
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn a high-capacity aluminium battery with a two-electron phenothiazine redox polymer as a positive electrode†
Gauthier
Studer
ab,
Alexei
Schmidt
bc,
Jan
Büttner
bcd,
Maximilian
Schmidt
 a,
Anna
Fischer
bcd,
Ingo
Krossing
*bcd and
Birgit
Esser
*ab
a,
Anna
Fischer
bcd,
Ingo
Krossing
*bcd and
Birgit
Esser
*ab
aInstitute of Organic Chemistry II and Advanced Materials, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: birgit.esser@uni-ulm.de; Web: https://www.esserlab.com
bFreiburg Materials Research Centre, University of Freiburg, Stefan-Meier-Str. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
cUniversity of Freiburg, Institute of Inorganic and Analytical Chemistry, Albertstraße 21, 79104 Freiburg, Germany Web: https://www.krossing-group.de
dCluster of Excellence livMatS@FIT – Freiburg Centre for Interactive Materials and Bioinspired Technologies, University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
First published on 22nd May 2023
Abstract
With aluminium being the most abundant metal in Earth's crust, rechargeable Al ion batteries (AIBs) hold great promise as next-generation energy storage devices. However, the currently used positive electrode materials suffer from low specific capacity, which limits the specific energies of these AIBs. Here, we present an organic redox polymer with two well-defined redox processes as a positive electrode material that overcomes these shortcomings. Cross-linked poly(3-vinyl-N-methylphenothiazine) with phenothiazine as a two-electron redox centre reversibly inserts [AlCl4]− ions at potentials of 0.81 and 1.65 V vs. Al|Al3+, delivers experimental specific capacities of up to 167 mA h g−1 in AIBs and surpasses graphite as a positive electrode material. After 5000 cycles at a 10C rate, this AIB retains 88% of its capacity. Even at a 100C rate, 64 mA h g−1 can be reversibly cycled, and the AIB returns to its original capacity without any changes at slower rates. This is the first report of a reversible two-electron redox process for a phenothiazine-based battery electrode material. With its high discharge voltage and specific capacity, and its excellent capacity retention at fast C-rates combined with flat charge/discharge plateaus, this AIB plays a major role in the development of rechargeable AIBs and will initiate further explorations of organic redox polymers as positive electrode materials, paving the way towards more sustainable energy storage devices.
Broader contextDue to the scarcity of lithium and transition metal oxides used in traditional batteries, there is a strong impetus to develop alternative battery technologies for applications ranging from small devices to large scale stationary storage of electricity. Since aluminium is one of the most widely available elements in the Earth's crust, Al-based batteries are considered promising candidates for such next-generation energy storage devices. However, to date, it remains a challenge to identify appropriate host electrode materials that reversibly insert (complex) aluminium ions. In this article, we demonstrate a strategy for designing such positive electrode materials. This strategy involves using an organic redox polymer as a positive electrode material, which reversibly inserts two [AlCl4]− ions with a specific capacity that surpasses that of graphite as a positive electrode material. In addition, it shows superior cyclability at fast C-rates. This concept could pave the way towards the development of advanced Al-based batteries and affordable energy storage devices. |
Introduction
Climate change and the increasing demand for electrical energy require the development of novel types of devices for the storage of renewable energy. While classical lithium-ion batteries1,2 might benefit from engineered electrode materials,3–5 next-generation batteries6,7 should rely on abundant elements, be safe and of low cost, use non-toxic materials and be easy to recycle. With 8.1 wt%, aluminium is the most abundant metal in the Earth's crust and its recycling is easy.8 Its high volumetric capacity of 8040 mA h cm−3 as a negative electrode material even exceeds that of lithium of 2046 mA h cm−3.9 In contrast to the latter, it can be reversibly stripped and deposited without forming dendrites,7,10 preventing short circuits. Favourably, the ionic liquid electrolytes currently used in Al batteries are non-flammable.11 Hence, rechargeable Al ion batteries (AIBs) hold great promise as storage devices.9,12,13 Yet, the development of rechargeable AIBs faces fundamental challenges and in particular lacks suitable positive electrode materials, leaving them still in their infancy.14–16Two types of charge storage mechanisms for positive electrodes are known for the Al(III) species formed upon cycling: storage as cationic (Al3+, [AlCl]2+, and [AlCl2]+) or anionic ([AlCl4]− and [Al2Cl7]−) (complex) aluminium ions. Only few examples have been reported where Al3+ ions were reversibly stored, probably due to their high charge density and low mobility.17–19 In some cases, monocationic [AlCl2]+ ions20,21 or dicationic [AlCl]2+ ions22–24 with reduced charge densities are stored. Therefore, the more favourable anion insertion mechanism is the main approach currently used. It proceeds with high reversibility, enabling fast charge/discharge rates and large operating potentials. The most studied positive electrode material (PEM) for [AlCl4]− storage is graphite, following the seminal report by Lin et al. in 2015.25 Al/graphite cells typically show a flat discharge potential of 1.8–1.9 V vs. Al|Al3+, good rate capability and cycling stability.26 However, the specific discharge capacities of graphite are limited to ca. 120 mA h g−1, with few reports on higher values, and specific energies‡ of up to 69 W h kg−1.27–29
Organic PEMs are excellent candidates for [AlCl4]− insertion, potentially enabling higher capacities and energy densities.30 Many p-type organic compounds can be reversibly oxidized at high potentials (up to 4 V vs. Li|Li+) and thereby store and release anions at fast charge/discharge rates.31 Organic PEMs were predominantly explored for Li-organic cells,32,33 but there are a few examples where p-type organics were used as PEMs in AIBs.34–36 The conductive polymers polypyrrole and polythiophene37 were first investigated and found to exhibit specific capacities of 30–100 mA h g−1 as well as specific energies‡ of around 45 W h kg−1. Polypyrenes38 exhibited slightly better performance with an average capacity of 100 mA h g−1 at 1.6–2.0 V vs. Al|Al3+ discharge voltage, while poly(3,4-ethylenedioxythiophene) (PEDOT)39 gave energy densities‡ of 50–64 W h kg−1 at a lower average discharge potential of 1.3 V. Recently, polymeric triarylamines were investigated, polymerized in situ within the electrode during electrochemical cycling from small molecule triarylamine precursors.40 A reversible capacity of 135 mA h g−1 was obtained, but with sloppy charge/discharge profiles around an average voltage of 1.1 V vs. Al|Al3+, resulting from the ill-defined structure of the organic PEM. A similar approach was used in another recent report on aminopyrenes, which were oligomerized in situ and exhibited specific discharge capacities of up to 195 mA h g−1 within a potential range of 0.1–2.2 V vs. Al|Al3+, but also associated with sloppy charge/discharge profiles.41 The performance of pyrenes could recently be boosted through combination with dihydrophenazines in conjugated microporous polymers, furnishing attractive capacities exceeding 200 mA h g−1 at potentials of 0.5 and 1.5 V vs. Al|Al3+.42
By contrast, (aliphatic) redox polymers are ideal candidates as organic PEMs:43 as opposed to conductive polymers, they contain electronically separated redox centres, resulting in well-defined redox processes, and their incorporation into a (cross-linked) polymer backbone renders them insoluble in battery electrolytes. Phenothiazine (PT) is a particularly well-suited p-type redox-active group, as it can undergo two reversible oxidations to a dication at relatively high redox potentials of 3.6 and 4.1 V vs. Li|Li+. PT-based polymers have shown excellent performance in Li-organic cells, regarding cycling stability and rate capability.44–48 Limiting their capacity, however, only the first redox process of each PT unit could be reversibly addressed in cells with LiPF6/carbonate-based electrolytes, which resulted in only lower (reversible) specific capacities of up to 112 mA h g−1 for poly(3-vinyl-N-methylphenothiazine) (PVMPT)44 or its cross-linked derivative X-PVMPT.46 Possibly, PT dications undergo irreversible side reactions with the carbonate-based electrolyte molecules, rendering the second redox process irreversible.
