd clusters and surface models have been also compared through experimental and theoretical methods on Pd30/Al2O3.3 Notably, for the carbidic Pd, the exact amount of C incorporation into the Pd lattice is still debated. Reported values of C-concentration on the PdCx do vary from 0.13 to 0.18.19,20 Several models for the C dissolution have been proposed in theoretical studies from ordered to completely random distribution in the Pd lattice. Seriani et al.21 have reported the formation of a metastable carbide that corresponds to 13–14% of C concentration in which hydrogen diffusion is hindered as compared to pristine Pd. This Pd6C metastable carbide formation has been further investigated through experimental22 and theoretical methods.23 In the recent study of He et al., C diffusion into the Pd(100) and Pd(111) surface models show zigzag trajectories for the migration pathways and a non-uniform distribution in the slab. Furthermore, exothermic behaviour for C migration is observed through the Pd(111) facet and endothermic for Pd(100) whilst the stability decreases with the insertion depth. Schuster et al. propose that the formation kinetics depend on the particle size whilst shape also influences hydrogen assisted reactions.24 The selectivity of the catalyst has also been reported to be affected by the particle size.25,26 Torres et al.27 have also examined the impact of different experimental conditions, showing that the dissolution of C is affected by the application of tensile stress on the surface. Additionally, the adsorption and conversion of hydrocarbons are affected by the surface morphology28 (flat vs. stepped), whilst similarly the expansion of Pd lattice on NPs close to the surface should be also considered since the formation of subsurface Pd carbides is expected to be hindered by the neighboring C–C repulsion. C incorporation in the Pd subsurface affects the adsorption type of species making the catalyst selective towards specific intermediates and products.29 It is therefore evident that the actual role of the carbide in selective hydrogenation depends on the formation process and distribution of C atoms into the pristine Pd. To this point, the aforementioned works have mostly focused on Pd bulk or surface/slab crystallographic arrangements, however, the formation mechanism in Pd NPs has not been reported.
In this study, we perform DFT calculations to investigate the PdCx formation on Pd nanoparticles of different crystallographic arrangements. Firstly, C surface residing and migration into the subsurface Pd octahedral interstitial sites is examined whilst the distribution of the dopant in the Pd host lattice is also considered through the interstitial-type diffusion via neighboring positions. Additional monitoring on the PdCx formation at increasing C-concentration is also considered to estimate the maximum amount for each structure. The aim of this work is to better understand the formation of Pd carbides and their role in catalysis.
2. Experimental
The pristine Pd and PdCx NPs structures are modelled through the linear – scaling DFT code ONETEP.30 The density matrix is constructed by using localized nonorthogonal Wannier functions (NGWFs) as expressed through a set of periodic sinc (p-sinc) functions.31 NGWFs are equivalent to plane waves for more conventional DFT codes and for our calculations the p-sinc basis set was set to a kinetic energy cut-off of 800 eV whilst for the exchange and correlation interactions, the formulation with the corrected density functional of Perdew, Burke and Ernzerhof (PBE)32 within the generalized gradient approximation (GGA) is applied with norm – conserving pseudopotentials. The density matrix and NGWFs are optimized via two separate loops. Since this work focuses on a metallic system, the Ensemble DFT (EDFT) method33 is employed with a Fermi-Dirac smearing of 0.1 eV under constant pressure conditions allowing the cells to relax in the minimum energy configuration. An NGWF radius of 9.0 Bohr has been used for all structures, whilst geometry optimization is performed at the Γ-point in cells of 18–22 Å.For the construction of NPs crystallographic arrangements, the ASE34 tool has been used with the average Pd–Pd bond for all optimized structures corresponding to an average of 2.74 Å. After determining the relative energies of reactants and products, the relaxed configurations of Pd NPs are then used as input for the transition states investigation along a minimum energy reaction path. For the estimation of the activation energy barriers, reaction energies and location of the transition state the Linear Synchronous Transit/Quadratic Synchronous Transit (LSTQST)35 protocol is employed. Initially, a rapid scan along the reaction/transition path is performed followed by a located minimization to find the most stable intermediate. The schematic representation of the Pd and PdCx cells is generated using the CrystalMaker36 software whilst for the reaction energies between different crystallographic configurations the following formula is used:
