
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico design of a lipid-like compound targeting KRAS4B-G12D through non-covalent bonds†
Huixia
Lu
 *ab,
Zheyao
Hu
b,
Jordi
Faraudo
a and
Jordi
Martí
*b
*ab,
Zheyao
Hu
b,
Jordi
Faraudo
a and
Jordi
Martí
*b
aInstitut de Ciencia de Materials de Barcelona (ICMAB-CSIC), Campus de la UAB, Bellaterra, Barcelona E-08193, Spain. E-mail: huixialu@icmab.es
bDepartment of Physics, Technical University of Catalonia-Barcelona Tech, B5-209 Northern Campus, Jordi Girona 1-3, 08034 Barcelona, Catalonia, Spain. E-mail: jordi.marti@upc.edu
First published on 21st November 2023
Abstract
One of the most common drivers in human cancer is the peripheral membrane protein KRAS4B, able to promote oncogenic signalling. To signal, oncogenic KRAS4B not only requires a sufficient nucleotide exchange, but also needs to recruit effectors by exposing its effector-binding sites while anchoring to the phospholipid bilayer where KRAS4B-mediated signalling events occur. The enzyme phosphodiesterase-δ plays an important role in sequestering KRAS4B from the cytoplasm and targeting it to cellular membranes of different cell species. In this work, we present an in silico design of a lipid-like compound that has the remarkable feature of being able to target both an oncogenic KRAS4B-G12D mutant and the phosphodiesterase-δ enzyme. This double action is accomplished by adding a lipid tail (analogous to the farnesyl group of the KRAS4B protein) to an previously known active compound (2H-1,2,4-benzothiadiazine, 3,4-dihydro-,1,1-dioxide). The proposed lipid-like molecule was found to lock KRAS4B-G12D in its GDP-bound state by adjusting the effector-binding domain to be blocked by the interface of the lipid bilayer. Meanwhile, it can tune GTP-bound KRAS4B-G12D to shift from the active orientation state to the inactive state. The proposed compound is also observed to stably accommodate itself in the prenyl-binding pocket of phosphodiesterase-δ, which impairs KRAS4B enrichment at the lipid bilayer, potentially reducing the proliferation of KRAS4B inside the cytoplasm and its anchoring at the bilayer. In conclusion, we report a potential inhibitor of KRAS4B-G12D with a lipid tail attached to a specific warhead, a compound which has not yet been considered for drugs targeting RAS mutants. Our work provides new ways to target KRAS4B-G12D and can also foster drug discovery efforts for the targeting of oncogenes of the RAS family and beyond.
1 Introduction
RAS proteins are peripheral membrane proteins that play a crucial role in the regulation of different signalling pathways in cells by cycling between their active GTP-bound state and their inactive GDP-bound state.1 RAS signalling pathways are of great interest in cancer therapy.2 Interestingly, a recent statistical analysis of cancer genetic databases revealed that ∼19% of all cancer cases contain RAS mutations.3 The major RAS isoforms are encoded by three genes, resulting in a total of four RAS proteins (KRAS4A, KRAS4B, HRAS, and NRAS). The catalytic domain (CD, res1–166) of KRAS4B is composed of the effector-binding domain (res1–86) and the allosteric domain (res87–166). Four regions border the nucleotide-binding pocket: the phosphate-binding loop (res10–17), switch I (SI, res30–38), switch II (SII, res60–76), and the base binding loops (res116–120 and res145–147).4KRAS4B and KRAS4A are distinguished by their C-terminal hypervariable region (HVR, res167–185). It has been observed that KRAS4B is about five fold more expressed in cell than KRAS4A,5 hence in the present work we have restricted ourselves to the simulation on KRAS4B. HVR preferentially binds the membrane in the liquid phase and spontaneously inserts its terminal farnesyl moiety (FAR) into the loosely packed phospholipid/cholesterol bilayers of the main structure of the plasma membrane (PM).6 The oncogenic mutants of KRAS4B are of particular interest due to its relatively high frequency in cancers, in particular in 17% of lung, 61% of pancreas, 20% of small intestine, 33% of large intestine, and biliary tract cancer cells.6–9 An analysis of mutation patterns across the RAS isoforms reveals that 66% of KRAS mutations occur at codon 12.8 However, up to date only two drugs (sotorasib, adagrasib) have been approved by the U.S. Food and Drug Administration for the treatment of cancers related to the KRAS-G12C mutation10,11 whereas, in order to inhibit the elusive KRAS-12D mutation, two compounds have been proposed (TH-Z835 and MRTX1133).12,13
Motivated by the high frequency of this mutation, we have performed in our lab extensive computational studies of the KRAS4B-G12D mutant.14–17 Our molecular dynamics (MD) simulations revealed useful information such as the main pathways between lipid bilayer anchored and released states of the protein and the preferential orientations of the protein at the bilayer. Importantly, simulations indicate that GTP-binding to KRAS4B has huge influence on the stabilisation of the protein and it can potentially help to open druggable pockets.14
It should be noted that the high frequency of this mutation makes it an ideal drug target. However, attempts to target mutant RAS proteins have proven challenging.18,19 One possible reason can be the lack of information of active structures of RAS proteins while the published crystal structures are in general obtained under experimental conditions. Independent groups have been working on exploring the conformational space of RAS proteins with the hope of filling this gap.14,20–24 Therefore, a natural continuation of these previous simulation studies is to employ the results described above to design compounds of potential interest to target KRAS4B. The question we will consider in this paper is the possibility of in silico design of a new molecule able to (i) lock KRAS4B in its GDP-bound inactive state and (ii) to act over the active GTP-bound KRAS4B stabilizing it into an orientation state (relative to the bilayer) that makes KRAS4B inactive. The technique that we will employ to consider this question will be microsecond scale MD simulations, which is well suited for in silico drug design.25 MD allows us to take into account important factors such as the effect of protein flexibility and the role of protein orientation with respect to the lipid bilayer which is a key factor as indicated by our previous studies.14–17 These factors cannot be included with other techniques such as docking calculations that consider only static structures.