Herein, we investigate cross-linked poly(3-vinyl-N-methylphenothiazine) (X-PVMPT) as the PEM in AIBs with a 1-ethyl-3-methylimidazolium (EMIm)-based chloroaluminate ionic liquid electrolyte. In this medium, both redox processes of each PT unit can be reversibly addressed (Fig. 1(a)). Notably, this is the first report of a reversible two-electron redox process for any phenothiazine-based battery electrode material.49 Hence, each PT unit in X-PVMPT can be oxidized from its neutral state A over the radical cation state C to the dication state D. In the intermediate oxidation state B, π*–π* interactions between neutral and oxidized PT units evolve.47 This is relevant for the Al/X-PVMPT battery, as will be discussed below. X-PVMPT-based electrodes insert [AlCl4]− or [Al2Cl7]− ions at average charge potentials of 0.81 and 1.65 V vs. Al|Al3+ with high reversibility and at fast charge/discharge rates. Experimental specific capacities of up to 167 mA h g−1 are accessible (theor.: 221 mA h g−1). Thus, the electrodes clearly surpass graphite as the PEM. Very recently, a phenoxazine-based polymer was reported as the PEM for Al batteries, but with up to 133 mA h g−1 the discharge capacities are lower than for X-PVMPT.50 In addition, X-PVMPT-based electrodes show excellent cycling stability, where 5000 cycles at a 10C rate proceeded under 88% retention of the initial specific capacity. The redox processes are well distinguishable with a small voltage hysteresis between the charge and discharge, even at high C-rates. This is a major advancement compared to other reported organic PEMs for AIBs.
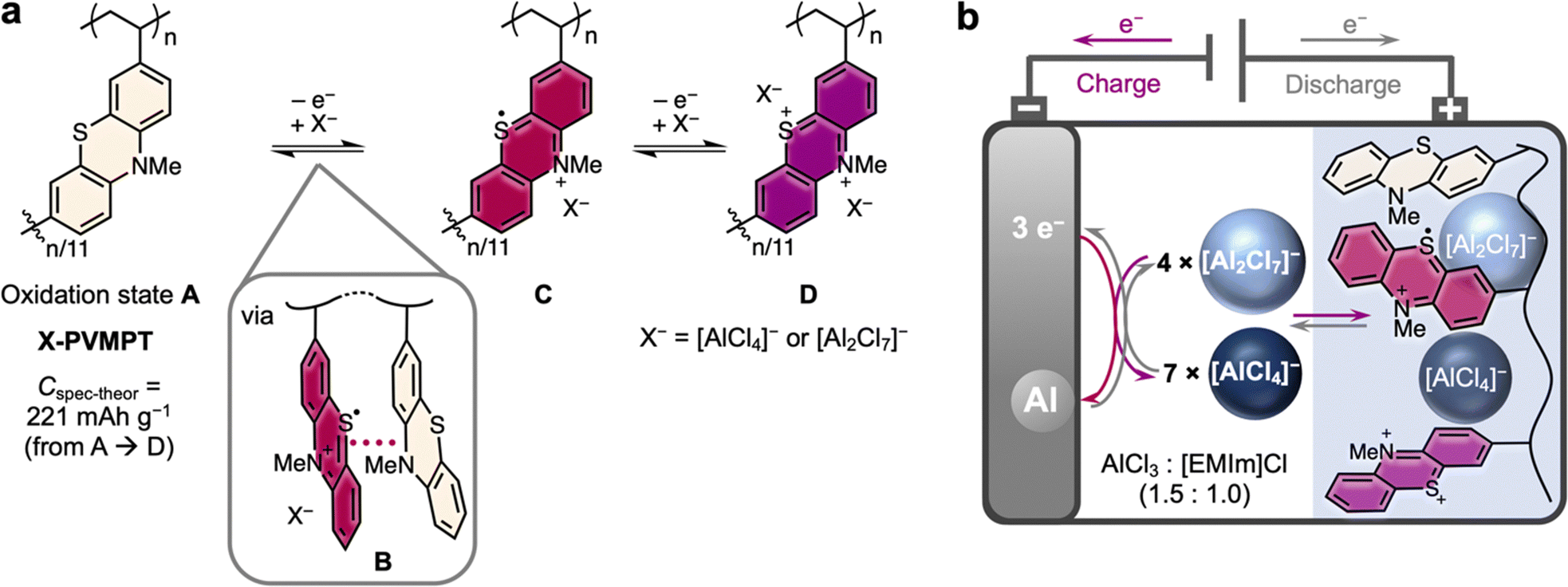 | ||
| Fig. 1 Concept of the Al/X-PVMPT battery. (a) Redox processes in the phenothiazine-based polymer X-PVMPT with oxidation states (A, B, C and D). (b) Schematic setup of the Al/X-PVMPT battery (the colours of the redox states of the PT units were chosen similarly to those experimentally observed). | ||
Results and discussion
Each phenothiazine unit in the polymer X-PVMPT can undergo a two-electron oxidation from the neutral state A to a dication Dvia the radical cation state C (Fig. 1(a)). This can be seen in cyclic voltammograms (CVs) of the non-crosslinked polymer PVMPT in solution47 and was further confirmed by DFT calculations (see the ESI† for details). Using isolated N-methylphenothiazine (MPT) as the redox-active subunit in X-PVMPT, the calculated potential difference between the first redox process (A → C) and the second redox process (C → D) amounts to 1.22 V. In the polymer X-PVMPT, cation-π*–π* interactions lead to the stabilization of the oxidized states and the occurrence of an intermediate oxidation state B, in which only every other PT unit is oxidized to a radical cation (Fig. 1(a)).47 For DFT calculations, we used a dimeric subunit of the polymer as a reference compound (MPT-dimer, see the ESI† for details). The occurrence of state B leads to a shift in the calculated redox potentials and a smaller potential difference between the two redox processes (oxidation states C and D) of only 0.64 V compared to MPT. This calculated potential difference correlates well with that measured in the CV in the Al/X-PVMPT cells of 0.76 V (e.g. in cycle 20, see Fig. 2(a) below). | ||
| Fig. 2 Electrochemical performance of Al/X-PVMPT batteries. (a) Cyclic voltammograms of X-PVMPT-based electrodes vs. Al, first 20 cycles (0.2 mV s−1). (b) Constant current cycling at varying C-rates, average over three cells with error bars (0.5C = 0.16 mA cm−2 and 100C = 31 mA cm−2).* (c) Constant current cycling at a 10C rate (3.1 mA cm−2), average over three cells with error bars.* (d) Charge/discharge profiles of selected cycles from plot c at a 10C rate. (* After 50 cycles of pre-conditioning at a 0.5C rate). | ||
A scheme of the Al/X-PVMPT battery is shown in Fig. 1(b). In the Al/X-PVMPT cells, the room temperature ionic liquid, EMIm chloride with 1.5 added equivalents of AlCl3 (AlCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[EMIm]Cl = 1.5:1), served as the electrolyte. Due to its wide electrochemical window, good ionic conductivity and relatively low viscosity, EMIm-chloroaluminates are the most used electrolytes in non-aqueous AIBs.12 Excess AlCl3 is required to form [Al2Cl7]− from [AlCl4]−, and both complex anions are needed for reversible Al plating and stripping on the negative electrode.51–53 The overall stoichiometry of the electrochemical reaction in the AIB is the following (PT indicates one redox-active phenothiazine subunit of X-PVMPT):
:[EMIm]Cl = 1.5:1), served as the electrolyte. Due to its wide electrochemical window, good ionic conductivity and relatively low viscosity, EMIm-chloroaluminates are the most used electrolytes in non-aqueous AIBs.12 Excess AlCl3 is required to form [Al2Cl7]− from [AlCl4]−, and both complex anions are needed for reversible Al plating and stripping on the negative electrode.51–53 The overall stoichiometry of the electrochemical reaction in the AIB is the following (PT indicates one redox-active phenothiazine subunit of X-PVMPT):
| 3PT0 + 8[Al2Cl7]− ⇌ 3PT˙+ + 4[Al2Cl7]− + 7[AlCl4]− + Al0 ⇌ 3PT2+ + 14[AlCl4]− + 2Al0 |
