
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the mechanism by which Gd(III) coiled coils achieve magnetic resonance relaxivity – a study into the water coordination chemistry†
S. L.
Newton
ab,
A.
Franke
cd,
A.
Zahl
c,
G.
Molinaro
a,
A.
Kenwright
 e,
D. J.
Smith
f,
I.
Ivanovic-Burmazovic
*cd,
M. M.
Britton
*a and
A. F. A.
Peacock
*a
e,
D. J.
Smith
f,
I.
Ivanovic-Burmazovic
*cd,
M. M.
Britton
*a and
A. F. A.
Peacock
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: m.m.britton@bham.ac.uk; a.f.a.peacock@bham.ac.uk
bPSIBS, School of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK
cDepartment Chemie und Pharmazie, Universität Erlangen-Nürnberg, D-91058, Erlangen, Germany
dDepartment of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstr. 5-13, 81377 München, Germany
eSchool of Chemistry, Durham University, Durham, DH1 3LE, UK
fSchool of Mathematics, University of Birmingham, Edgbaston, B15 2TT, UK
First published on 24th October 2023
Abstract
A class of Gd(III) coiled coils achieve high MRI relaxivity, in part due to their slow rotational correlation time. However, extending their length is unable to further enhance performance, as the mechanism by which relaxivity is achieved is dominated by the presence of three inner sphere waters in rapid exchange, through an associative mechanism.
Complexes of Gd(III) are routinely used to enhance contrast in magnetic resonance imaging (MRI) to aid in clinical diagnosis. However, the relaxivity, a measure of efficiency, of most clinical contrast agents (CAs) is not optimal, in part due to their small size and rapid tumbling. One strategy for enhancing relaxivity has been to increase the rotational correlation (tumbling) time (τR), through the design of macromolecular Gd(III) complexes,1 based on for example, polymers,2 nanotubes,3,4 nanodiamonds,5 dendrimers,6 polysacharides,7,8 viral capsids,9 liposomes10 and peptides/proteins.11–13 Of relevance to this work are the reports of Gd(III) coiled coils, a class of artificial miniature metalloproteins, that exhibit high relaxivity compared to conventional small molecule CAs.14–17
Linear translation of the Gd(III) binding site towards the coiled coil N-terminus yields Gd(MB1-1)3, a more stable, highly hydrated (determined for the Tb(III) analogue) and higher relaxivity complex.15 Though the promising relaxivity is in part thought to be due to their larger size and slower tumbling, the mechanism by which relaxivity is achieved is not well understood. Specifically, the true hydration state of the bound Gd(III) [crystallographic evidence is lacking] and the rate of water exchange, key parameters that govern relaxivity, are unknown.
Herein we report a systematic study of the impact of peptide length on the structure and function of Ln(III) coiled coils based on MB1-1 (Fig. 1). The relationship between Gd(III) coordination chemistry, coupled with a detailed analysis of the water exchange chemistry at the Gd(III) centre, and MR relaxivity, is investigated.