| Ereaction = Eproducts − Ereactants |
3. Results and discussion
3.1 Crystallographic arrangements
Initially, we considered the NPs crystallographic arrangements to explore the carbidisation process on different facets. For our calculations, the PdCx NPs are modelled using truncated octahedral Pd38, octahedral Pd44 and cuboctahedral Pd55 (Fig. 1a–c). The geometries have been considered as appropriate to represent a range of nanoparticle structures from both experimental and computational approaches. Additionally, we aim to account for the effect of different facets on the carbidisation mechanisms through their coexistence on the same particle whilst, a comparison of outcomes for a range of different structures was required to validate results. | ||
| Fig. 1 Schematic representation for (a) Pd38 truncated octahedron (b) Pd44 octahedron (c) Pd55 cuboctahedron (d) C residing on the [111] facet of Pd38 (e) C residing on the [111] facet of Pd44 (f) C residing on the [111] facet of Pd55 (g) C bonding on the [111] facet of Pd38 (h) C bonding on the [111] facet of Pd44 (i) C bonding on the [111] facet of Pd55 (j) C bonding on the [100] facet of Pd38 (k) C bonding on the [100] facet of Pd55 and (l) C in the Pd octahedral site. Pd atoms for grey spheres and C atoms for brown spheres. | ||
All NPs have been modelled as isolated structures in conditions of vacuum to avoid interactions with their periodic images. Initially, we assessed how C resides on the surface of Pd NPs. Therefore, we investigated the preferential configurations of the surface Pd–C interactions as an initial process from C exposure, prior to the interstitial carbide formation. Fig. 1d–f represents the preferential configurations for C on Pd38, Pd44 and Pd55 on the [111] facet. For all structures, an individual C atom will bind with three surface Pd atoms (hollow site) with the average C–Pd bond length being 1.85 Å, 1.87 Å and 1.86 Å for Pd38, Pd44 and Pd55, respectively (Fig. 1g–i). In addition, C binds with four surface Pd atoms on the [100] facet as shown in Fig. 1j and k whilst the average C–Pd bond length is 1.98 Å and 1.99 Å for Pd38 and Pd55, respectively. For the interstitial formation, each C atom is in the middle of the Pd octahedral site which is the minimum energy configuration,37 binding with six Pd atoms (Fig. 1l). The structural changes of the Pd NPs surface due to binding with an individual C are summarized in Table 1.
| [111] | Pd–Pd (Å) – pristine | Pd–Pd (Å) – surface C | ||||
|---|---|---|---|---|---|---|
| Bond | AB | BC | CA | AB | BC | CA |
| Pd38 | 2.72 | 2.72 | 2.74 | 2.87 | 2.95 | 2.92 |
| Pd44 | 2.77 | 2.69 | 2.69 | 2.96 | 2.91 | 2.91 |
| Pd55 | 2.75 | 2.75 | 2.75 | 3.02 | 3.01 | 3.02 |
| [100] | Pd–Pd (Å) – pristine | Pd–Pd (Å) – surface C | ||||||
|---|---|---|---|---|---|---|---|---|
| Bond | AB | BC | CD | DA | AB | BC | CD | DA |
| Pd38 | 2.75 | 2.75 | 2.75 | 2.75 | 2.79 | 2.79 | 2.79 | 2.79 |
| Pd55 | 2.75 | 2.75 | 2.75 | 2.75 | 2.79 | 2.79 | 2.82 | 2.82 |
3.2 Carbidisation processes
After establishing the initial and final positions of C, we investigated the process of migration from the surface into the interior of the particle via transition state search calculations. In previous work, Crespo-Quesada et al.38 showed on cubic and octahedral NPs, that the carbidisation rate on Pd [111]/octahedral NPs was lower as compared with Pd [100]/cubes and attributed this to the stronger binding of acetylene with the [100] 4-fold sites. In their study, which did not include transition state searches, DFT calculations were performed on Pd36Cn (n = 1, 2 or 6). This provided useful insights, however it is limited in terms of different shapes and sizes of NP. In this work, we look at the actual mechanism of carbidisation in NPs of different shapes and sizes, alongside how the extent of C incorporation (∼2–20%) affects the carbidisation process.Our initial expectation was that C resides initially on the surface of both [100] and [111] facets for Pd38/Pd55. We found that C will preferentially migrate into the Pd NP interstitial sites through the [111] facet. Fig. 2 present the surface and interstitial diffusion mechanisms for Pd38, Pd44 and Pd55, respectively as assessed through the transition states investigation. Firstly, for all Pd NPs, the reaction energy for the C migration through the [111] facet is exothermic. The activation energy barriers for C incorporation through the [111] facet correspond to 35.7 kJ mol−1, 19.3 kJ mol−1 and 21.2 kJ mol−1 for Pd38, Pd44 and Pd55, respectively.