Recently, benzene was shown to be an additive that reduces the viscosity of this ionic liquid electrolyte, yet maintains its ability to reversibly deposit Al.55 We reasoned that the more polar fluorinated benzenes with strong sp2-C–F bonds could be superior candidates as additives due to their higher polarity but maintained stability towards Lewis-acid-induced side reactions. Hence, we investigated fluorobenzene, 1,2-difluorobenzene, 1,2,3-trifluorobenzene, 1,2,3,4-tetrafluorobenzene, pentafluorobenzene, and hexafluorobenzene as additives. However, compared to the pristine AlCl3:[EMIm]Cl (1.5:1) electrolyte, adding the fluorinated benzenes did not lead to a significant improvement. In some cases, slightly lower overpotentials for Al deposition were observed; but this effect was minor, and we therefore proceeded using the pristine electrolyte (see the ESI,† Section 1 with Fig. S1–S10 for details).
Electrochemical performance
To evaluate the electrochemical performance of X-PVMPT in AIBs, composite electrodes were fabricated containing 50 wt% X-PVMPT, 45 wt% acetylene black as conductive additive and 5 wt% PVDF as a binder on molybdenum discs as the current collector (Fig. S14, ESI† shows the electrochemical stability window of the Mo current collector). Metallic Al was used as the counter and reference electrodes, and electrochemical measurements were performed in Swagelok-type cells (Fig. S13, ESI†). We optimized the potential range to 0.30–2.20 V vs. Al|Al3+ to obtain the maximum capacity from X-PVMPT as the active electrode material, but avoid the partially irreversible and slower insertion and adsorption of chloroaluminate anions on the carbon additive (see the ESI† for details, Fig. S15–S17). Measurements using only acetylene black on the molybdenum current collector at a similar current density as applied to the X-PVMPT electrodes at 10C rate cycling (3.1 mA cm−2) showed a specific charge and discharge capacity of less than 10 mA h g−1 (Fig. S18 and S19, ESI†), confirming that only X-PVMPT showed significant electrochemical activity in the investigated potential range of 0.30–2.20 V vs. Al|Al3+.The CVs of the first 20 cycles of an Al/X-PVMPT cell at 0.2 mV s−1 (Fig. 2(a)) show two well separated redox processes, which correspond to the two oxidations of each PT unit in the polymer X-PVMPT (A to C and C to D, see Fig. 1(a)). The first redox process (A to C) is centred at E1/2 = 0.79 V vs. Al|Al3+ and characterized by a small peak-to-peak separation of 91 mV. This underlines the reversibility and faradaic nature of this redox event. Between cycles 1 and 20 the potential remains constant. The second redox process (C to D), on the other hand, shows a gradual change over the first 20 cycles. In an anodic (oxidative) scan direction, the peak potential in the first cycle appears at 1.91 V vs. Al|Al3+, and in further cycles it is split into two peaks with peak potentials of 1.58 and 1.84 V vs. Al|Al3+. In a cathodic (reductive) scan direction, the peak potential for the second redox event remains constant over 20 cycles at 1.52 V vs. Al|Al3+. This change in the anodic scan direction indicates an activation process occurring in the PEM, which might be related to rearrangement processes within the polymer, allowing for stabilizing cation-π*–π*-interactions between PT units to form and the occurrence of the intermediate oxidation state B (Fig. 1(a)).47 This results in stabilization and therefore a decrease in the redox potential of the second oxidation step over the initial cycles (from E1/2 = 1.88 V to E1/2 = 1.55 V vs. Al|Al3+), furnishing a final potential difference of 0.76 V between the two redox waves. We also observed this activation process in galvanostatic measurements, where only after a preconditioning period with 50 cycles at 0.5C the maximum performance of the X-PVMPT electrodes was obtained (see Fig. S20, ESI†). The differential capacity plots of the pre-conditioning cycles show a profile similar to the CVs from Fig. 2(a), where the second redox process (oxidation state C to D, see Fig. 1) is also split into two peaks initially, but merges into one peak after ca. 20 cycles, see Fig. S21 and S22, ESI†. Such pre-cycling has also been reported to be required for other AIBs.56
Fig. 2(b) shows the constant current cycling data of Al/X-PVMPT cells at different C-rates (average of three cells shown) after 50 cycles of pre-conditioning at a 0.5C rate. At a 0.5C rate, equivalent to a current density of 0.16 mA cm−2 or 0.11 A g−1 of the active material, a high average specific discharge capacity of 167 mA h g−1 was obtained. This corresponds to 76% of the theory (221 mA h g−1) and shows that most of the PT-units in X-PVMPT participate in the two consecutive reversible redox processes (cf.Fig. 1(a)). Such reversible behaviour for the two redox reactions of each PT unit was never reported before.49 We assume that the discrepancy between the theoretical and experimentally accessible specific capacities is related to the occurrence of oxidation state B (see Fig. 1(a)) as the discharged form of the polymer, based on previous studies on PVMPT in Li-based half cells47,57 and on our SEM/EDX studies discussed below. In state B, every other PT unit is still in its radical cation form, which means that only 1.5 electrons are transferred per PT unit during discharge, and consecutively in all following charge/discharge cycles. This corresponds well with the observation that 76% of the theoretical specific capacity is accessible in the experiment, assuming that cycling occurs between oxidation states B and D, and also with the fact that the plateau at lower potential corresponding to the oxidation from B to C is associated with a smaller capacity than the plateau at higher potential, corresponding to the oxidation from C to D (Fig. 2(d)).