The compound considered in this work (designed as LIG1) is shown in Fig. 1, together with the chemical structures of FAR and a small molecule, 3,4-dihydro-1,2,4-benzothiadiazine-1,1-dioxide (C7H8N2O2S, DBD), including a typical snapshot of sys2 are illustrated here. There we can see that LIG1 is a chimeric molecule made by the addition of a lipid tail to DBD. The rationale behind the design of this chimeric compound is based on our MD simulations. DBD is a bicyclic heterocyclic benzene derivative featuring amphiphilic properties and providing an opportunity as pharmacophore/warhead during the process of drug design. It can produce a wide variety of derivatives26,27 an it has been long used in the human therapy as diuretic,28,29 antiviral,30–33 anti-inflammatory34 or anticancer35,36 agent, among others, due to its strong pharmacological activity.37 Prior research on DBD derivatives binding to RAS proteins also provides the possibility of playing a role as inhibitors of RAS proteins.17 Hence, we have decided to adopt DBD as the initial template to be used in the design of the new potential inhibitor reported here.
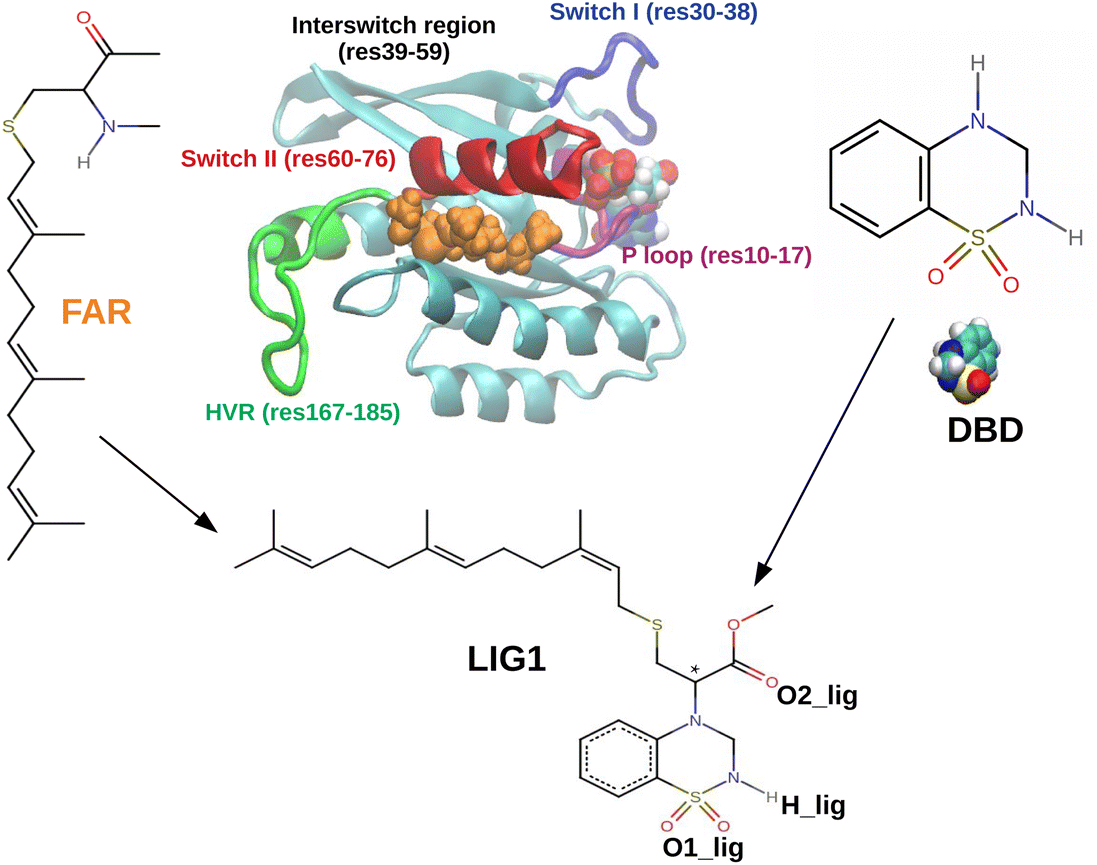 | ||
| Fig. 1 Scheme illustrating the concept behind the design of the proposed compound LIG1, showing the schemes of the relevant compounds considered in our study and an snapshot of the KRAS4B protein obtained in MD simulations in water in absence of a lipid bilayer. Chiral carbon of LIG1 indicated by a star here. The snapshot emphasizes the binding of the protein terminal farnesyl moiety (FAR, shown in orange) with the switch II region (in red) of the catalytic domain of the protein. This binding spontaneously appears in absence of a phospholipid bilayer. The chemical structures shown here correspond to the farnesyl moiety, the DBD molecule and the proposed chimeric compound made by adding a FAR tail to DBD. | ||
Neverthless our MD simulations will show that DBD alone is not able to establish stable interactions with KRAS4B (see the section “Results”). However, this molecule can be easily modified to fit into a binding pocket by adding an appropriately designed hydrophobic tail. The key for this design comes from another observation from MD simulations. KRAS4B is a peripheral membrane protein that binds to the lipid bilayer due to the insertion of its terminal farnesyl moiety located in the C-terminal hypervariable region of the protein. Interestingly, in the specific case in which the lipid bilayer is not present, our MD simulations show that this terminal farnesyl moiety is able to bind spontaneously to the switch II region of the catalytic domain of KRAS4B, as shown in Fig. 1 (see further details in the section “Results”). This serendipitous observation suggests our design for LIG1. We propose LIG1 build by the addition of a FAR tail to DBD (see Fig. 1). The objective of the MD simulations reported in this paper is precisely to investigate the interactions of LIG1 with KRAS4B.
By applying MD simulations in the microsecond time scale, we will gain insight into the role of LIG1 and DBD small-molecules might play for KRAS4B-G12D in its GTP-bound and GDP-bound states. Exploring whether LIG1 can non-covalently help to inhibit KRAS4B-G12D might provide wider application to different RAS-driven cancers. In addition, we will show that it is essential to consider KRAS4B at PM to properly account for its functioning.38
Our MD studies presented here suggest another possible interaction of interest of LIG1. As shown in Fig. 2, the solubilization factor PDEδ has been reported to play a role in sequestering KRAS4B from the cytosol by binding the FAR tail and, thereby, enhancing its diffusion throughout the cell and finally releasing KRAS4B to bind with the PM.39,40 PDEδ inhibition was proved to decrease the RAS-mediated signalling. Our MD simulations also show that LIG1 stably accommodates in the prenyl-binding pocket of PDEδ, therefore LIG1 may be able to impair KRAS4B enrichment at the bilayer. The two possible interactions of LIG1, as identified by our MD simulations are shown schematically in Fig. 2.