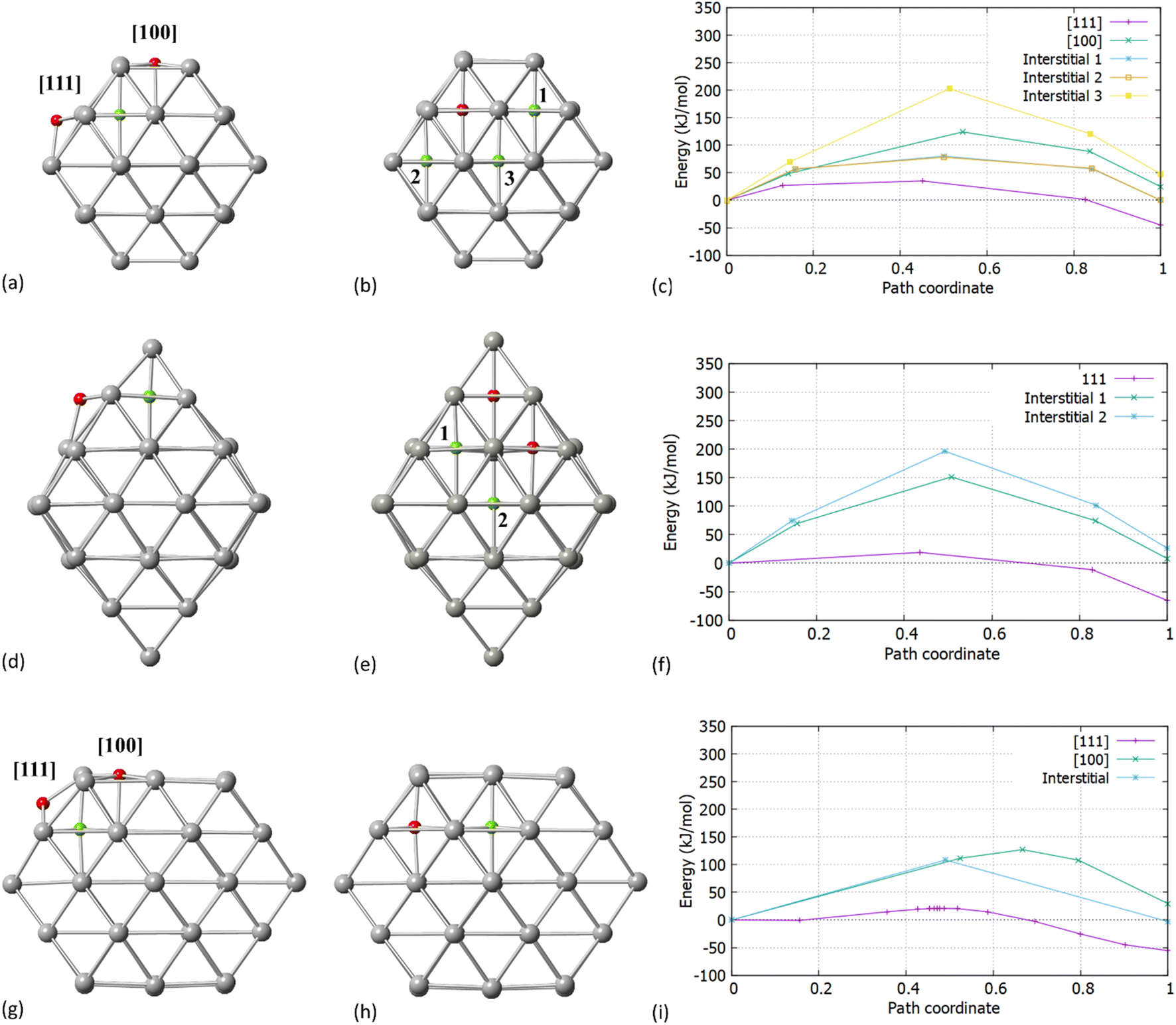 | ||
| Fig. 2 Activation energy barriers for the carbidisation mechanisms in Pd NPs (a) transition states for C migration in Pd38 through the [100] and [111] facets (b) interstitial-type diffusion of C for neighboring sites in Pd38 (c) activation energies for surface and interstitial-type mobilities in Pd38 (d) transition states for C migration in Pd44 through the [100] and [111] facets (e) interstitial-type diffusion of C for neighboring sites in Pd44 (f) activation energies for surface and interstitial-type mobilities in Pd44 (g) transition states for C migration in Pd55 through the [100] and [111] facets (h) interstitial-type diffusion of C for neighboring sites in Pd55 and (i) activation energies for surface and interstitial-type mobilities in Pd55. Red spheres correspond to initial positions of C and green spheres to final positions of C. | ||
When we considered the cuboctahedral Pd55 and truncated octahedral Pd38 NPs, the insertion reaction from the [100] facet into the particle is endothermic and requires a higher activation energy barrier of 124.5 kJ mol−1 and 127.4 kJ mol−1 for Pd38 and Pd55, respectively, making C insertion less energetically favourable. We expect that even if the [100] facet strongly binds with hydrocarbons, it will limit the reaction process and have a minor contribution to the total carbidisation rate. In contrast, the exothermic behavior for [111] as represented in Fig. 2c and i shows that C atoms will migrate into the NP through the [111] facet and reside in the octahedral Pd interstitial sites. Additionally, the calculated reaction energies for the mechanism are −45.05 kJ mol−1, −65.45 kJ mol−1 and −55.42 kJ mol−1 for Pd38, Pd44 and Pd55, respectively. This demonstrates that the process is slightly more favoured for the octahedral Pd44. Interestingly, considering the shape difference for Pd38, Pd44 and Pd55 there is a relatively small activation energy barrier for carbidisation in all cases for the [111] facet. The results on the activation energy barriers for C migration are in good agreement (36.6–40.5 kJ mol−1) with previously reported values on slab models.23 The energies of reaction and relevant barriers considering C incorporation from the surface into the NP for all structures are summarized in Table 2.