The X-PVMPT-based electrodes showed excellent rate performance. Even at a high 20C rate, corresponding to current densities of 6.2 mA cm−2 and 4.4 A g−1 of the active material, the reversible specific discharge capacities amount to record-breaking 136 mA h g−1 (see comparative Table S7 in the ESI†).30,58,59 At 50 and 100C (11 and 22 A g−1, respectively) rates, the specific discharge capacities declined more strongly, but still reached significant average values of 105 and 64 mA h g−1, respectively. Reducing the current to a 1C rate refurbished the initial specific capacity and allowed stable cycling for a further 50 cycles at an average capacity of 166 mA h g−1, which demonstrates that no decomposition of the active material took place, even at rates as high as 100C. The decline in specific capacity at rates above 20C may result from several factors: from the differential capacity plots (Fig. S23, ESI†), it follows that the two redox processes of X-PVMPT are centred at 0.80 and 1.55 V vs. Al|Al3+ at a slow 0.5C rate with small peak-to-peak separations of 48 mV and 47 mV, respectively. These values do not significantly change up to a rate of 20C. Starting from 50C, however, the battery experiences a greater polarization with peak separations of 307 mV and 256 mV for the two redox processes and increased Ohmic losses resulting from the internal resistance of the cell. This can cause the cell to prematurely reach the pre-defined cutoff potential before the active material has completed the electrochemical process, as is well visible in the charge/discharge curves in Fig. S24 (ESI†). These overpotentials at high C-rates can be rationalized by the lack of electronic conductivity of X-PVMPT as an aliphatic polymer and by limitations in ion transport processes at very high rates.31
The specific energies of the Al/X-PVMPT cells, taking into account the masses of active materials in electrodes and electrolytes, reach up to 30 W h kg−1, but particularly impressive are the power densities of up to 7000 W kg−1, clearly surpassing those of Al/graphite batteries (see Section 2.3 in the ESI† and Fig. S35).28,29,60
We next investigated the long-term cycling performance of Al/X-PVMPT cells at a high 10C rate – equal to a current density of 3.1 mA cm−2 or 2.2 A g−1 of the active material (Fig. 2(c)). An average of three cells was measured, demonstrating the reproducibility of the obtained data (for individual plots see Fig. S25, ESI†). From the initial average specific discharge capacity of 151 mA h g−1, 88% was retained after 5000 cycles at a 10C rate. This corresponds to a capacity fade of only 0.0024% per cycle, demonstrating excellent long-term cycling stability of the Al/X-PVMPT cells at this high current density, surpassing all other reported Al-organic batteries.40,56 The charge/discharge voltage profiles at a 10C rate (Fig. 2(d)) show two plateaus, representing the two well-defined redox processes of each PT unit with average discharge potentials of 1.48 and 0.74 V vs. Al|Al3+ (for differential capacity plots see Fig. S26, ESI†). Such flat charge/discharge plateaus are rarely observed in Al-organic batteries using p-type PEMs, in particular at such high current densities,40,41 and highlight the excellent suitability of X-PVMPT, if paired with EMIm-chloroaluminate as the electrolyte, as the PEM for AIBs.
Hence, herein we report the first reversible two-electron redox process for a PT-based battery electrode material, probably enabled by the ionic liquid electrolyte. Furthermore, regarding the three combined parameters of discharge voltage, specific capacity and capacity retention at fast C-rates, the X-PVMPT-electrodes reported herein outperform all other known organic PEMs in AIBs.30 The redox processes are well distinguishable with a small voltage hysteresis between the charge and discharge, even at high C-rates, advancing this system over other organic PEMs for AIBs.