 | ||
| Fig. 2 LIG1 may block KRAS4B signalling pathways through two means, resulting KRAS4B in its GDP-bound inactive states and an impaired enrichment of KRAS4B at the inner leaflet of PM. Figure created with BioRender.com (2023). | ||
2 Results and discussion
In this work we have performed MD simulations at the microsecond scale (in total 22.31 μs) in different situations to investigate the role of the phospholipid bilayer, LIG1, and DBD play in the dynamics behaviour of KRAS4B-G12D. Details of designed MD simulations are summarized in Table 1. We considered simulations of KRAS4B with different ligands (DBD and LIG1) and in different conditions (bound to a lipid bilayer and in NaCl solution), and one KRAS4B-G12D anchored to PM without considering any ligand (sys0) was taken as the control group.| Systems | Protein | Ligand | State | Environment | No. of atoms | Simulation time |
|---|---|---|---|---|---|---|
| sys0 | KRAS4B | — | GTP | PM | 112![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 618 618 |
2000 ns |
| sys1 | KRAS4B | DBD | GTP | PM | 99429 |
1500 ns |
| sys1-rep | KRAS4B | DBD | GTP | PM | 99429 |
2500 ns |
| sys2 | KRAS4B | DBD | GTP | Solution | 98560 |
1770 ns |
| sys2-rep | KRAS4B | DBD | GTP | Solution | 98560 |
1040 ns |
| sys3 | KRAS4B | LIG1 | GTP | PM | 110691 |
4000 ns |
| sys4 | KRAS4B | LIG1 | GDP | PM | 110767 |
3000 ns |
| sys5 | PDEδ | LIG1 | — | Solution | 72976 |
2000 ns |
| sys6 | — | LIG1 | — | PM | 48808 |
500 ns |
We have considered sys6 to explore the dynamics and its binding free-energy of LIG1 with PM. We have also run additional simulations of 4 μs to understand the effect of isomerization of LIG1 on KRAS4B-G12D, details shown in the ESI.† More information can be found in section “Methods”.
2.1 Structural characteristics of the oncogenic KRAS4B
We start by discussing the results of sys1 and sys2 in Table 1 that correspond to the interaction of Kras4B with DBD. From Fig. S4A and C,† we can see that DBD is seldom bound with CD of KRAS4B, knowing the radius of KRAS4B backbone is around 17 Å: in two replica trajectories run in this work. In other words, DBD is unable to establish stable interactions with KRAS4B, which makes DBD alone impossible to act as a KRAS4B-G12D inhibitor. In Fig. 3 we show the structural results for KRAS4B with considering DBD or LIG1 (sys1 to sys4 in Table 1). High flexibility of SI and SII both in solution and at the lipid bilayer environments was also observed in all systems here. | ||
| Fig. 3 LIG1 reduces the flexibility of switch regions for both states of KRAS4B: averaged root mean-square fluctuation (RMSF) profiles (A and B) and root mean-square difference (RMSD) of the catalytic domain of KRAS4B along the simulation time (C). In all cases only Cα atoms of KRAS4B were considered. | ||
For sys2, SI and SII regions both show significantly higher flexibility than other systems where PM is considered (sys1, sys3 and sys4, see Fig. 3A). According to structural data41 the switch regions do not stay permanently in the stabilized conformations. Instead, an increasing body of evidence suggests that switch regions display highly flexible dynamics.42–45 We observed that switch regions of all systems exhibit a much flexible property than other moieties of CD, which is in agreement with the experimental atomic displacement parameter of switch regions, also known as B-factor that indicates the atomic fluctuations in the crystal structures,46,47 indicating excellent simulation convergence. Through a compound (LIG1) binding with KRAS4B, flexibility of SII is largely reduced, moreover, the RMSF value of SI of sys4 is higher than that of sys3, which agrees with other published works.14,16,47,48 Using the crystal structure as a reference, we can see that the RMSD trajectories of KRAS4B stay around 3 Å for sys1, sys2 and sys4, higher than that of sys3 (around 1.8 Å), indicating that LIG1 binding with GTP-bound KRAS4B-G12D helps it stabilize its structure and reduces its flexibility more than its GDP-bound state, see Fig. 3C. LIG1 shows higher influence on the activity of KRAS4B and stronger interactions between LIG1 and SII for GTP-bound than that of the GDP-bound KRAS4B-G12D. And the results here can be verified by a smaller distance of center of mass between LIG1 and SII for the GTP-bound KRAS (around 5 Å) than that for the GDP-bound case (around 8 Å) in Fig. S4D of ESI.† It shows that LIG1 presents certain variant effect on different guanosine-bound KRAS4B-G12D mutant.
We continued to explore the corresponding atomic mechanisms. The local structure of how LIG1 establishes in the SII pocket in sys3 and sys4 can be analyzed by means of normalized radial distribution functions, see Fig. 4 and 5.
 | ||
| Fig. 4 A–C and E–G: Selected averaged radial distribution functions g(r) of LIG1 with actives sites of CD of GTP-bound KRAS4B-G12D (sys3). Sites of protein residues refer to active sites of their side chains, except that “O_G75” represents its oxygen atom of the peptide bond of the backbone. Contributions of oxygen/hydrogen atoms that share the negative/positive charge of side chains have been averaged. And “O1_lig”, “O2_lig”, and “H_lig” stand for active sites of LIG1 indicated in Fig. 1. D: Snapshots of typical LIG1-CD bonds. H: Snapshots of side chain of Y64 switches between residues H95 and Q99 by forming established hydrogen-bond with active nitrogen of H59 (E) and active oxygen atom of Q99 (F) at different frames along the simulation time. Here LIG1 is shown in van der Waals representation for the sake of clarity. Binding sites have been highlighted in solid purple lines. Panels D and H made with Chimera.49 | ||
 | ||
| Fig. 5 A–C and E–G: Selected averaged radial distribution functions g(r) of LIG1 with actives sites of CD of GDP-bound KRAS4B-G12D (sys4). D and H: Snapshots of typical LIG1-CD bonds. Binding sites have been highlighted in solid purple lines. D and H made with Chimera.49 | ||
As can be seen from Fig. 4 and 5, LIG1 is able to accommodate itself into SII pockets by means of hydrogen-bonds (HBs) between its DBD motif with residues G75 (Fig. 4B and 5E) and K104 (Fig. 4A and 5B) in both systems. The signature of HB in lipids and protein systems is a high peak around 1.6–2.1 Å of variable intensity.50,51 Strong and long-lasting hydrogen-bond between “H_lig” and “O_G75” is observed in both cases, which helps LIG1 stably interact with SII region. Especially for KRAS4B-G12D in its GTP-bound state, side chain of Y64 has been found to form stable hydrogen bonding with Q99 and H95, respectively, see Fig. 4F–H. At GDP-bound state, LIG1 was found to be able to interact with residues K167 and H166 of KRAS4B through hydrogen bonds (see Fig. 5A, C, and D). Side chains of residues R68 of SII and Q99 of α-helix-3 can rotate to form a closed SII pocket, contributing the hydrophobic cavity shown in Fig. 5D. These result in a much reduced flexibility in the structure of SII with a smaller RMSF for the GTP-bound KRAS4B than its GDP-bound state in the case of LIG1 binding into SII pocket, see Fig. 3B.