| [111] | Eact (kJ mol−1) | Ereaction (kJ mol−1) |
|---|---|---|
| Pd38 | 35.7 | −45.05 |
| Pd44 | 19.3 | −65.45 |
| Pd55 | 21.2 | −55.42 |
| [100] | Eact (kJ mol−1) | Ereaction (kJ mol−1) |
|---|---|---|
| Pd38 | 124.5 | 24.68 |
| Pd55 | 127.4 | 29.54 |
Whilst the first part of our calculations focused on the C migration from the surface to interstitial site, we then considered the C mobility into the NP via examining interstitial-type diffusion mechanisms. For Pd38, a corresponding energy barrier of 77.2 kJ mol−1 was found when migrating through neighboring sites at the subsurface region. Additionally, a considerable increase in the activation energy is observed when migrating towards the core of the particle (193.5 kJ mol−1) as shown in Fig. 2c. After C insertion into a subsurface interstitial site, C will preferentially move through nearest positions close to the surface rather than further diffuse into the interior of the Pd lattice. This is an important finding when considering the maximum amount of C that can be incorporated into a given NP; higher C loadings require full occupation of the octahedral interstitial sites, however, there is a strong preference for C atoms to reside in the subsurface area. For octahedral Pd44, as shown in Fig. 2e and f a higher activation energy barrier of 151.5 kJ mol−1 and 196.8 kJ mol−1 is required for C migration towards the subsurface and the core, respectively. With these higher barriers it is expected that C will reside on the initially occupied positions with limited mobility into neighboring sites. The C subsurface diffusion mechanisms, as well as the C–C repulsion forces have been considered in this study as design criteria on pre-formed carbidic structures to investigate the carbidisation mechanisms at increasing C concentrations. A criterion we take into account when we design carbidised nanoparticles is to allow at least an unoccupied interstitial site between neighboring carbon atoms. It is also observed that at higher concentrations, interstitial subsurface C binds with less than 6 Pd atoms, residing towards the surface of the NP.
3.3 Carbide formation at higher C-concentration