Charge storage mechanism
A closer look at the charge/discharge profiles in Fig. 2(d) shows that the plateau at a lower potential (average 0.74 V vs. Al|Al3+) favourably contributes only to a smaller amount of the specific capacity than the plateau at a higher potential (average 1.48 V vs. Al|Al3+). Hence, a larger amount of charge can be stored in the X-PVMPT electrodes at the higher potential than at the lower potential, which indicates that not all PT units take part in both redox processes A to C and C to D (see Fig. 1). To shed light into this cycling behaviour, we performed constant current cycling measurements at a 0.5C rate (Fig. 3(a)) and investigated both charged and discharged electrodes using scanning electron microscopy (SEM) measurements combined with energy-dispersive X-ray (EDX) analyses (Fig. 3(b)). These measurements provide insight into the presence of chloroaluminate anions within the positive electrode in the different states of charge.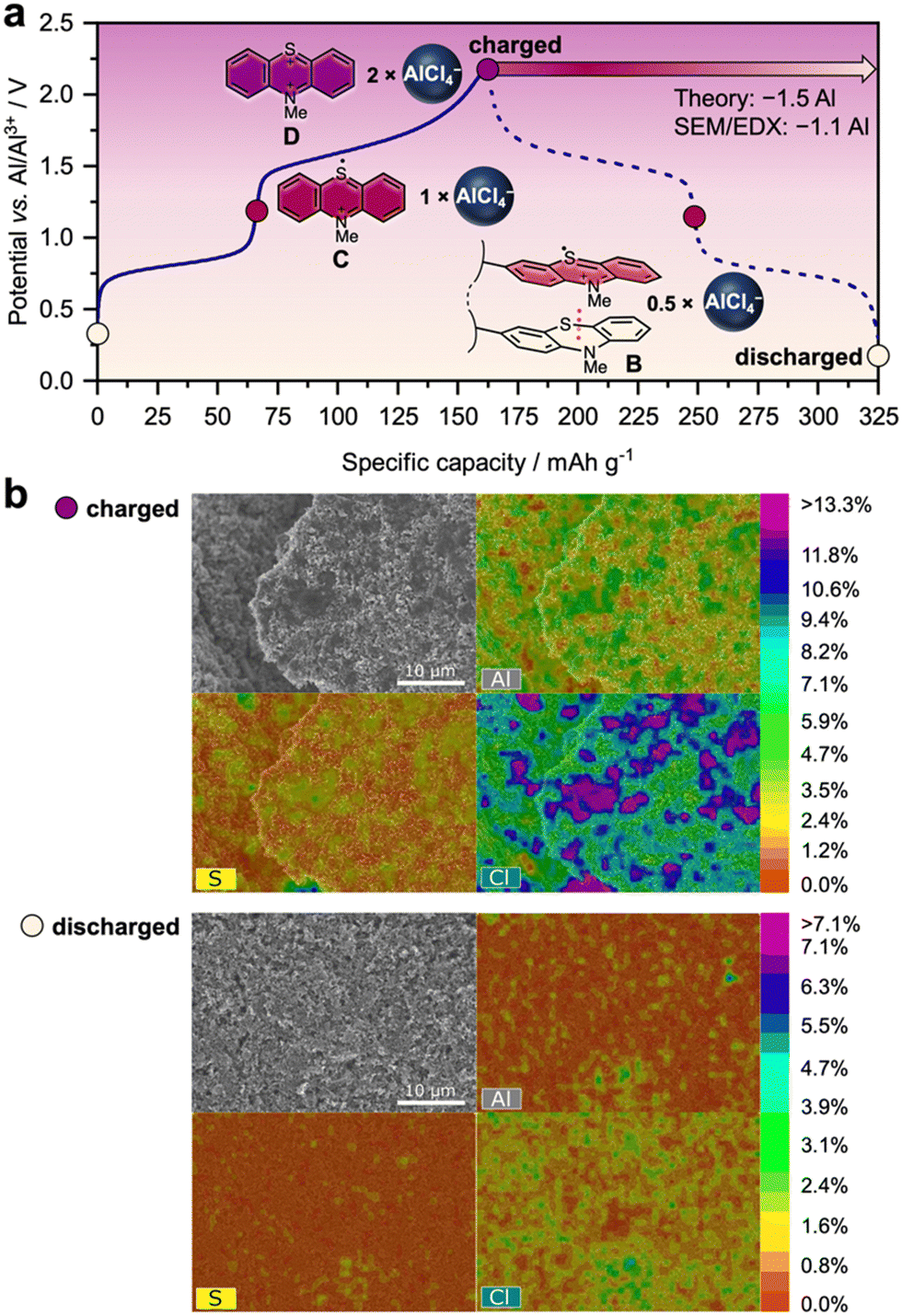 | ||
| Fig. 3 Charge/discharge mechanism of Al/X-PVMPT batteries. (a) Charge/discharge profiles (at a 0.5C rate, after 50 cycles of pre-conditioning) with correlated oxidation states of the PT units and required [AlCl4]− ions for counterbalancing of charges (assuming that [AlCl4]− is the predominant inserting chloroaluminate ion based on its smaller size than [Al2Cl7]−).54 (b) SEM images of the X-PVMPT electrodes in the charged (2.2 V vs. Al|Al3+) and discharged (to 0.3 V vs. Al|Al3+) states with atomic concentration heat map overlays based on EDX spectroscopy. | ||
To remove additional electrolyte on the surface, the electrodes were carefully rinsed with o-difluorobenzene before the SEM/EDX measurements; nevertheless, traces of electrolyte residuals cannot be fully excluded, and hence, the quantification of the Al/S ratio is not a quantitative representation of the state of charge (SOC) and should not be over interpreted.
During the constant current cycling measurement at a 0.5C rate shown in Fig. 3(a) (51st cycle at a 0.5C rate), the plateaus during charge are centred at 0.81 and 1.65 V vs. Al|Al3+ (slightly differing from the potentials obtained at a 10C rate in Fig. 2(d)) and correspond to specific charge capacities of 66 mA h g−1 and 98 mA h g−1, respectively, a total of 164 mA h g−1 for the complete charge cycle. Assuming that charge and discharge occur between oxidation states B and D (Fig. 1(a)), two equivalents of chloroaluminate anions ([AlCl4]−/[Al2Cl7]−) are required in the fully charged state D of X-PVMPT to balance two positive charges on each PT unit, while 0.5 equivalents remain in the discharged state B (see also Fig. 3(a)).
SEM/EDX spectra of the charged electrode show that significant amounts of Al and Cl are indeed present (Fig. 3(b)). Their localization correlates well with that of the sulphur atoms from the PT units, indicating their role as charge-stabilizing counter anions. In the discharged electrode, on the other hand, the Al and Cl concentrations are decreased, but both elements are still present. A quantification shows that the ratio of Al/S changes from 3.5 to 2.4 during discharge. Assuming that [AlCl4]− is the predominant inserting chloroaluminate ion based on its smaller size than [Al2Cl7]−,54 this would correspond to a loss of 1.1 [AlCl4]− ion equivalents relative to each PT unit in X-PVMPT. This correlates well with the expected value of 1.5 [AlCl4]− ions, based on the observed specific discharge capacity of 164 mA h g−1. This corresponds to a 74% utilization of the theoretical specific capacity for the 2-electron oxidation of each PT unit (221 mA h g−1) and cycling between oxidation states B and D of X-PVMPT. Hence, the polymer is not reduced back to its neutral state during discharge, but retains some [AlCl4]− (or [Al2Cl7]−) ions in the discharged state, as expected for oxidation state B. The additional amounts of Al and Cl present in the electrode in both the charged and discharged forms might be due to a solid–electrolyte interphase (SEI) layer, which formed in the initial cycles, as also observed by Nann and coworkers.61 Furthermore, due to an excess of electrolyte employed, certain amounts of electrolyte with chloroaluminate ions might also still be present in the discharged electrode.