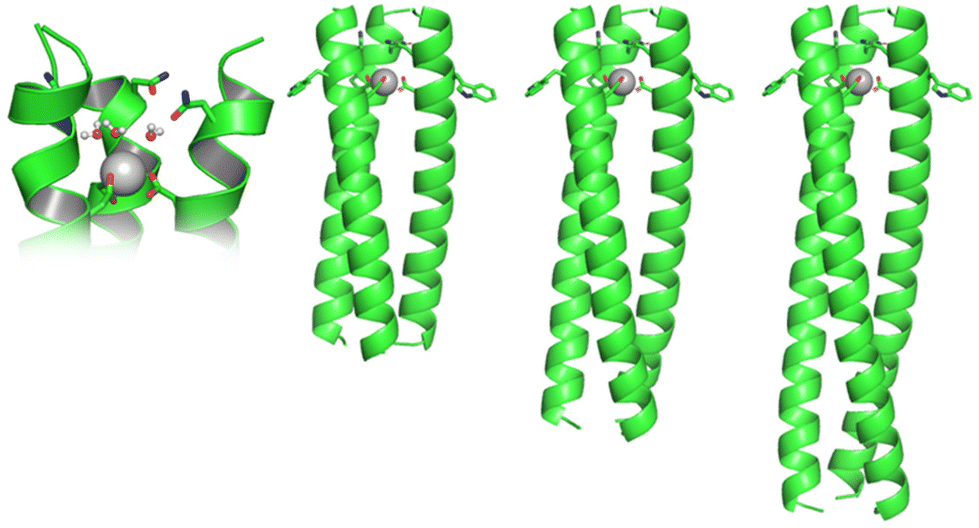 | ||
| Fig. 1 Cartoon of a hypothetical short (MB1-1S), medium (MB1-1) and long (MB1-1L) Gd(III) coiled coil, and a close-up of the proposed binding site with Gd(III) bound to three inner sphere water molecules. | ||
These findings have implications for the design of Gd(III) peptides/proteins for MRI applications, as well as for metallo coiled coils and their aqua complexes, for applications in biotechnology more widely, such as catalysis. Furthermore, these studies provide important opportunities for insight into the burgeoning field of lanthanide biochemistry.18–21 This includes an example where Gd (though inefficient) supports the growth of a lanthanide dependent methanotroph.22
To fully understand the mechanism by which these Gd(III) coiled coils achieve high relaxivity, the relationship between molecular rotation, the water coordination chemistry of the bound Gd(III) and relaxivity, has been investigated through a series of three peptides, designed to form Gd(III) coiled coils of varying length (Ac-G IAANEWK DAAIEQK (IAAIEQK)x G-NH2), see Fig. 1 and S1.† The short (MB1-1S; x = 2), previously reported medium (MB1-1; x = 3)15 and long (MB1-1L; x = 4) peptides, based on four, five and six heptads, respectively, all feature a Gd(III) binding site generated by adjacent Asn and Asp layers, towards the N-terminus (top).
Given that MB1-1 was the most stable and well folded of the coiled coils which featured the destabilising Asn3Asp3 binding site,15 it was reasoned that the fifth heptad may not be essential. However, MB1-1S, which consists of four heptads, is found to be largely unfolded in the apo state, with only a small change upon the addition of GdCl3 (Fig. 2A). This likely reflects that the Asn3Asp3 binding site, despite being located towards a terminus, interrupts two hydrophobic packing layers in two adjacent heptads, leaving only two fully intact heptads, which are unable to effectively compensate. We therefore conclude that a minimum of five heptads are required for the folding of coiled coils which feature the Asn3Asp3 binding site, even in the presence of Ln(III). Intriguingly, though metallo coiled coils as short as three heptads are known,23 all reported Ln(III) coiled coils to date, consist of five heptads.14–16,24–28 These longer lengths likely reflect the destabilising nature of negatively charged Ln(III) binding sites. Given that MB1-1S failed to form a folded coiled coil, even in the presence of Gd(III), its Ln(III) coordination chemistry is not discussed further, and instead attention is directed towards comparisons between the folded MB1-1 and MB1-1L peptides.