2.2 Role of PM in the activity of KRAS4B
To observe the effects of DBD and LIG1 on protein dynamics, we calculated the cross-correlation of the atomic fluctuations obtained from their MD trajectories. The transmission of the allosteric signal within the protein shall couple motion between active site residues.52From Fig. 6, a similar pattern of positive and negative correlations in motion were observed for systems of KRAS4B anchoring into PM. DBD is considered in sys1, positive correlated motion is still observed in its effector-binding domain, which is marked in blue box in Fig. 6A, even the mutated KRAS4B presents in the accumulated inactive orientation states. In Fig. 6C and D, we can observe that LIG1 directly binding with KRAS4B effectively diminished correlations in the effector-binding motif, whereas the increased negative correlated motion between the helix3 (res87–104) and the effector-binding domain is observed for GDP-bound protein. However, when solvated in the aqueous environment, it is obvious to notice that both negative and positive correlations in motion have been lost, which agrees with Jang's work,53 indicating a decreased cross-talk in the structure of KRAS4B. This may indicate that PM is crucial for KRAS4B's dynamics in order to transmit signal across its structure. We have also taken a close investigation on the cross-correlation of switch regions, see Fig. S6 of ESI,† in which we can observe that strong correlations in motion in the interswitch regions are conserved in all systems in PM environment. Effector-binding regions which are important for effector recruitment have lost correlations in motion for KRAS4B in solution environment. Only for KRAS4B at the bilayer with proper orientation positive or negative correlated motions can be observed. Our results are in good agreement with the published work.53 Together, KRAS4B anchoring to PM is crucial to fulfil its physiological function. Therefore LIG1 binding into SII pocket of mutated KRAS4B inhibit its proper biological function by interfering the corresponding cross-talk throughout its structure.
 | ||
| Fig. 6 A–D: Comparison of correlation analysis of the motion during the final 1000 ns MD simulation of different systems studied. Highly (anti)correlated motion are orange or red (blue). Correlations within effector-binding domain are marked in blue boxes. | ||
2.3 LIG1 targeting KRAS4B-G12D in both GDP/GTP-bound states
It has been shown that in addition to FAR insertion into the PM, the CD of KRAS4B adopts three rapidly interconverting orientation states through interactions with the anionic PM via (1) helix structures α-helix 3–5 on the allosteric domain while the effector-binding domain is solvent accessible (active state, OS1), (2) β-strands 1–3 on the effector-binding domain while effector-binding domain is occluded by the PM (inactive state, OS2), or (3) the intermediate orientation state (OS0).15,54,55 Proper orientation states of KRAS4B on lipid bilayer directly affect its signalling activity. Here we have adopted the well-defined parameter Θ14,15,55 to describe the orientation of KRAS4B at the PM.We have only investigated the distribution of the orientations of KRAS4B-G12D on the ionic PM under different conditions for three systems (sys0, sys3, and sys4), in order to study the influence of LIG1 on such orientation states. The angle Θ is defined as the angle between the bilayer normal Z-direction and a vector running the Cα atoms of the residue K5 and the residue V9 which belong to the first strand β1 in the structure of KRAS4B, see Fig. 7A. Fig. 7B shows the contact area between CD with the interface of bilayer, indicating the interactions between them. Without considering any ligand, the peak of the probability profile of the corresponding contact area locates in the higher region (sys0), followed by GTP-bound KRAS4B-G12D (sys3) and GDP-bound mutant (sys4), indicating a decreasing in the interactions for the major orientation states (active OS1 for sys0 and sys3, inactive OS2 for sys4) for KRAS4B-G12D on the PM of systems considered here. In general, KRAS4B tends to establishes more interactions with the PM in order to bind stably with the PM while exposing its effector-binding domain towards cytoplasm to stay in its active orientation state.
 | ||
| Fig. 7 A: Definition of the orientation angle Θ. Representations are the same as in Fig. 1, except that the direction between residues Cα K5 and V9 is shown in orange arrow. B: Distributions of contact area between CD and the PM. C: Distributions of the orientations of KRAS4B on the phospholipid bilayer in the cases of LIG1 and DBD molecules. D: Typical orientation states for KRAS4B-G12D on phospholipid bilayer observed along the trajectories. | ||
In Fig. 7C, without considering any ligand in the system (sys0), we can learn that KRAS4B-G12D mainly locates on the PM while exposing its effector-binding domain, that is in its active OS1 orientation state, while KRAS4B-G12D staying in its inactive orientation state was also found to be possible, and it agrees with a published work indicating that the GTP-bound KRAS4B has been reported to exist mostly in its active state.53 However, LIG1 binding into SII helps the GTP-bound KRAS4B-G12D to be populated averagely in its three states (active, intermediate, and inactive states), showing much less activity for sys3, comparing to that of sys0. It is clearly revealed here that LIG1 binding with KRAS4B of sys3 is able to trigger a population shift toward the inactive state hindering the effector-binding site and switching the catalytic domain orientation of KRAS4B. Meanwhile, LIG1 is shown to be able to lock GDP-bound KRAS4B-G12D to majorly exhibit the inactive orientation at the PM while effectively hiding the effector-binding interfaces, inhibiting the guanine nucleotide exchange, resulting in a mostly GDP-bound state such that the following signalling pathways are inhibited, under circumstance of sys3 and sys4 sharing the same initial orientation and conformation in the structure.