To further investigate the experimentally confirmed limitations of C concentration in Pd NPs, we examined the activation energy barriers and reaction energies of carbidisation in C-doped Pd structures. As shown in Fig. 3, we considered the relevant transition mechanisms, as demonstrated through the first part of our calculations, from the pristine Pd to pre-formed interstitial carbidic NPs for different C-concentrations per atom fraction (e.g., 1 C atom in Pd38 NP corresponds to 2.6% concentration for the PdC system). We observed an increase of the average Pd–Pd bond length with respect to C concentration for all structures as summarized in Table 3. The values obtained through DFT are in agreement with previously published experimental results.2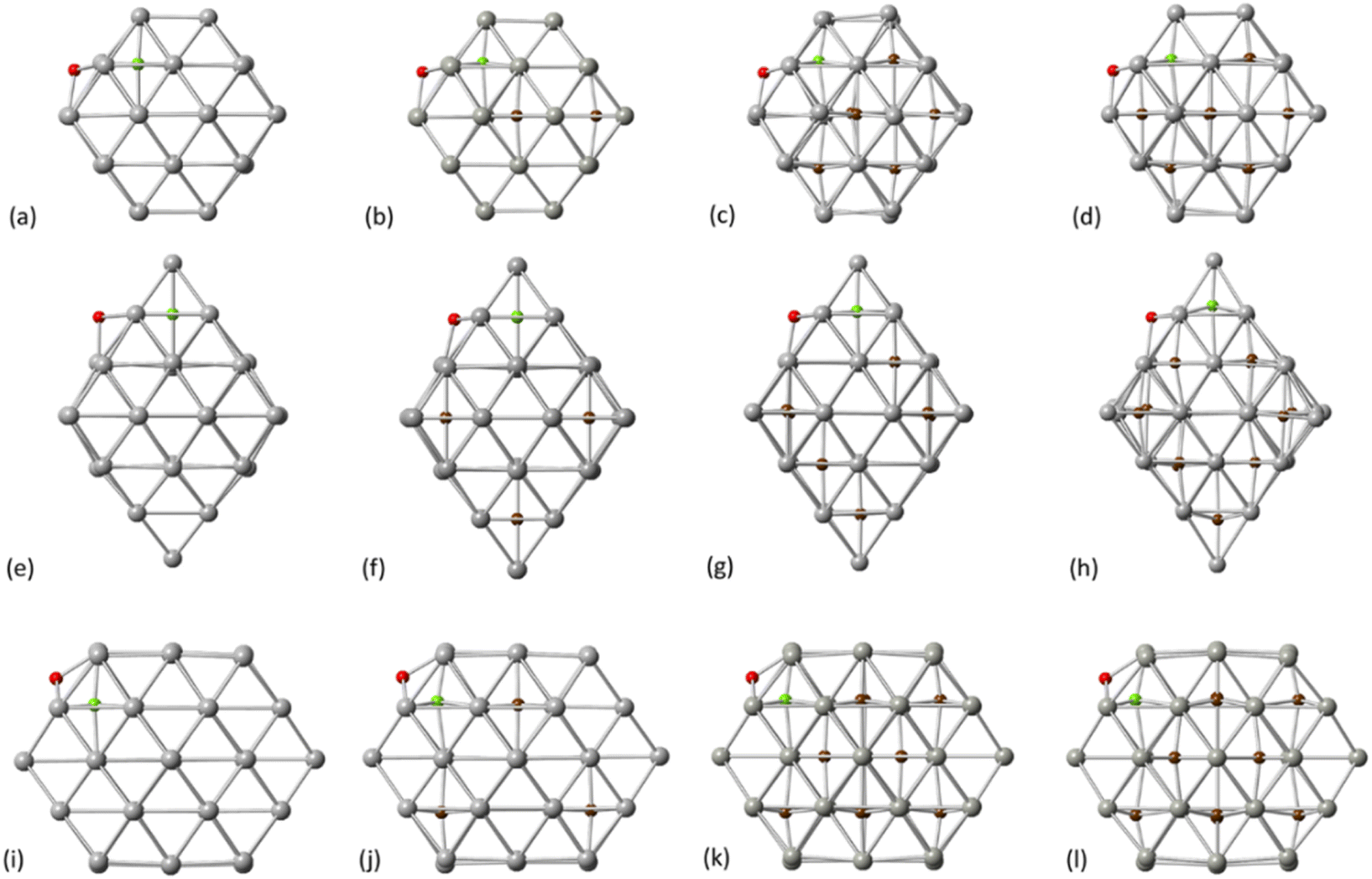 | ||
| Fig. 3 Transition states for the carbidisation mechanisms at increasing C concentration for Pd38 – (a) 2.6% (b) 9.5% (c) 15.6% and (d) 20.8%, Pd44 – (e) 2.2% (f) 8.3% (g) 15.4% and (h) 21.4%, Pd55 – (i) 1.8% (j) 8.3% (k) 15.4% and (l) 20.3%. Red spheres correspond to initial positions of C and green spheres to final positions of C. | ||
| Pd38 | Pd44 | Pd55 | |||
|---|---|---|---|---|---|
| C (%) | Pd–Pd (Å) | C (%) | Pd–Pd (Å) | C (%) | Pd–Pd (Å) |
| 0.0 | 2.74 | 0.0 | 2.72 | 0.0 | 2.73 |
| 2.6 | 2.75 | 2.2 | 2.74 | 1.8 | 2.74 |
| 9.5 | 2.79 | 8.3 | 2.79 | 8.3 | 2.79 |
| 15.6 | 2.83 | 15.4 | 2.84 | 15.4 | 2.85 |
| 20.8 | 2.88 | 21.4 | 2.87 | 20.3 | 2.87 |
The increasing accommodation of C in the Pd lattice was examined via the reaction energies between the relevant surface and subsurface configurations. Furthermore, the Pd6C formation at approximately 13% of C doping is considered with each Pd atom to be bound with no more than one C atom.21 Thermodynamically, it is expected that above a maximum carbidisation threshold, C will reside on the surface rather than migrating into the subsurface region. As shown in Fig. 4, the energy profiles for C incorporation at low concentrations correspond to exothermic reactions. In this case, the final state (products) is more thermodynamically favourable than the initial (reactants), leading to the formation of interstitial Pd carbide. As more C is introduced in the host Pd lattice, the reaction becomes less exothermic and eventually turns to endothermic above a specific concentration for each structure. The maximum C amount that is approximately 14% for Pd38 and 11% for Pd44/Pd55, shows that where the reaction energy becomes positive, further C incorporation is unfavourable and thus C will reside on the surface rather than migrate into the particle.