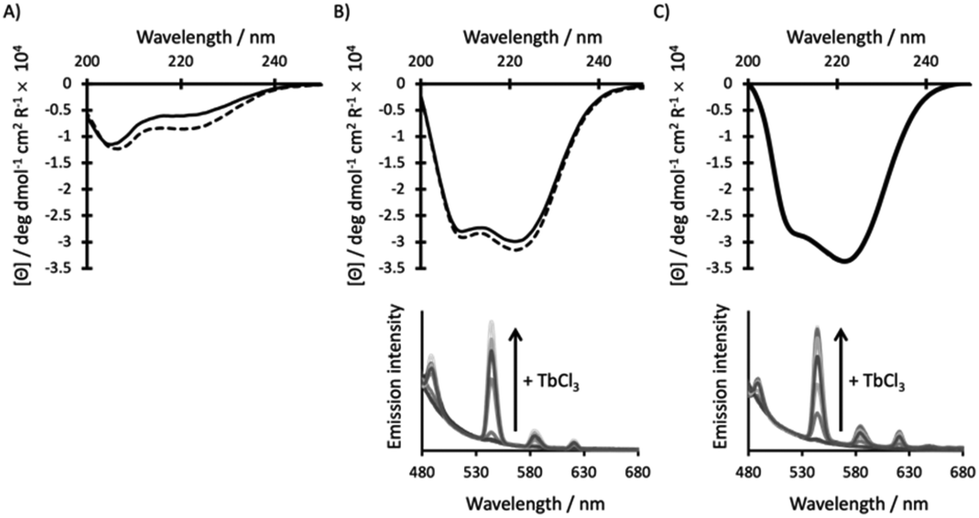 | ||
| Fig. 2 CD (top) and emission (bottom) spectra of 30 μM (A) MB1-1S, (B) MB1-1 and (C) MB1-1L peptide monomer, in the absence (solid line) and presence (dashed line) of 10 μM GdCl3 (top) or increasing aliquots of TbCl3 (bottom), in 10 mM HEPES buffer pH 7.0. λexc = 280 nm. | ||
The addition of a heptad, lengthening the coiled coil from five (MB1-1) to six (MB1-1L), has no significant impact on the extent of peptide folding for either the apo or Gd(III) complex, as determined from the CD spectra (Fig. 2B and C, S2†). Though these findings suggest that the extra heptad does not induce further coiled coil folding, it has previously been reported to enhance stability with respect to denaturation, by ∼5 kcal mol−1.23 Despite a shift in the chemical denaturation profiles that supports enhanced stability on addition of the 6th heptad (Fig. S3†), we were unable to fit our data reliably due to the lack of a clear baseline for the unfolding transition. Similarly, thermal unfolding profiles of MB1-1 and MB1-1L, do not show a clear baseline for the unfolding transition, but do importantly show that the Gd(MB1-1)3 and Gd(MB1-1L)3 complexes remain folded at biologically relevant temperature (37 °C), see Fig. S4.† Our data are, therefore, consistent with no significant enhancement in folding, but a qualitive enhancement in stability, on extending the coiled coil length.
Tb(III) luminescence experiments (Fig. 2 and S5†) complement the CD titrations as they provide important insight into the Ln(III) coordination chemistry. Regardless of coiled coil length, titrations are consistent with one Tb(III) ion saturating a binding site generated by a three stranded coiled coil, and with the same apparent affinity (within error, Table 1). Importantly, any enhancement in peptide stability achieved by lengthening the peptide, has not translated into a significant change in binding affinity. The current binding constants (apparent log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka ∼5.0) are considerably lower than current clinical MRI contrast agents,29 and would require significant enhancement prior to translation into a clinical setting.
Ka ∼5.0) are considerably lower than current clinical MRI contrast agents,29 and would require significant enhancement prior to translation into a clinical setting.