To further shed light into the charge storage mechanism of the Al/X-PVMPT batteries, we performed electrochemical kinetics studies. Evaluation of scan-rate dependent CVs allows differentiating between diffusion- and surface-controlled redox processes. The scan-rate dependent CVs of X-PVMPT electrodes are plotted in Fig. 4(a) and show an increase of the anodic (peaks 1 and 2) and cathodic (Peaks 3 and 4) peak currents with the scan rate. We use the known relationship between the peak current i and the scan rate ν
| i = kνb, | (1) |
 | ||
| Fig. 4 Electrochemical kinetics of Al/X-PVMPT batteries. (a) CVs at various scan rates. (b) Logarithmic plots of the peak current vs. the scan rate. (c) Diffusion- and surface-controlled fractions of the redox reaction. (d) Diffusion coefficients for [AlCl4]− from GITT measurements (0.5C rate, SOC = state of charge). | ||
The surface-controlled process contribution was further investigated using eqn (2) proposed by Kim et al.63
| Itot (V) = k1ν + k2ν0.5 | (2) |
Furthermore, the small size and non-coordinating nature of the [AlCl4]− counterions enable their fast diffusion within the positive electrode material, see also the diffusion coefficients plotted in Fig. 4(d).
Hence, ion diffusion does take place close to the redox sites, as clearly visible from the shapes of the CVs in Fig. 4(a) and it is not a limiting factor. The amorphous morphology of the cross-linked X-PVMPT also contributes, which can increase the available space for ions to move and enhance the ionic conductivity, resulting in minor diffusion limitations, as discussed for the other p-type network polymer in the literature.64 This observation explains the excellent rate capability of the Al/X-PVMPT batteries, as the redox reactions in the positive electrode have no diffusion limitations.
We further evaluated the kinetics using the galvanostatic intermittent titration technique (GITT) (Fig. S34, ESI†). The measurement was performed after a pre-conditioning step of 50 cycles at 0.5C. The GITT protocol consisted of alternating 6 min long constant current pulses at 0.5C and rest steps of 1 h to allow for relaxation of the system. During charge, the insertion of the first [AlCl4]− ion (SOC below 50%) has a relatively fast kinetics with a diffusion coefficient D on the order of 10−9 cm2 s−1, while that of the second [AlCl4]− ion (SOC 50–100%) is associated with a smaller diffusion coefficient of ca. 10−10–10−11 cm2 s−1 (Fig. 4(d)). In the discharge, the first [AlCl4]− ion reinserts quickly with D ≈ 10−9 cm2 s−1 (SOC 100–50%), and the second ion slightly slower with D ≈ 10−9–10−10 cm2 s−1 (SOC below 50%). The magnitude of these diffusion coefficients clearly indicates diffusion of the ions within the amorphous polymer phase. They are in a similar range as those reported for Al-graphite batteries, which lie in the order of D ≈ 10−8–10−9 cm2 s−1.65 In conjunction with the strongly surface-controlled process, they contribute to the high rate capability of the X-PVMPT electrodes.
Conclusions
In summary, we have investigated cross-linked poly(3-vinyl-N-methylphenothiazine) (X-PVMPT) as the organic positive electrode material for the insertion of [AlCl4]− ions in rechargeable Al batteries using an EMIm chloroaluminate electrolyte. Unprecedented for phenothiazine-based battery electrode materials, both redox processes of each phenothiazine unit could be used with this electrolyte and delivered experimental specific capacities of up to 167 mA h g−1. This exceeds that of graphite in Al-graphite batteries, favourably further paired with excellent cyclability and rate capability. Hence, 5000 cycles at a 10C rate was achieved with 88% capacity retention, outperforming all other organic electrode materials for Al batteries. At a high current density of 4.4 A g−1, the X-PVMPT-electrodes delivered a record specific discharge capacity of 136 mA h g−1. Kinetic mechanistic investigations confirmed the reversible [AlCl4]− ion insertion during charging and showed that this process is not diffusion-limited, accounting for the excellent rate capability of the electrodes. Our study constitutes a major advance in the development or rechargeable Al batteries and will initiate further explorations of organic redox polymers as positive electrode materials together with ionic liquid-based electrolytes in such cells, paving the way towards more sustainable energy storage devices.Experimental
Materials and electrode preparation
The crosslinked X-PVMPT polymer with 10 mol% of the crosslinker was synthesized using the same procedure as previously reported.46 Its thermal analysis data can be found in Fig. S11 and S12 (ESI†). Composite electrodes were prepared using 50 wt% X-PVMPT, 45 wt% carbon black (acetylene black, Alfa Aesar, 100% compressed, 99.9+%, 75 m2 g−1, bulk density 170–230 g l−1) and 5 wt% PVdF (Kynar® HSV 900, Arkema). The components were mixed and dispersed in N-methyl-2-pyrrolidone (NMP, Acroseal®, Thermo scientific, 99.5%, stored over molecular sieves), using a planetary centrifugal mixer (ARM 310, Thinky mixer). The electrode formulation was then cast onto molybdenum disks as current collectors (thickness: 1 mm, diameter: 12 mm, 99.9%, Goodfellow). The resulting coated disks were dried at ambient pressure for 12 h at 60 °C in a drying oven and then in in vacuo (10−3 mbar, 60 °C, 24 h). The active material (X-PVMPT) mass loadings of the electrodes laid between 1.2 and 1.7 mg cm−2.Cell assembly
Electrochemical experiments were performed using a perfluoroalkoxy (PFA)-based Swagelok® three electrode cell setup (Fig. S13, ESI†). All cells were assembled in an Ar-filled Glovebox with H2O and O2 levels of <0.1 ppm. The fabricated X-PVMPT-based electrodes were used as working electrodes (WEs, diameter: 12 mm). Polished aluminium disks were used as counter electrodes (CEs, thickness: 1 mm, diameter: 12 mm, 99.999%, Goodfellow), and polished aluminium wire were as reference electrodes (RE, diameter: 1 mm, 99.999%, Goodfellow). Glass fibre separators (diameter: 13 mm, GF/D, Whatman™, Cytiva) were placed between the WE and RE and between the RE and CE and soaked with 100 μL of electrolyte (AlCl3 (99.999%, Sigma Aldrich): [EMIm]Cl (>98%, iolitec), 1.5:1.0).