 | ||
| Fig. 4 Transition states for the carbidisation processes at increasing C concentrations in (a) Pd38 – the left panel shows the activation energies for C concentration at 2.6%, 9.5%, 15.6% and 20.8% and the right panel shows the reaction energies (b) Pd44 – the left panel shows the activation energies for C concentration at 2.2%, 8.3%, 15.4% and 21.4% and the right panel shows the reaction energies and (c) Pd55 – the left panel shows the activation energies for C concentration at 1.8%, 8.3%, 15.4% and 20.3% and the right panel shows the reaction energies. | ||
4. Conclusions
In this work, we performed DFT calculations to gain insights on the intrinsic mechanisms of carbidisation in Pd NPs; a process yet not fully understood but important to the catalytic performance of the material in a range of applications. The geometries chosen to represent the Pd NPs structures are the truncated octahedral Pd38, octahedral Pd44 and cuboctahedral Pd55, aiming to account the carbidisation process for both the [100] and [111] facets of the NP structure.Initially, an individual C atom binds with three surface Pd atoms for the [111] facet and four surface Pd atoms for the [100] facet. In addition, the minimum energy interstitial sites are octahedral where C binds with six Pd atoms in the interior of the particle. Upon insertion of C, minor lattice distortion occurs and the transition results to the occupation of the Pd octahedral interstitial site in the subsurface area, thus our simulations firstly focused on one single C incorporation in the pristine lattice. It is shown that for Pd38 and Pd55, carbidisation preferentially occurs through the [111] facet where the mechanism corresponds to an exothermic reaction with low activation energy values of 21.2–35.7 kJ mol−1. In contrast, for the same mechanism through the [100] facet, an endothermic profile is observed on the reaction with activation energy barriers of more than 120 kJ mol−1. Furthermore, the activation energy barriers for interstitial-type diffusion of C through neighbouring sites in the interior of the particle, show preferential residing to the subsurface region (exothermic) rather than closer to the core (endothermic). It is therefore shown that C will migrate from the surface into the octahedral interstitial sites up to relatively limited concentration since it will mainly reside in the subsurface area. Carbidisation at higher concentrations was also investigated using pre-formed carbidic Pd structures to estimate the maximum doping limitations. The distribution of C atoms in the available octahedral sites for low concentrations is within the subsurface area with at least one unoccupied interstitial site between them. Reaction energies for the same mechanism are exothermic up to a 11–14% for all structures whilst beyond this point, further incorporation is energetically unfavourable confirming the limitations of C concentration in Pd NPs.
This study is a first step towards understanding the carbide phase formation that takes place during catalytic reactions with Pd NPs. Future work through experimental and computational studies should further investigate the formation and the role of Pd carbide NPs in heterogeneous catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AK, KM and RV are grateful to EPSRC (grant number EP/V000691/1) for postdoctoral funding. We are grateful to The Materials and Molecular Modelling MMM Hub (EPSRC grant number EP/T022213/1) for access to the Young supercomputer, the UKCP consortium (EPSRC grant number EP/P022030/1) for access to the ARCHER2 supercomputer and the University of Southampton for access to the Iridis5 supercomputer.Notes and references
- M. Armbrüster, M. Behrens, F. Cinquini, K. Föttinger, Y. Grin, A. Haghofer, B. Klötzer, A. Knop-Gericke, H. Lorenz, A. Ota, S. Penner, J. Prinz, C. Rameshan, Z. Révay, D. Rosenthal, G. Rupprechter, P. Sautet, R. Schlögl, L. Shao, L. Szentmiklósi, D. Teschner, D. Torres, R. Wagner, R. Widmer and G. Wowsnick, How to Control the Selectivity of Palladium-based Catalysts in Hydrogenation Reactions: The Role of Subsurface Chemistry, ChemCatChem, 2021, 4, 1048 CrossRef.
- W. Jones, P. P. Wells, E. K. Gibson, A. Chutia, I. P. Silverwood, C. R. A. Catlow and M. Bowker, Carbidisation of Pd Nanoparticles by Ethene Decomposition with Methane Production, ChemCatChem, 2019, 11, 4334 CrossRef CAS.
- L. P. L. Gonçalves, J. Wang, S. Vinati, E. Barborini, X.-K. Wei, M. Heggen, M. Franco, J. P. S. Sousa, D. Y. Petrovykh, O. S. G. P. Soares, K. Kovnir, J. Akola and Y. V. Kolen'ko, Combined experimental and theoretical study of acetylene semi-hydrogenation over Pd/Al2O3, Int. J. Hydrogen Energy, 2020, 45, 1283 CrossRef.
- A. L. Bugaev, O. A. Usoltsev, A. A. Guda, K. A. Lomachenko, I. A. Pankin, Y. V. Rusalev, H. Emerich, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, Palladium Carbide and Hydride Formation in the Bulk and at the Surface of Palladium Nanoparticles, J. Phys. Chem. C, 2018, 122, 12029 CrossRef CAS.
- J. A. McCaulley, Temperature dependence of the Pd K-edge extended X-ray absorption fine structure of PdCx, Phys. Rev. B, 1993, 47, 4873 CrossRef CAS PubMed.
- H. A. Aleksandrov, L. V. Moskaleva and Z.-J. Zhao, Duygu Basaran, Zhao-Xu Chen, Donghai Mei, Notker Rösch, Ethylene conversion to ethylidyne on Pd(111) and Pt(111): A first-principles-based kinetic Monte Carlo study, J. Catal., 2012, 285, 187 CrossRef CAS.