2.4 Interactions of LIG1 with PDEδ
Signal transduction of KRAS4B is strongly linked to its enrichment at the PM.56 LIG1 shares the same FAR motif as full-length KRAS4B, so we have investigated whether LIG1 can act as the PDEδ inhibitor to impair KRAS4B enrichment at PM.In Fig. 8, the averaged value of RMSD profile is of around 1.9 Å, a low value together with the corresponding RMSF profile, showing a steady structure of PDEδ due to the anti-parallel beta strands in its structure. Fig. 8D shows clearly that LIG1 accommodates itself steadily in the cavity formed by the hydrophobic side chains of related residues. Meanwhile, LIG1 can also establish hydrogen bonds with side chains T149 and A112 of PDEδ through its active sites of the inimo and sulfonyl groups of the DBD moiety, indicating high binding affinity between LIG1 and PDEδ protein. Taken together, LIG1 might inhibit the pathways related to KRAS4B in both ways. To better understand this point, we will report calculations of binding affinities in the next section.
 | ||
| Fig. 8 A, B and C: RMSD trajectories, RMSF plot and sequence of PDEδ, D: detailed hydrophobic interactions of LIG1 (tan sticks) and PDEδ, E: superimposed structure of PDEδ of the crystal structure (blue) and last (red) frame of production runs for 2000 ns simulation time showing no dramatic changes in the structure of PDEδ, F: hydrogen bonding between LIG1 and T149 and A112 of PDEδ. H atoms of T149, A112, and LIG1 not shown for the sake of clarity. | ||
2.5 Binding affinities of LIG1 with KRAS4B-G12D and PDEδ
Here we investigated the binding affinity between LIG1 to KRAS4B-G12D in its different guanosine-bound states. In Table 2, the binding free-energies of LIG1 to the phospholipid bilayer and proteins calculations were achieved employing the adaptive biasing force (ABF) method57 in its NAMD58 formulation and implementation.59 More details are reported in “Methods”.| System | Protein | Ligand | Orientation state | ΔG (kcal mol−1) |
|---|---|---|---|---|
| sys3 | GTP | LIG1 | Active | 80.6 |
| sys3 | GTP | LIG1 | Inactive | 69.4 |
| sys4 | GDP | LIG1 | Inactive | 113.5 |
| sys5 | PDEδ | LIG1 | — | 88.2 |
| sys6 | — | LIG1 | — | 63.6 |
For GTP-bound KRAS4B in its inactive orientation state, it takes LIG1 69.4 kcal mol−1 to depart from the SII, which is easier than that in the active state (80.6 kcal mol−1). For the inactive state of KRAS4B of sys4, the binding free-energy required for LIG1 to depart from KRAS4B-G12D is revealed to be 113.5 kcal mol−1, which is much higher than that of KRAS in GTP-bound state of sys3. This is due to the steric hindrance effect of the biological bilayer, blocking the exit of LIG1. For sys4, in the majorly accumulated orientation state for KRAS4B (cosΘ ∼0.7), LIG1 is well-wrapped in the closed pocket composed by SII and the PM, which makes it almost impossible for LIG1 to be off-target from KRAS4B-G12D mutant. We have also investigated the energy required for amphiphilic LIG1 from the state of deeply locating in the PM to the state of being solvated in the aqueous region is of 63.6 kcal mol−1 (sys6). However, it is much more difficult for LIG1 to escape from the cavity of PDEδ to be released into the aqueous region (88.2 kcal mol−1) than that of amphiphilic LIG1 departure from the state of deeply locating in the PM to the state of being solvated in the aqueous region (63.6 kcal mol−1). The observation here agrees with that LIG1 helps lock KRAS4B-G12D in its GDP-bound inactive orientation state. We have also run additional simulations to explore whether the isomer of LIG1 has difference influence on the dynamics of KRAS4B-G12D on the PM. From Table 2 of ESI† we can observe that the binding affinities are similar for LIG1 and its isomer, although some variations have been found due to the orientations of KRAS4B-G12D for both GTP/GDP-bound states. From our results, we conclude that activities of the two enantiomers will be qualitatively different because their influence on orientations of KRAS are opposite, shown in Fig. S1E of ESI:† its isomer does not have effective action of deactivation whereas LIG1 enhances the inactive states for KRAS4B-G12D on the PM.
3 Methods
3.1 MD simulation details
In total 9 model systems were constructed to systematically examine how the small-molecule DBD, lipid-like compound LIG1 and its isomer can affect KRAS4B-G12D's orientation and corresponding signalling pathways. We have run 2 μs, 4 μs, 2.8 μs, 4 μs, 3 μs, and 2 μs for systems of interest, such as sys0, sys1, sys2, sys3, sys4, and sys5, respectively. In order to investigate the binding free-energy of LIG1 with lipid bilayer, we have run 500 ns of simulation time for sys6 to well equilibrate the system. The isomer of LIG1 have also been explored and the results are shown in Tables 1 and 2 of ESI.† Along with this work, we have also tested the possibility of DBD working as the warhead to target the KRAS4B-G12D, with the setup characteristics for all simulations listed in Table 1.Anionic phospholipid bilayer constituted by DOPC (56%), DOPS (14%) and cholesterol (30%) were considered for all systems. These lipid bilayer systems contains a total of 308 lipid molecules fully solvated by TIP3P water molecules in potassium chloride solution at the human body concentration (0.15 M). DBD molecule was localized near KRAS4B at the same side of PM while solvated by water molecules and KRAS4B was initially set anchoring to the bilayer at the beginning of simulations in all phospholipid bilayer systems, see Fig. S7.† For water system, DBD was placed around 30 Å away from KRAS4B along the Y axis.