| Peptide | % Foldinga | LogKTbb |
#H2OTbc |
τ
Rd/ns |
r
1e/mM−1 s−1 (7 T) |
r
2e/mM−1 s−1 (7 T) |
r
1e/mM−1 s−1 (1 T) |
r
2e/mM−1 s−1 (1 T) |
|
|---|---|---|---|---|---|---|---|---|---|
| Apo | 1 Eq. Gd3+ | ||||||||
| a Determined by CD spectroscopy, see ESI.† b Apparent binding constant for the Tb3+ complex as determined by fluorescence spectroscopy. c Number of coordinated waters as determined by luminescence emission of the Tb3+ complex. d Theoretical rotational correlation time around the minor axis. e Relaxivity as determined by NMR spectroscopy on the Gd3+ complexes. f Data from ref. 15. | |||||||||
| MB1-1S | 20 ± 1 | 33 ± 4 | — | — | — | — | — | — | — |
| MB1-1 | 80 ± 6f | 83 ± 7f | 5.3 ± 0.1f | 3.1 ± 0.2f | 7 | 10.0 ± 1.5f | 89.3 ± 16.8f | 64.3 ± 3.2 | 87.6 ± 0.9 |
| MB1-1L | 84 ± 2 | 85 ± 2 | 5.0 ± 0.2 | 3.7 ± 0.7 | 10 | 10.9 ± 0.8 | 81.8 ± 5.4 | 67.4 ± 4.2 | 96.5 ± 3.2 |
Further interrogation of the Tb(III) coordination environments, revealed similar Tb(III) hydration states (see Table 1 and Fig. S6†), when complexed to MB1-1 and MB1-1L, respectively. The 6th heptad, introduced ca. 4–5 nm away from the binding site at the opposite end of the coiled coil, does not have a significant impact on coiled coil folding, Tb(III) affinity, or water penetration and coordination, but does slightly enhance the coiled coils stability (vide supra). We hypothesised that, as for Tb(III), the Gd(III) complexes of MB1-1 and MB1-1L would feature the same coordination chemistry. However, the Gd(MB1-1)3 and Gd(MB1-1L)3 complexes could differ in their rotational correlation times.
Extending the coiled coil length yields complexes of differing molecular weight, 12.2 kDa and 14.4 kDa for Gd(MB1-1)3 and Gd(MB1-1L)3, respectively. Hydrodynamic calculations yielded theoretical rotational correlation times around the minor axis of 7 and 10 ns for the Gd(MB1-1)3 and Gd(MB1-1L)3 complexes, respectively, and have been used to predict longitudinal relaxivity (r1) at two field strengths (clinically more relevant 1 T and higher field at 7 T), as a function of water exchange rate, see Fig. S7.†
Relaxivity data for Gd(III) coiled coils recorded for the first time at low field (1 T), shows a significant increase in the longitudinal relaxivity (r1) compared to data at high field (7 T), see Fig. S8† and Table 1. Relaxivity at this clinically more relevant field, r1 > 60 mM−1 s−1 (1 T), is highly competitive when compared to typical single digit values reported for small molecule clinical Gd(III) CAs.30
Despite differences in molecular weight and theoretical differences in tumbling rates, the experimental relaxivities for Gd(MB1-1)3 and Gd(MB1-1L)3 are the same (within error) at both low and high field strength (Fig. S8† and Table 1). At low field, slowing rotational correlation time is generally considered an effective strategy for enhancing relaxivity. Given that changes in tumbling on this timescale is not the limiting factor, a greater understanding of the Gd(III)-water coordination chemistry is thus required.
To determine the water exchange chemistry at the bound Gd(III), variable temperature 17O NMR studies were performed. The temperature dependence of the reduced transverse relaxation rates of Gd(MB1-1)3 and Gd(H2O)8 (aqueous GdCl3), show that both complexes operate in the fast water exchange regime (Fig. S9†). Fitting the number of inner sphere water molecules yielded values of 3.5 ± 0.6 and 7.7 ± 1.3 for the Gd(MB1-1)3 and aqua Gd(III) complexes, respectively. The former is in good agreement with the value obtained spectroscopically for the Tb(MB1-1)3 analogue, see Table 1.