- C. Hamill, H. Driss, A. Goguet, R. Burch, L. Petrov, M. Daous and D. Rooney, Mild temperature palladium-catalyzed ammoxidation of ethanol to acetonitrile, Appl. Catal., 2015, 506, 261 CrossRef CAS.
- A. Borodziński and G. C. Bond, Selective Hydrogenation of Ethyne in Ethene-Rich Streams on Palladium Catalysts, Part 2: Steady-State Kinetics and Effects of Palladium Particle Size, Carbon Monoxide, and Promoters, Catal. Rev.: Sci. Eng., 2008, 50, 379 CrossRef.
- R. Pellegrini, G. Agostini, E. Groppo, A. Piovano, G. Leofanti and C. Lamberti, 0.5wt.% Pd/C catalyst for purification of terephthalic acid: Irreversible deactivation in industrial plants, J. Catal., 2011, 280, 150 CrossRef CAS.
- S. M. Rogers, C. R. A. Catlow, C. E. Chan-Thaw, A. Chutia, N. Jian, R. E. Palmer, M. Perdjon, A. Thetford, N. Dimitratos, A. Villa and P. P. Wells, Tandem Site- and Size-Controlled Pd Nanoparticles for the Directed Hydrogenation of Furfural, ACS Catal., 2017, 7, 2266 CrossRef CAS.
- G. F. Tierney, S. Alijani, M. Panchal, D. Decarolis, M. Briceno de Gutierrez, K. M. H. Mohammed, J. Callison, E. K. Gibson, P. B. J. Thompson, P. Collier, N. Dimitratos, E. Crina Corbos, F. Pelletier, A. Villa and P. P. Wells, Controlling the Production of Acid Catalyzed Products of Furfural Hydrogenation by Pd/TiO2, ChemCatChem, 2021, 13, 5121 CrossRef CAS.
- N. K. Nag, A Study on the Formation of Palladium Hydride in a Carbon-Supported Palladium Catalyst, J. Phys. Chem. B, 2001, 105, 5945 CrossRef CAS.
- Y. Liu, Y. He, D. Zhou, J. Feng and D. Li, Catalytic performance of Pd-promoted Cu hydrotalcite-derived catalysts in partial hydrogenation of acetylene: effect of Pd–Cu alloy formation, Catal. Sci. Technol., 2016, 6, 3027 RSC.
- Y. Zhou, W. Baaziz, O. Ersen, P. A. Kots, E. I. Vovk, X. Zhou, Y. Yang and V. V. Ordomsky, Decomposition of Supported Pd Hydride Nanoparticles for the Synthesis of Highly Dispersed Metallic Catalyst, Chem. Mater., 2018, 30, 8116 CrossRef CAS.
- O. A. Usoltsev, A. Y. Pnevskaya, E. G. Kamyshova, A. A. Tereshchenko, A. A. Skorynina, W. Zhang, T. Yao, A. L. Bugaev and A. V. Soldatov, Dehydrogenation of Ethylene on Supported Palladium Nanoparticles: A Double View from Metal and Hydrocarbon Sides, Nanomaterials, 2020, 10, 1643 CrossRef CAS PubMed.
- D. Teschner, E. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, H. Sauer, A. Knop-Gericke, R. Schlögl, M. Chamam, A. Wootsch, A. S. Canning, J. J. Gamman, S. D. Jackson, J. McGregor and L. F. Gladden, Alkyne hydrogenation over Pd catalysts: A new paradigm, J. Catal., 2006, 242, 26 CrossRef CAS.
- Y. Liu, F. Fu, A. McCue, W. Jones, D. Rao, J. Feng, Y. He and D. Li, Adsorbate-Induced Structural Evolution of Pd Catalyst for Selective Hydrogenation of Acetylene, ACS Catal., 2020, 10, 15048 CrossRef CAS.
- M. D. Rötzer, M. Krause, M. Huber, F. F. Schweinberger, A. S. Crampton and U. Heiz, Ethylene hydrogenation on supported Pd nanoparticles: Influence of support on catalyst activity and deactivation, J. Catal., 2021, 397, 90 CrossRef.
- S. B. Ziemecki, G. A. Jones, D. G. Swartzfager, R. L. Harlow and J. Faber Jr., Formation of interstitial Pd-C Phase by interaction of ethylene, acetylene and Carbon monoxide with Palladium, J. Am. Chem. Soc., 1985, 107, 4547 CrossRef CAS.
- R. Guo, Q. Chen, X. Li, Y. Liu, C. Wang, W. Bi, C. Zhao, Y. Guo and M. Jin, PdCx nanocrystals with tunable compositions for alkyne semihydrogenation, J. Mater. Chem. A, 2019, 7, 4714 RSC.