Crystal structure of KRAS4B with partially disordered hypervariable region (pdb 5TB5), GTP (pdb 5VQ2) and GDP (pdb 5xco) were used to generate full length GTP-bound/GDP-bound KRAS4B proteins using Chimera, and missing residues in the flexible HVR were added while all the systems were prepared using CHARMM-GUI and GTP-bound configuration for KRAS4B was prepared by replacing GDP by the GTP molecule. Parameters of GDP, GTP, and DBD were directly adopted from the CHARMM36m FF topology files and the structure of LIG1 and its isomer were prepared through ligand reader & modeller of CHARMM-GUI web-based tool.60,61 The crystal structure (pdb 5TAR) was used to generate the full-length PDEδ, interactions between FAR tail and PDEδ were conserved in the crystal structure, see in Fig. 8E using Chimera. All pdb files were downloaded from RCSB PDB Protein Data Bank.62 Structures of complexes of LIG1 targeting into SII pocket of KRAS4B-G12D in its GTP or GDP-bound states were generated using Chimera by superimposing LIG1 to FAR of KRAS4B of sys2 while FAR has inserted well into the SII pocket and well established interactions between FAR and SII of sys2 were conserved for LIG1 of sys3 and sys4. Before generating all MD inputs in this work using CHARMM-GUI membrane builder,63 we have solvated all the complex of protein–ligands were solvated in the NaCl aqueous box with a concentration of 150 mM to gradually relax the structures of complexes we have prepared. All systems were firstly minimized for 10000 steps with a timestep of 1.0 fs while fixing the backbone atoms of the protein (KRAS4B and PDEδ), GTP, GDP, magnesium ion, and all atoms of the LIG1 and its isomer. Then they were equilibrated for 4 × 125000 steps with a timestep of 1.0 fs while gradually turning off the restraint on the backbone of KRAS4B during equilibration. Relaxing the structure of complexes was performed using NAMD 2.14 package.58,64 And the CHARMM36m force field (FF)65 was adopted for lipid–lipid and lipid–protein interactions. The N-termini and C-termini of KRAS4B were set as –NH3+ and –COCH3 groups, respectively.
All-atom MD simulations were conducted using the AMBER20.66 All systems were energy minimized for 20000 steps followed by five 250 ps equilibrium runs while gradually reducing the harmonic constraints on the systems. Minimization steps for replicated systems of sys1 and sys2 were set to be 5000 and 10000 steps, rather than 2000 steps, to generate independent production trajectories. Production runs were performed with NPT ensemble, Langevin dynamics with the collision frequency 1.0 ps−1 was used for temperature regulation (300 K) and Monte Carlo barostat was used for the pressure regulation (1 bar), respectively. Semiisotropic pressure scaling with constant surface tension for xy plane is used in statistical ensembles for simulating liquid interfaces. The time step was set to be 4 fs and the frames were saved by every 25000 steps for analysis. The particle mesh Ewald method was used to calculate the electrostatic interaction, and the van der Waals interactions were calculated using a cutoff of 12 Å. Periodic boundary conditions in three directions of space have been taken.
3.2 MD simulation analysis
The contact area through monitoring the solvent-accessible surface area along the trajectories for different structures is calculated using Tcl scripts by VMD.67 Only Cα atoms of protein molecules were used to calculate the root mean squared deviation (RMSD) and root mean square fluctuation (RMSF) profiles. The normalized covariance (correlation) of the MD simulations was performed by using CARMA,68 which gave rise to a residue–residue correlation map. A set of nodes in the correlation map with connecting edges, which in this case amino acid nodes were centered Cα atoms. Here we define that the edges between nodes whose residues are within 4.5 Å for at least 75% of the frames analyzed. Intra-molecular correlations between residues were measured by normalizing the cross correlation matrix of atomic fluctuations over the length of the simulation.69 If two residues move in the same (opposite) direction in most the frames, the motion is considered as (anti-)correlated, and the correlation value is close to −1 or 1. If the correlation value is close to zero, these two residues are considered uncorrelated. We used the classical adaptive biasing force (ABF) method in NAMD57,58 to describe the binding affinity of LIG1 and its isomer with KRAS, LIG1 with PDEδ, and LIG1 with the lipid bilayer using chosen variables. In this work, we took the distance of the center of mass of two groups as the variable for all systems related to LIG1 and its isomer (results reported in ESI†). For LIG1/isomer related systems, group 1 (all atoms of LIG1/isomer) and group 2 (backbone atoms of a protein domain (res60–157) that contains SII of KRAS4B) were defined to describe the distance separating LIG1/isomer from the SII of KRAS4B-G12D. For PDEδ, group 2 was taken as all the backbone atoms of PDEδ. The corresponding translocation of LIG1 from the phospholipid bilayer was defined as the projection onto the z-axis of the distance separating the center of mass of LIG1 molecule from that of all the phosphorus atoms of the bilayer. The reaction pathway spanning approximately 30 Å – i.e. from the “LIG1 bound into KRAS/PDEδ” state to the “LIG1 solvated in aqueous region” state, has been broken down into six consecutive 5 Å wide windows. Each of them started from a configuration in which the corresponding variable is in the relevant interval. A bin width of 0.1 Å, the boundary potentials with a force constant of 10 kcal mol−1 Å−1, and a threshold of 1000 force samples were adopted to obtain a reasonable estimate of the force applied along the chosen variable. All related files including an example used to calculate the binding free-energy of this work can be found in our repository “anticancer-drug-KRAS4B-G12D” at Github ICMAB Softmatter.704 Conclusions
In this work we describe an “in silico” designed lipid-like compound, LIG1. Our MD simulations predict that LIG1 directly interacts with KRAS4B-G12D in both guanosine-bound states and its regulator PDEδ through hydrophobic force by its FAR tail and hydrogen bonds by the DBD motif to inhibit KRAS4B's abnormal signalling activities. Our observations also suggest that KRAS4B anchoring to PM is crucial to conserve its intramolecular communication between different residues/domains to fulfil its physiological function, hence, considering PM during its corresponding drug discovery for KRAS4B and other membrane oncogene proteins is essential and that has been neglected for a long time.LIG1 reveals to be able to lock GDP-bound KRAS4B-G12D in its inactive orientations at the PM while effectively hiding the effector-binding interfaces with very high binding affinity, which leads to inhibit the guanine nucleotide exchange. Meanwhile, LIG1 reveals to trigger a population shift from its active state to its inactive states for GTP-bound KRAS4B-G12D. Therefore, the following signalling pathways in which GTP-bound KRAS4B-G12D involves are hindered. Our study shows that LIG1 directly targets PDEδ which inhibits the KRAS4B-PDEδ interaction with the binding free-energy of 88.2 kcal mol−1 and impairs KRAS4B enrichment at the phospholipid bilayer, thereby may suppress the in vitro and in vivo proliferation of KRAS4B-dependent cancer cells, further deepening our understanding of drug-KRAS4B recognition. It would be interesting to examine the molecular mechanisms/interactions between LIG1 and its derivatives to target different oncogenic KRAS4B mutants, or other RAS species, since up to date lipid-like compounds have not been considered to target RAS mutants. In this work we report the dynamic properties of SII pocket of KRAS4B upon LIG1 binding, which will be neglected by docking method used in virtual screen. Results presented here call for in-depth experimental investigations.