Electrochemical measurements
Cyclic voltammetry and constant current measurements were performed using a MPG-2 battery testing system (Biologic Science Instruments), after a constant current pre-cycling/conditioning step of 50 cycles at 0.5C.SEM/EDX
Scanning electron microscopy characterization was performed using a Hitachi field emission gun scanning electron microscope (FEG-SEM) SU8220 operated at an acceleration voltage of 6 kV. EDX measurements were performed with the same acceleration voltage at a working distance of roughly 16 mm and recorded using a Bruker X-Flash detector. The software used for the data evaluation was Bruker esprit 2.5.1.221.Author contributions
B. E., I. K. and G. S. designed the concept of this project. B.E. and I.K. directed the research. G. S. performed the synthesis of X-PVMPT, all cell fabrication, and electrochemical and kinetics measurements. A. S. conducted the experiments using additives for the ionic liquid electrolyte. A. F. and J. B. designed the SEM/EDX experiments, and J. B. and G. S. performed the SEM/EDX experiments. M. S. performed the DFT calculations, coached by I. K. B. E. and G. S. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work contributed to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe) and was funded by the German Research Foundation (DFG) under Project ID 390874152 (POLiS Cluster of Excellence, EXC 2154) and 441215516 (SPP 2248 – Polymer-based batteries). Financial support through the German Federal Environmental Foundation (DBU, graduate fellowship for G. S.) is gratefully acknowledged as well as financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2193/1 – 390951807 (livMatS Cluster of Excellence) and through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster), the Eva Maria Stihl Stiftung in the project Saltus! and the state of Baden-Württemberg through bwHPC.Notes and references
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- T. Kim, W. Song, D.-Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- J. Li, Z. Zhu, Y. Huang, F. Wang and M.-S. (Jie Tang) Balogun, Mater. Today Energy, 2022, 26, 101001 CrossRef CAS.
- Y. Huang, H. Yang, T. Xiong, D. Adekoya, W. Qiu, Z. Wang, S. Zhang and M.-S. Balogun, Energy Storage Mater., 2020, 25, 41–51 CrossRef.
- X. Yao, C. Li, R. Xiao, J. Li, H. Yang, J. Deng and M.-S. Balogun, Small, 2022, 18, 2204534 CrossRef CAS PubMed.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- Committee TGAR, Global aluminium recycling: A cornerstone of sustainable development, 2009.
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J.-F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS PubMed.
- H. Chen, H. Xu, B. Zheng, S. Wang, T. Huang, F. Guo, W. Gao and C. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 22628–22634 CrossRef CAS PubMed.
- M. Galiński, A. Lewandowski and I. Stępniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- B. Craig, T. Schoetz, A. Cruden and C. Ponce de Leon, Renewable Sustainable Energy Rev., 2020, 133, 110100 CrossRef CAS.
- H. Yang, H. Li, J. Li, Z. Sun, K. He, H. Cheng and F. Li, Angew. Chem., Int. Ed., 2019, 58, 11978–11996 CrossRef CAS PubMed.
- T. Leisegang, F. Meutzner, M. Zschornak, W. Münchgesang, R. Schmid, T. Nestler, R. A. Eremin, A. A. Kabanov, V. A. Blatov and D. C. Meyer, Front. Chem., 2019, 7, 1–21 CrossRef PubMed.
- Y. Zhang, S. Liu, Y. Ji, J. Ma and H. Yu, Adv. Mater., 2018, 30, 1706310 CrossRef PubMed.
- F. Wu, H. Yang, Y. Bai and C. Wu, Adv. Mater., 2019, 31, 1806510 CrossRef PubMed.
- L. Zhou, Z. Zhang, L. Cui, F. Xiong, Q. An, Z. Zhou, X.-F. Yu, P. K. Chu and K. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100354 CrossRef CAS.
- Y. Hu, D. Ye, B. Luo, H. Hu, X. Zhu, S. Wang, L. Li, S. Peng and L. Wang, Adv. Mater., 2018, 30, 1703824 CrossRef PubMed.
- C. Legein, B. J. Morgan, F. Fayon, T. Koketsu, J. Ma, M. Body, V. Sarou-Kanian, X. Wei, M. Heggen, O. J. Borkiewicz, P. Strasser and D. Dambournet, Angew. Chem., Int. Ed., 2020, 59, 19247–19253 CrossRef CAS PubMed.
- X. Peng, Y. Xie, A. Baktash, J. Tang, T. Lin, X. Huang, Y. Hu, Z. Jia, D. J. Searles, Y. Yamauchi, L. Wang and B. Luo, Angew. Chem., Int. Ed., 2022, 61, e202203646 CAS.
- X. Han, S. Li, W. Song, N. Chen, H. Chen, S. Huang and S. Jiao, Adv. Energy Mater., 2021, 11, 2101446 CrossRef CAS.
- D.-J. Yoo, M. Heeney, F. Glöcklhofer and J. W. Choi, Nat. Commun., 2021, 12, 2386 CrossRef CAS PubMed.
- J. Bitenc, N. Lindahl, A. Vizintin, M. E. Abdelhamid, R. Dominko and P. Johansson, Energy Storage Mater., 2020, 24, 379–383 CrossRef.
- N. Lindahl, J. Bitenc, R. Dominko and P. Johansson, Adv. Funct. Mater., 2020, 2004573 CrossRef CAS.
- X. Xiao, M. Wang, J. Tu, Y. Luo and S. Jiao, ACS Sustainable Chem. Eng., 2019, 7, 16200–16208 CrossRef CAS.
- G. A. Elia, I. Hasa, G. Greco, T. Diemant, K. Marquardt, K. Hoeppner, R. J. Behm, A. Hoell, S. Passerini and R. Hahn, J. Mater. Chem. A, 2017, 5, 9682–9690 RSC.
- D.-Y. Y. Wang, C.-Y. Y. Wei, M.-C. C. Lin, C.-J. J. Pan, H.-L. L. Chou, H.-A. A. Chen, M. Gong, Y. Wu, C. Yuan, M. Angell, Y.-J. J. Hsieh, Y.-H. H. Chen, C.-Y. Y. Wen, C.-C. C.-W. W. Chen, B.-J. J. Hwang, C.-C. C.-W. W. Chen and H. Dai, Nat. Commun., 2017, 8, 14283 CrossRef CAS PubMed.
- S. Wang, K. V. Kravchyk, F. Krumeich and M. V. Kovalenko, ACS Appl. Mater. Interfaces, 2017, 9, 28478–28485 CrossRef CAS PubMed.