Assuming three inner sphere waters for Gd(MB1-1)3, the fits to the Swift-Connick equation (see ESI†) yield the rate (6.4 × 10−8 s−1, giving a water residence time of 1.56 ns), enthalpy (ΔH‡ 18.0 ± 0.4 kJ mol−1) and entropy activation parameters (ΔS‡ −15.9 ± 1.3 J K−1 mol−1) for water exchange at the bound metal in Gd(MB1-1)3, see Table 2. Measuring kex as a function of pressure yields an activation volume, ΔV‡, of −0.5 ± 0.4 cm3 mol−1, Fig. S10.†
| Gd(H2O)3(MB1-1)3 | Gd(H2O)8 | |
|---|---|---|
| k 298ex (s−1) | 6.4 × 108 | 9.8 × 108 |
| ΔH‡ (kJ mol−1) | 18.0 ± 0.4 | 16.3 ± 0.4 |
| ΔS‡ (J K−1 mol−1) | −15.9 ± 1.3 | −18.0 ± 1.3 |
| ΔV‡ (cm3 mol−1) | −0.5 ± 0.4 | (−3.3 ± 0.2)31 |
| #H2OGd | 3.5 ± 0.6 | 7.7 ± 1.3 |
The water residency time was found to be short (1.56 ns) and of the same order of magnitude as that measured for free aqueous Gd(III) (1.02 ns). In contrast, coordination of various polyamine carboxylate ligands to Gd(III) can slow down the water exchange processes by up to two orders of magnitude (k298ex = 8 × 108 s−1 and k298ex ∼ 106 s−1 for the aqua and chelated complexes, respectively).30 However, the nature of the coordinating ligand has a huge impact. For example, the presence of more O− ligands, such as those provided by MB1-1, compared to N-donor atoms, labilises coordinated water molecules.31 In the case of the Gd(MB1-1)3 complex, the water exchange rates are only slightly slower than that of Gd(H2O)8. Not only is this much faster than for small molecule Gd(III) complexes, but it is also fast compared to a water residence time of 10 ns, reported for a Gd(III) binding site engineered onto the surface of a rat adhesion protein.11
The activation enthalpy for water exchange at the Gd(III) centre of Gd(MB1-1)3 is similar to that obtained for the Gd(H2O)8 control and significantly lower than those reported for other Gd(III) CAs (∼50 kJ mol−1).30 This, together with the negative values of activation entropy, indicates that the coordination sphere of Gd(III) in Gd(MB1-1)3 must be less crowded (max 8-coordinate) than in most small molecule chelated complexes.30 We propose that the water exchange processes at the Gd(MB1-1)3 complex follows an associatively activated interchange mechanism (Ia), which is in line with the obtained negative values of ΔS‡ and small negative value of activation volume.
NMR measurements also reveal that data can be best fit with scalar coupling constants of ∼1.5 MHz for Gd(MB1-1)3, compared to the lower values of 0.8 and ∼0.6 MHz for the free aqua and chelated Gd(IIII) complexes. This would be consistent with a shorter distance between the Gd(III) and the oxygen of exchanging water (rGd–O) for Gd(MB1-1)3 compared to the aqua and chelated small molecule Gd(III) complexes, further supporting a non-crowded Gd(III) coordination sphere.
This work for the first time explores the mechanism by which a new class of Gd(III) complexes, based on the coiled coil, achieve competitive MR relaxivity, when compared to clinical CAs. This is in part due to their large size and slow rotational correlation time. The coiled coil can be extended, to further slow the tumbling of the complex, without impacting on the coordination chemistry of the metal binding site. However, it is not a strategy by which relaxivity can be further enhanced. The MR relaxivity is achieved by rapid exchange of three inner sphere waters, through an associative mechanism, at a coordinatively unsaturated Gd(III) site. The water exchange rate is almost as fast as for free Gd(III). Predicting the impact of rotational correlation time (τR) on r1 for Gd(III) bound to three waters with a water residency time of 1.56 ns, is shown in Fig. S11,† and demonstrates that no significant improvement can be achieved by extending the coiled coil further. Relaxivity enhancement could hypothetically be achieved by increasing the water residency time. For example, the binding site could be redesigned by altering the sterics and hydrogen bonding opportunities presented by second coordination sphere residues.