Author contributions
H. L. and J. M. designed the study. H. L. performed the simulations, analyzed the data, and generated the figures. All authors took part in writing, discussing and revising the manuscript.Conflicts of interest
The authors declare that they have no competing financial interests.Acknowledgements
We thank financial support by the Margarita Salas grant which is funded by the European Union – NextGenerationEU awarded to the Universitat Politecnica de Catalunya (Huixia Lu). We also thank financial support by grant PID2021-124297NB-C32 and PID2021-124297NB-C33 funded by MCIN/AEI/10.13039/501100011033, “ERDF A way of making Europe” and given by the “European Union NextGenerationEU/PRTR”. We also acknowledge financial support from Generalitat de Catalunya – AGAUR (grants 2021 SGR 01519 and 2021 SGR 01411). ICMAB is supported by the Spanish Government through the “Severo Ochoa” Program for Centers of Excellence in R&D (CEX2019-000917-S). We thank the CESGA supercomputing center for computer time and technical support at the Finisterrae III supercomputer and the Barcelona Supercomputing Center for support on projects BCV-2023-3-0004 and BCV-2023-3-0005. Molecular graphics made with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. Zheyao Hu is a Ph.D. fellow from the China Scholarship Council (grant 202006230070).References
- W. Kolch, D. Berta and E. Rosta, Biochem. J., 2023, 480, 1–23 CrossRef CAS PubMed.
- J. Downward, Nat. Rev. Cancer, 2003, 3, 11–22 CrossRef CAS PubMed.
- I. Prior, in Ras Variant Biology and Contributions to Human Disease, ed. I. Rubio and I. Prior, Springer US, New York, NY, 2021, pp. 3–18 Search PubMed.
- J. M. Ostrem and K. M. Shokat, Nat. Rev. Drug Discovery, 2016, 15, 771–785 CrossRef CAS PubMed.
- P. Liu, Y. Wang and X. Li, Acta Pharm. Sin. B, 2019, 9, 871–879 CrossRef PubMed.
- H. Jang, S. Abraham, T. Chavan, B. Hitchinson, L. Khavrutskii, N. Tarasova, R. Nussinov and V. Gaponenko, J. Biol. Chem., 2015, 290, 9465–9477 CrossRef CAS PubMed.
- G. Hobbs, C. Der and K. Rossman, J. Cell Sci., 2016, 129, 1287–1292 CAS.
- E. Pleasance, R. Cheetham, P. Stephens, D. McBride, S. Humphray, C. Greenman, I. Varela, M.-L. Lin, G. Ordóñez and G. Bignell, et al. , Nature, 2010, 463, 191–196 CrossRef CAS PubMed.
- I. Prior, P. Lewis and C. Mattos, Cancer Res., 2012, 72, 2457–2467 CrossRef CAS PubMed.
- C. Sheridan, Nat. Biotechnol., 2021, 39, 1032–1034 CrossRef CAS PubMed.
- S. Dhillon, Drugs, 2023, 83, 275–285 CrossRef CAS PubMed.
- X. Wang, S. Allen, J. F. Blake, V. Bowcut, D. M. Briere, A. Calinisan, J. R. Dahlke, J. B. Fell, J. P. Fischer and R. J. Gunn, et al. , J. Med. Chem., 2021, 65, 3123–3133 CrossRef PubMed.
- Z. Mao, H. Xiao, P. Shen, Y. Yang, J. Xue, Y. Yang, Y. Shang, L. Zhang, X. Li and Y. Zhang, et al. , Cell Discovery, 2022, 8, 5 CrossRef CAS PubMed.
- H. Lu and J. Martí, Nanoscale, 2022, 14, 3148–3158 RSC.
- H. Lu and J. Martí, Membranes, 2020, 10, 364 CrossRef CAS PubMed.
- H. Lu and J. Martí, J. Phys. Chem. Lett., 2020, 11, 9938–9945 CrossRef CAS PubMed.
- Z. Hu and J. Marti, Int. J. Mol. Sci., 2022, 23, 13865 CrossRef CAS PubMed.
- A. G. Stephen, D. Esposito, R. K. Bagni and F. McCormick, Cancer Cell, 2014, 25, 272–281 CrossRef CAS PubMed.
- A. D. Cox, S. W. Fesik, A. C. Kimmelman, J. Luo and C. J. Der, Nat. Rev. Drug Discovery, 2014, 13, 828–851 CrossRef CAS PubMed.
- J. Chen, S. Zhang, W. Wang, L. Pang, Q. Zhang and X. Liu, J. Chem. Inf. Model., 2021, 61, 1954–1969 CrossRef CAS PubMed.
- J. Chen, L. Wang, W. Wang, H. Sun, L. Pang and H. Bao, Comput. Biol. Med., 2021, 135, 104639 CrossRef CAS PubMed.
- P. Prakash and A. A. Gorfe, J. Phys. Chem. B, 2019, 123, 8644–8652 CrossRef CAS PubMed.
- S. Vatansever, B. Erman and Z. H. Gümüş, Comput. Struct. Biotechnol. J., 2020, 18, 1000–1011 CrossRef CAS PubMed.
- D. Liu, Y. Mao, X. Gu, Y. Zhou and D. Long, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024725118 CrossRef CAS.
- M. De Vivo, M. Masetti, G. Bottegoni and A. Cavalli, J. Med. Chem., 2016, 59, 4035–4061 CrossRef CAS PubMed.
- Z. Hu and J. Marti, Membranes, 2022, 12, 331 CrossRef CAS PubMed.
- Z. Hu, J. Martí and H. Lu, J. Chem. Phys., 2021, 155, 154303 CrossRef CAS PubMed.
- M. M. Platts, Br. Med. J., 1959, 1, 1565 CrossRef CAS PubMed.
- R. M. Taylor and T. H. Maren, J. Pharmacol. Exp. Ther., 1963, 140, 249–257 CAS.
- A. Martinez, A. I. Esteban, A. Castro, C. Gil, S. Conde, G. Andrei, R. Snoeck, J. Balzarini and E. De Clercq, J. Med. Chem., 1999, 42, 1145–1150 CrossRef CAS PubMed.