- K. V. Kravchyk, S. Wang, L. Piveteau and M. V. Kovalenko, Chem. Mater., 2017, 29, 4484–4492 CrossRef CAS.
- Z. Yang, F. Wang, P. Meng, J. Luo and C. Fu, Energy Storage Mater., 2022, 51, 63–79 CrossRef.
- C. N. Gannett, L. Melecio-Zambrano, M. J. Theibault, B. M. Peterson, B. P. Fors and H. D. Abruña, Mater. Rep.: Energy, 2021, 1, 100008 CAS.
- B. Esser, F. Dolhem, M. Becuwe, P. Poizot, A. Vlad and D. Brandell, J. Power Sources, 2021, 482, 228814 CrossRef CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS PubMed.
- J. Xie and Q. Zhang, Small, 2019, 15, 1805061 CrossRef PubMed.
- H. Ye and Y. Li, Energy Fuels, 2021, 35, 7624–7636 CrossRef CAS.
- K. Qin, J. Huang, K. Holguin and C. Luo, Energy Environ. Sci., 2020, 13, 3950–3992 RSC.
- N. S. Hudak, J. Phys. Chem. C, 2014, 118, 5203–5215 CrossRef CAS.
- M. Walter, K. V. Kravchyk, C. Böfer, R. Widmer and M. V. Kovalenko, Adv. Mater., 2018, 30, 1705644 CrossRef PubMed.
- T. Schoetz, B. Craig, C. Ponce de Leon, A. Bund, M. Ueda and C. T. J. Low, J. Energy Storage, 2020, 28, 101176 CrossRef.
- G. Wang, E. Dmitrieva, B. Kohn, U. Scheler, Y. Liu, V. Tkachova, L. Yang, Y. Fu, J. Ma, P. Zhang, F. Wang, J. Ge and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202116194 CrossRef CAS PubMed.
- Y. Kong, C. Tang, C. Lei, A. K. Nanjundan, S. Chen, N. Ahmed, D. Rakov, A. Du, X. Huang and C. Yu, Nano Energy, 2022, 102, 107727 CrossRef CAS.
- W. Ma, L. Luo, X. Huang, P. Dong, Y. Chen, C. Zhang, F. Huang, J. Jiang and Y. Cao, Adv. Energy Mater., 2023, 13, 2203253 CrossRef CAS.
- B. Esser, Org. Mater., 2019, 01, 063–070 CrossRef CAS.
- V. Perner, D. Diddens, F. Otteny, V. Küpers, P. Bieker, B. Esser, M. Winter and M. Kolek, ACS Appl. Mater. Interfaces, 2021, 13, 12442–12453 CrossRef CAS PubMed.
- F. Otteny, G. Studer, M. Kolek, P. Bieker, M. Winter and B. Esser, ChemSusChem, 2020, 13, 2232–2238 CrossRef CAS PubMed.
- F. Otteny, M. Kolek, J. Becking, M. Winter, P. Bieker and B. Esser, Adv. Energy Mater., 2018, 8, 1802151 CrossRef.
- M. Kolek, F. Otteny, P. Schmidt, C. Mück-Lichtenfeld, C. Einholz, J. Becking, E. Schleicher, M. Winter, P. M. Bieker and B. Esser, Energy Environ. Sci., 2017, 10, 2334–2341 RSC.
- P. Acker, J. S. Wössner, G. Desmaizieres and B. Esser, ACS Sustainable Chem. Eng., 2022, 10, 3236–3244 CrossRef CAS.
- F. Otteny, G. Desmaizieres and B. Esser, in Redox Polymers for Energy and Nanomedicine, ed. N. Casado and D. Mecerreyes, Royal Society of Chemistry, 2020, pp. 166–197 Search PubMed.
- Z. Yang, X. Huang, P. Meng, M. Jiang, Y. Wang, Z. Yao, J. Zhang, B. Sun and C. Fu, Angew. Chem., Int. Ed., 2023, 62, e202216797 CrossRef CAS PubMed.
- C. Ferrara, V. Dall’Asta, V. Berbenni, E. Quartarone and P. Mustarelli, J. Phys. Chem. C, 2017, 121, 26607–26614 CrossRef CAS.
- P. R. Gifford and J. B. Palmisano, J. Electrochem. Soc., 1988, 135, 650–654 CrossRef CAS.
- P. K. Lai and M. Skyllas-Kazacos, J. Electroanal. Chem. Interfacial Electrochem., 1988, 248, 431–440 CrossRef CAS.
- W. R. Carper, C. E. Keller, N. A. Evangelos and M. E. Zandler, Main Group Chem., 1996, 1, 257–263 CrossRef CAS.
- Y. Park, D. Lee, J. Kim, G. Lee and Y. Tak, Phys. Chem. Chem. Phys., 2020, 22, 27525–27528 RSC.
- D. J. Kim, D.-J. Yoo, M. T. Otley, A. Prokofjevs, C. Pezzato, M. Owczarek, S. J. Lee, J. W. Choi and J. F. Stoddart, Nat. Energy, 2019, 4, 51–59 CrossRef CAS.
- M. Kolek, F. Otteny, J. Becking, M. Winter, B. Esser and P. Bieker, Chem. Mater., 2018, 30, 6307–6317 CrossRef CAS.
- E. Faegh, B. Ng, D. Hayman and W. E. Mustain, Nat. Energy, 2021, 6, 21–29 CrossRef CAS.
- H. Chen, H. Xu, S. Wang, T. Huang, J. Xi, S. Cai, F. Guo, Z. Xu, W. Gao and C. Gao, Sci. Adv., 2017, 3, eaao7233 CrossRef PubMed.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324–328 CrossRef CAS PubMed.
- N. Canever, F. R. Hughson and T. Nann, ACS Appl. Energy Mater., 2020, 3, 3673–3683 CrossRef CAS.
- H. Lindström, S. Södergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
- H.-S. Kim, J. B. Cook, H. Lin, J. S. Ko, S. H. Tolbert, V. Ozolins and B. Dunn, Nat. Mater., 2017, 16, 454–460 CrossRef CAS PubMed.
- B. M. Peterson, C. N. Gannett, L. Melecio-Zambrano, B. P. Fors and H. Abruña, ACS Appl. Mater. Interfaces, 2021, 13, 7135–7141 CrossRef CAS PubMed.
- D. Han, M. Cao, N. Li, D. She, W. Song, H. Chen, S. Jiao and D. Fang, Chin. J. Chem., 2021, 39, 157–164 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee00235g |
| ‡ Including the masses of active materials and electrolytes. |
| This journal is © The Royal Society of Chemistry 2023 |