The experimentally determined water exchange rate, and an understanding of the water coordination chemistry at a protein bound Ln(III), has important implications for the development of MRI contrast agents. It also provides important insight into the behaviour of native sites in Ln(III) biochemistry, a rapidly growing area of research. For example, though not clear if biologically relevant, the X-ray crystal structure of methylacidiphilum fumariolicum SolV methanol dehydrogenase (pdb 4MAE) was found to contain solvent coordinated to the bound Ce at the active site.22 Finally, to the best of our knowledge this is the first report of the water exchange rate at a M–OH2 site within a coiled coil, and as such could have important implications for metallo peptide and protein design more widely.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SLN gratefully acknowledges financial support from the EPSRC through a studentship from the PSIBS Doctoral Training Centre (EP/F50053X/1), and sponsorship from the Royal Society of Chemistry NMR Discussion Group. We thank the University of Birmingham and EPSRC grant EP/K039245/1 for support of this research. IIB and AF thank the DFG for supporting a high-pressure NMR facility through the programme Major Research Instrumentation as per Art. 91b GG as well as Ludwig-Maximilians-Universität München and its Faculty of Chemistry and Pharmacy.References
- C.-T. Yang, P. Padmanabhan and B. Z. Gulyás, RSC Adv., 2016, 6, 60945–60966 RSC.
- Q. Xu, L. Zhu, M. Yu, F. Feng, L. An, C. Xing and S. Wang, Polymer, 2010, 51, 1336–1340 CrossRef CAS.
- B. Sitharaman, K. R. Kissell, K. B. Hartman, L. A. Tran, A. Baikalov, I. Rusakova, Y. Sun, H. A. Khant, S. J. Ludtke, W. Chiu, S. Laus, É. Tóth, L. Helm, A. E. Merbach and L. J. Wilson, Chem. Commun., 2005, 3915–3917, 10.1039/B504435A.
- A. Servant, I. Jacobs, C. Bussy, C. Fabbro, T. da Ros, E. Pach, B. Ballesteros, M. Prato, K. Nicolay and K. Kostarelos, Carbon, 2016, 97, 126–133 CrossRef CAS.
- L. M. Manus, D. J. Mastarone, E. A. Waters, X.-Q. Zhang, E. A. Schultz-Sikma, K. W. MacRenaris, D. Ho and T. J. Meade, Nano Lett., 2010, 10, 484–489 CrossRef CAS PubMed.
- W. C. Floyd, P. J. Klemm, D. E. Smiles, A. C. Kohlgruber, V. C. Pierre, J. L. Mynar, J. M. J. Fréchet and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 2390–2393 CrossRef CAS PubMed.
- C. B. Sirlin, D. R. Vera, J. A. Corbeil, M. B. Caballero, R. B. Buxton and R. F. Mattrey, Acad. Radiol., 2004, 11, 1361–1369 CrossRef PubMed.
- T. Courant, V. G. Roullin, C. Cadiou, M. Callewaert, M. C. Andry, C. Portefaix, C. Hoeffel, M. C. de Goltstein, M. Port, S. Laurent, L. V. Elst, R. Muller, M. Molinari and F. Chuburu, Angew. Chem., Int. Ed., 2012, 51, 9119–9122 CrossRef CAS PubMed.
- E. A. Anderson, S. Isaacman, D. S. Peabody, E. Y. Wang, J. W. Canary and K. Kirshenbaum, Nano Lett., 2006, 6, 1160–1164 CrossRef CAS PubMed.
- G. J. Strijkers, W. J. M. Mulder, R. B. van Heeswijk, P. M. Frederik, P. Bomans, P. C. M. M. Magusin and K. Nicolay, Magn. Reson. Mater. Phys., Biol. Med., 2005, 18, 186–192 CrossRef CAS PubMed.