- N. Seriani, F. Mittendorfer and G. Kresse, Carbon in palladium catalysts: A metastable carbide, J. Chem. Phys., 2010, 132, 024711 CrossRef PubMed.
- R. Schuster, M. Bertram, H. Runge, S. Geile, S. Chung, V. Vonk, H. Noei, A. Poulain and Y. Lykhach, Andreas Stierle and Jörg Libuda, Metastability of palladium carbide nanoparticles during hydrogen release from liquid organic hydrogen carriers, Phys. Chem. Chem. Phys., 2021, 23, 1371 RSC.
- Y. He and C. Wu, Equilibrium distribution of dissolved carbon in PdCx: Density functional theory and canonical Monte Carlo simulations, J. Phys. Chem. C, 2021, 125(38), 20930 CrossRef CAS.
- P. Mäki-Arvela and D. Yu, Murzin Effect of metal particle shape on hydrogen assisted reactions, Appl. Catal., 2021, 618, 118140 CrossRef.
- F. Jiang, J. Cai, B. Liu, Y. Xu and X. Liu, Particle size effects in the selective hydrogenation of cinnamaldehyde over supported palladium catalysts, RSC Adv., 2016, 6, 75541 RSC.
- Y. F. Han, D. Kumar, C. Sivadinarayana, A. Clearfield and D. W. Goodman, The Formation of PdCx over Pd-Based Catalysts in Vapor-Phase Vinyl Acetate Synthesis: Does a Pd–Au Alloy Catalyst Resist Carbide Formation?, Catal. Lett., 2004, 94, 131 CrossRef CAS.
- D. Torres, F. Cinquini and P. Sautet, Pressure and Temperature Effects on the Formation of a Pd/C Surface Carbide: Insights into the Role of Pd/C as a Selective Catalytic State for the Partial Hydrogenation of Acetylene, J. Phys. Chem. C, 2013, 117(21), 11059 CrossRef CAS.
- J. Andersin, N. Lopez and K. Honkala, DFT Study on the complex reactions networks in the conversion of ethylene to ethylidyne on flat and Stepped Pd, J. Phys. Chem. C, 2009, 113(19), 8278 CrossRef CAS.
- A. Garcia-Ortiz, J. D. Vidal, S. Iborra, M. J. Climent, J. Cored, D. Ruano, V. Pérez-Dieste, P. Concepción and A. Corma, Synthesis of a hybrid Pd0/Pd-carbide/carbon catalyst material with high selectivity for hydrogenation reactions, J. Catal., 2020, 389, 706 CrossRef CAS.
- J. C. A. Prentice, J. Aarons, J. C. Womack, A. E. A. Allen, L. Andrinopoulos, L. Anton, R. A. Bell, A. Bhandari, G. A. Bramley, R. J. Charlton, R. J. Clements, D. J. Cole, G. Constantinescu, F. Corsetti, S. M.-M. Dubois, K. K. B. Duff, J. M. Escartín, A. Greco, Q. Hill, L. P. Lee, E. Linscott, D. D. O'Regan, M. J. S. Phipps, L. E. Ratcliff, Á. Ruiz Serrano, E. W. Tait, G. Teobaldi, V. Vitale, N. Yeung, T. J. Zuehlsdorff, J. Dziedzic, P. D. Haynes, N. D. M. Hine, A. A. Mostofi, M. C. Payne and C.-K. Skylaris, The ONETEP linear-scaling density functional theory program, J. Chem. Phys., 2020, 152, 174111 CrossRef CAS PubMed.
- C.-K. Skylaris, A. A. Mostofi, P. D. Haynes, O. Diéguez and M. C. Payne, Nonorthogonal generalized Wannier function pseudopotential plane-wave method, Phys. Rev. B, 2002, 66, 035119 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- Á. Ruiz-Serrano and C.-K. Skylaris, A variational method for density functional theory calculations on metallic systems with thousands of atoms, J. Chem. Phys., 2013, 139, 054107 CrossRef PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The Atomic Simulation Environment-A Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, A generalized synchronous transit method for transition state location, Comput. Mater. Sci., 2003, 28, 250 CrossRef CAS.
- CrystalMaker®: a crystal and molecular structures program for Mac and Windows, 9.0.3, CrystalMaker Software Ltd, Oxford, England, 2014, https://www.crystalmaker.com Search PubMed.
- M. Maciejewski and A. Baker, Incorporation and reactivity of carbon in palladium, Pure Appl. Chem., 1995, 67, 1879 CrossRef CAS.
- M. Crespo-Quesada, S. Yoon, M. Jin, A. Prestianni, R. Cortese, F. Cárdenas-Lizana, D. Duca, A. Weidenkaff and L. Kiwi-Minsker, Shape-Dependence of Pd Nanocrystal Carburization during Acetylene Hydrogenation, J. Phys. Chem. C, 2015, 119, 1101 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07462a |
| This journal is © The Royal Society of Chemistry 2023 |