
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaterials engineering strategies for cancer vaccine adjuvant development
Xuanbo
Zhang
ab,
Bowei
Yang
a,
Qianqian
Ni
*acd and
Xiaoyuan
Chen
 *acde
*acde
aDepartments of Diagnostic Radiology, Surgery, Chemical and Biomolecular Engineering, and Biomedical Engineering, Yong Loo Lin School of Medicine and College of Design and Engineering, National University of Singapore, Singapore 119074, Singapore. E-mail: qqian.ni@nus.edu.sg; chen.shawn@nus.edu.sg
bDepartment of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang, Liaoning 110016, China
cClinical Imaging Research Centre, Centre for Translational Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117599, Singapore
dNanomedicine Translational Research Program, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Singapore
eInstitute of Molecular and Cell Biology, Agency for Science, Technology, and Research (A*STAR), 61 Biopolis Drive, Proteos, Singapore, 138673, Singapore
First published on 4th April 2023
Abstract
Cancer vaccines have emerged as a powerful new tool for cancer immunotherapy. Adjuvants are vaccine ingredients that enhance the strength, velocity, and duration of the immune response. The success of adjuvants in achieving stable, safe, and immunogenic cancer vaccines has generated enthusiasm for adjuvant development. Specifically, advances in materials science are providing insights into the rational design of vaccine adjuvants for topical cancer immunotherapy. Here, we outline the current state of materials engineering strategies, including those based on molecular adjuvants, polymers/lipids, inorganic nanoparticles, and bio-derived materials, for adjuvant development. We also elaborate on how these engineering strategies and the physicochemical features of the materials involved influence the effects of adjuvants.
 Xuanbo Zhang | Xuanbo Zhang is in pursuit of his PhD in Shenyang Pharmaceutical University, under the supervision of Prof. Cong Luo and Prof. Jin Sun. He is currently a joint PhD student in the Yong Loo Lin School of Medicine, National University of Singapore, under the supervision of Prof. Xiaoyuan (Shawn) Chen and Dr Qianqian Ni. His research interests focus on adjuvants, vaccine design and mRNA delivery. |
 Bowei Yang | Bowei Yang is now a PhD student in the Yong Loo Lin School of Medicine, National University of Singapore, under the supervision of Dr Qianqian Ni and Prof. Xiaoyuan (Shawn) Chen. His research interests focus on mRNA delivery and immunotherapy. |
 Qianqian Ni | Dr Qianqian Ni received her PhD degree from an 8 year clinical medicine program at Nanjing University in 2018. After postdoctoral training in the Laboratory of Molecular Imaging and Nanomedicine at the National Institutes of Health (NIH), she joined the National University of Singapore (NUS) as Assistant Professor in the Department of Diagnostic Radiology in 2021. Her major research interest is devoted to the development of nucleic acid nanomedicines for DNA/RNA delivery and cancer immunotherapy. |
 Xiaoyuan Chen | Prof. Xiaoyuan (Shawn) Chen received his PhD degree in chemistry from the University of Idaho (1999). After being a faculty at the University of Southern California and Stanford University and then Senior Investigator/Lab Chief at the National Institutes of Health, he is now Nasrat Muzayyin Professor in medicine and technology, Yong Loo Lin School of Medicine and Faculty of Engineering, National University of Singapore. His current research interests are mainly theranostics (radiotheranostics, nanotheranostics, immunotheranostics, magnetotheranostics, phototheranostics, etc.) that are clinically translatable. He has published over 900 papers and numerous books (H-index = 178). |
Key learning points1. Physicochemical engineering of molecular adjuvants could improve targeted delivery and elicit a robust immune response with minimal toxic side effects.2. Synthetic self-adjuvanting materials could coordinate with subunit antigens to augment adaptive antitumor immunity with a simplified vaccine manufacturing process. 3. Intrinsic physicochemical features such as the topological characteristics of inorganic nanoparticles could be harnessed to develop novel and more effective cancer vaccine adjuvants. 4. Bio-derived materials obtained via genetic or chemical manipulation provide safer and potent vaccine adjuvants in cancer immunotherapy. |
1. Introduction