- J. J. Yang, J. Yang, L. Wei, O. Zurkiya, W. Yang, S. Li, J. Zou, Y. Zhou, A. L. W. Maniccia, H. Mao, F. Zhao, R. Malchow, S. Zhao, J. Johnson, X. Hu, E. Krogstad and Z.-R. Liu, J. Am. Chem. Soc., 2008, 130, 9260–9267 CrossRef CAS PubMed.
- P. Caravan, J. M. Greenwood, J. T. Welch and S. J. Franklin, Chem. Commun., 2003, 2574–2575, 10.1039/B307817E.
- A. S. Krishnan, A. A. Neves, M. M. de Backer, D.-E. Hu, B. Davletov, M. I. Kettunen and K. M. Brindle, Radiology, 2008, 246, 854–862 CrossRef PubMed.
- M. R. Berwick, D. J. Lewis, A. W. Jones, R. A. Parslow, T. R. Dafforn, H. J. Cooper, J. Wilkie, Z. Pikramenou, M. M. Britton and A. F. A. Peacock, J. Am. Chem. Soc., 2014, 136, 1166–1169 CrossRef CAS PubMed.
- M. R. Berwick, L. N. Slope, C. F. Smith, S. M. King, S. L. Newton, R. B. Gillis, G. G. Adams, A. J. Rowe, S. E. Harding, M. M. Britton and A. F. A. Peacock, Chem. Sci., 2016, 7, 2207–2216 RSC.
- L. N. Slope, M. G. Hill, C. F. Smith, P. Teare, F. J. de Cogan, M. M. Britton and A. F. A. Peacock, Chem. Commun., 2020, 57, 3729–3732 RSC.
- A. M. Webster and A. F. A. Peacock, Chem. Commun., 2021, 57, 6851–6862 RSC.
- Y. Hibi, K. Asai, H. Arafuka, M. Hamajima, T. Iwama and K. Kawai, J. Biosci. Bioeng., 2011, 111, 547–549 CrossRef CAS PubMed.
- E. Skovran and N. C. Martinez-Gomez, Science, 2015, 348, 862 CrossRef CAS PubMed.
- L. J. Daumann, Angew. Chem., Int. Ed., 2019, 58, 12795–12802 CrossRef CAS PubMed.
- E. R. Featherston and J. A. Cotruvo, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868, 118864 CrossRef CAS PubMed.
- A. Pol, T. R. M. Barends, A. Dietl, A. F. Khadem, J. Eygensteyn, M. S. M. Jetten and H. J. M. Op den Camp, Environ. Microbiol., 2014, 16, 255–264 CrossRef CAS PubMed.
- D. Ghosh, K.-H. Lee, B. Demeler and V. L. Pecoraro, Biochemistry, 2005, 44, 10732–10740 CrossRef CAS PubMed.
- P. Teare, C. F. Smith, S. J. Adams, S. Anbu, B. Ciani, L. J. C. Jeuken and A. F. A. Peacock, Dalton Trans., 2018, 47, 10784–10790 RSC.
- L. N. Slope, O. J. Daubney, H. Campbell, S. A. White and A. F. A. Peacock, Angew. Chem., Int. Ed., 2021, 60, 24473–24477 CrossRef CAS PubMed.
- W. D. Kohn, C. M. Kay and R. S. Hodges, J. Pept. Res., 1998, 51, 9–18 CrossRef CAS PubMed.
- W. D. Kohn, C. M. Kay, B. D. Sykes and R. S. Hodges, J. Am. Chem. Soc., 1998, 120, 1124–1132 CrossRef CAS.
- A. Kashiwada, K. Ishida and K. Matsuda, Bull. Chem. Soc. Jpn., 2007, 80, 2203–2207 CrossRef CAS.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- D. H. Powell, O. M. N. Dhubhghaill, D. Pubanz, L. Helm, Y. S. Lebedev, W. Schlaepfer and A. E. Merbach, J. Am. Chem. Soc., 1996, 118, 9333–9346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt02909c |
| This journal is © The Royal Society of Chemistry 2023 |