- A. Martinez, C. Gil, A. Castro, C. Perez, M. Witvrouw, C. Pannecouque, J. Balzarini and E. De Clercq, Antiviral Chem. Chemother., 2001, 12, 347–351 CrossRef CAS PubMed.
- D. Das, J. Hong, S.-H. Chen, G. Wang, L. Beigelman, S. D. Seiwert and B. O. Buckman, Bioorg. Med. Chem., 2011, 19, 4690–4703 CrossRef CAS PubMed.
- V. M. Patil, S. Prakash Gupta, S. Samanta and N. Masand, Med. Chem., 2012, 8, 1099–1107 CAS.
- A. Tait, A. Luppi, A. Hatzelmann, P. Fossa and L. Mosti, Bioorg. Med. Chem., 2005, 13, 1393–1402 CrossRef CAS PubMed.
- A. Kamal, M. N. A. Khan, Y. Srikanth, K. S. Reddy, A. Juvekar, S. Sen, N. Kurian and S. Zingde, Bioorg. Med. Chem., 2008, 16, 7804–7810 CrossRef CAS PubMed.
- X. Ma, J. Wei, C. Wang, D. Gu, Y. Hu and R. Sheng, Eur. J. Med. Chem., 2019, 170, 112–125 CrossRef CAS PubMed.
- A. P. Larsen, P. Francotte, K. Frydenvang, D. Tapken, E. Goffin, P. Fraikin, D.-H. Caignard, P. Lestage, L. Danober and B. Pirotte, et al. , ACS Chem. Neurosci., 2016, 7, 378–390 CrossRef CAS PubMed.
- K. Y. Lee, M. Ikura and C. B. Marshall, Angew. Chem., 2023, 135(18), e202218698 CrossRef.
- B. Papke, S. Murarka, H. A. Vogel, P. Martín-Gago, M. Kovacevic, D. C. Truxius, E. K. Fansa, S. Ismail, G. Zimmermann and K. Heinelt, et al. , Nat. Commun., 2016, 7, 11360 CrossRef CAS PubMed.
- S. Dharmaiah, L. Bindu, T. H. Tran, W. K. Gillette, P. H. Frank, R. Ghirlando, D. V. Nissley, D. Esposito, F. McCormick and A. G. Stephen, et al. , Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6766–E6775 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- M. Khaled, A. Gorfe and A. Sayyed-Ahmad, J. Phys. Chem. B, 2019, 123, 7667–7675 CrossRef CAS PubMed.
- G. Tu, Q. Liu, Y. Qiu, E. L.-H. Leung and X. Yao, Int. J. Mol. Sci., 2022, 23, 13845 CrossRef CAS PubMed.
- T. Pantsar, Comput. Struct. Biotechnol. J., 2020, 18, 189–198 CrossRef CAS PubMed.
- T. Pantsar, S. Rissanen, D. Dauch, T. Laitinen, I. Vattulainen and A. Poso, PLoS Comput. Biol., 2018, 14, e1006458 CrossRef PubMed.
- K. Trueblood, H.-B. Bürgi, H. Burzlaff, J. Dunitz, C. Gramaccioli, H. Schulz, U. Shmueli and S. Abrahams, Acta Crystallogr., Sect. A: Found. Crystallogr., 1996, 52, 770–781 CrossRef.
- T. Pantsar, Sci. Rep., 2020, 10, 11992 CrossRef CAS PubMed.
- A. Kapoor and A. Travesset, Proteins: Struct., Funct., Bioinf., 2015, 83, 1091–1106 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- H. Lu and J. Martí, J. Chem. Phys., 2018, 149, 164906 CrossRef PubMed.
- H. Liu, H. Zhang and B. Jin, Spectrochim. Acta, Part A, 2013, 106, 54–59 CrossRef CAS PubMed.
- A. Sethi, J. Eargle, A. A. Black and Z. Luthey-Schulten, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6620–6625 CrossRef CAS PubMed.
- H. Jang, A. Banerjee, T. S. Chavan, S. Lu, J. Zhang, V. Gaponenko and R. Nussinov, FASEB J., 2016, 30, 1643 CrossRef CAS PubMed.
- M. T. Mazhab-Jafari, C. B. Marshall, M. J. Smith, G. M. Gasmi-Seabrook, P. B. Stathopulos, F. Inagaki, L. E. Kay, B. G. Neel and M. Ikura, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6625–6630 CrossRef CAS PubMed.
- P. Prakash, D. Litwin, H. Liang, S. Sarkar-Banerjee, D. Dolino, Y. Zhou, J. F. Hancock, V. Jayaraman and A. A. Gorfe, Biophys. J., 2019, 116, 179–183 CrossRef CAS PubMed.
- M. Schmick, A. Kraemer and P. I. Bastiaens, Trends Cell Biol., 2015, 25, 190–197 CrossRef CAS PubMed.
- E. Darve, D. Rodríguez-Gómez and A. Pohorille, J. Chem. Phys., 2008, 128, 114120 CrossRef.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. Henin, G. Fiorin, C. Chipot and M. L. Klein, J. Chem. Theory Comput., 2010, 6, 35–47 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- S. Kim, J. Lee, S. Jo, C. L. Brooks III, H. S. Lee and W. Im, CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules, 2017 Search PubMed.
- A. Kouranov, L. Xie, J. de la Cruz, L. Chen, J. Westbrook, P. Bourne and H. Berman, Nucleic Acids Res., 2006, 34, D302–D305 CrossRef CAS PubMed.
- S. Jo, J. Lim, J. Klauda and W. Im, Biophys. J., 2009, 97, 50–58 CrossRef CAS PubMed.
- J. C. Phillips, D. J. Hardy, J. D. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin and W. Jiang, et al. , J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- J. Huang and A. MacKerell Jr., J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- N. M. Glykos, J. Comput. Chem., 2006, 27, 1765–1768 CrossRef CAS PubMed.
- T. Ichiye and M. Karplus, Proteins: Struct., Funct., Bioinf., 1991, 11, 205–217 CrossRef CAS PubMed.
- H. Lu, Z. Hu, J. Faraudo and J. Marti, Github, 2023, https://github.com/soft-matter-theory-at-icmab-csic, accessed: date accessed Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr04513g |
| This journal is © The Royal Society of Chemistry 2023 |