In recent years, cancer immunotherapy has become a feasible strategy for cancer treatment.1 Among various types of cancer immunotherapies, cancer vaccines, which leverage tumor antigens and adjuvants to cultivate the immune system for cancer cell identification and elimination, are emerging as a promising approach.2 As a major component of a cancer vaccine, the adjuvant primarily enhances the strength, velocity and durability of the immune response to the vaccine.3,4 The concept of adjuvants was proposed in 1925, when Gaston Ramon discovered that sterile additives can increase antibody production in animals.3,5 In the following year, Alexander Glenny first reported that aluminum salt-precipitated diphtheria toxoid induced stronger immune potency, demonstrating the adjuvant effect of aluminum salts in immunotherapy.3,6–8 Driven by vaccine adjuvants, cancer vaccines work via the processing and presentation of tumor antigens in antigen-presenting cells (APCs) followed by the activation of antigen-specific T cells to recognize and kill cancer cells.9,10 Although several aluminum adjuvants have been approved by the US Food and Drug administration (FDA) since 1939, the development of adjuvants has been exceptionally slow.5 To date, only a few adjuvant ingredients such as MF59 and AS04 have been approved (Table 1), with numerous other adjuvants still in early-phase clinical or pre-clinical studies.3| Adjuvant | Composition | Representative vaccines |
|---|---|---|
| These data were collected from: https://www.cdc.gov/vaccinesafety/concerns/adjuvants.html. | ||
| Aluminum | Aluminum hydroxide, potassium aluminum sulfate | Anthrax, HepB (Engerix-B), HPV (Gardasil 9) |
| AS01B | Monophosphoryl lipid A (MPL) and QS-21 | Zoster vaccine (Shingrix) |
| AS04 | MPL + aluminum salt | Human papillomavirus, HPV (Cervarix) |
| CpG 1018 | Cytosine phosphoguanine (CpG) | HepB (Heplisav-B) |
| Matrix-MTM | Saponins | COVID-19 vaccine (Novavax COVID-19 Vaccine) |
| MF59 | Oil in water emulsion composed of squalene | Influenza (Fluad and Fluad Quadrivalent) |
One of the major challenges that hinder cancer vaccine development is the low immunogenicity of tumor antigens; tumor-associated antigens (TAAs) are likely to induce T-cell central tolerance, while neoantigens derived from mutated epitopes are patient specific and difficult to identify.11 Adjuvants that elicit effective immune responses in cancer immunotherapy have thus been pursued. Unlike traditional licensed preventive vaccines, in which adjuvants target well-defined foreign antigens, adjuvants in therapeutic cancer vaccines are required to boost the desired immune response to weak tumor antigens.12 Adjuvants can typically be classified into two categories, vehicles (e.g., emulsions and liposomes) and immunostimulants (e.g., cytokines and bacterial exotoxins), according to their functions. In cancer vaccines, adjuvants can be added to the vaccine formulation and co-administered with tumor antigens for delivery; the adjuvant material will then assist subunit tumor antigens in inducing robust and durable innate and adaptive immunity as well as antigen-specific immune responses (Fig. 1).9,11,13 Alternatively, the local immune microenvironment of tumor tissues could be broadly activated to generate tumor cell death; in this emerging idea, in situ vaccination would facilitate the availability of tumor antigens.10 In this way, adjuvant-activated APCs can leverage antigens from dying tumor cells after cancer treatment (e.g., conventional radiotherapy, chemotherapy, or oncolytic virus treatment) to provoke cytotoxic T lymphocyte (CTL) responses and subsequently lead to cell death (Fig. 1).10,14 In fact, such an in situ cancer vaccine strategy has been widely studied and is also known as immunogenic cell death (ICD)-based cancer treatment. This strategy pursues the highest level of exposure to immunogenic substances such as calreticulin and ATP to activate local anti-tumor immunity in the process of killing tumor cells, thereby achieving a better tumor treatment effect and immune memory.15 However, due to the limited immunogenicity of in situ vaccines, the development of adjuvants in cancer immunotherapy has largely relied on novel and functional adjuvant materials that enable targeted delivery to tumor tissues and strengthen the immunogenicity of the tumor microenvironment (TME) via the in situ activation of immune cells.11,14
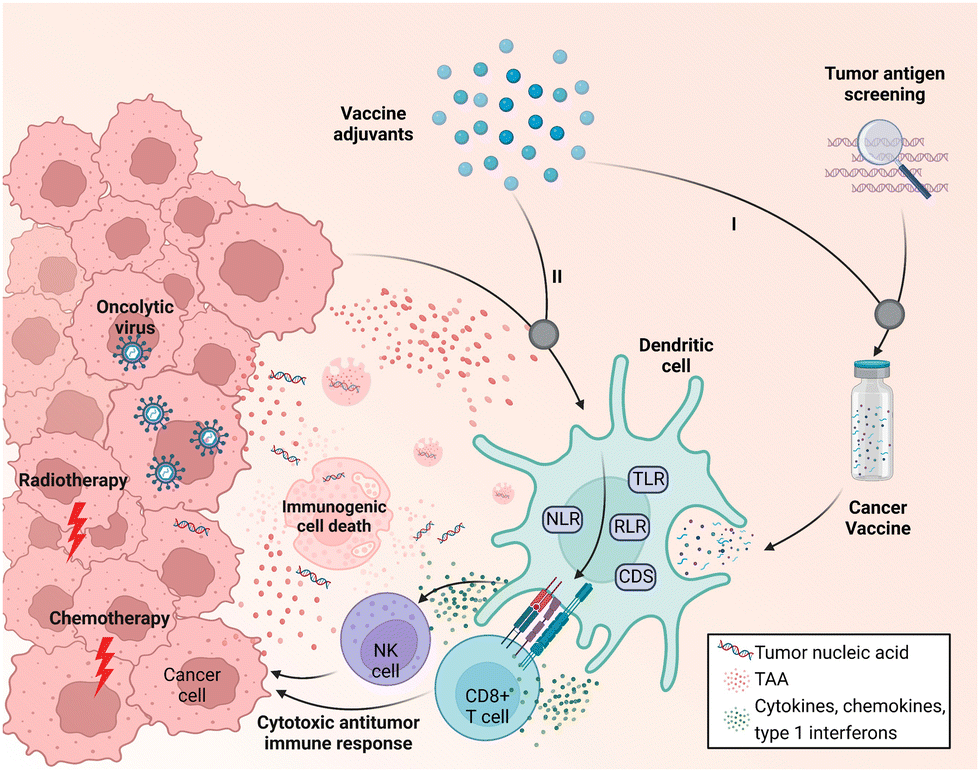 | ||
| Fig. 1 Schematic of the two working modes of cancer vaccine adjuvants. (I) Adjuvants are additives in the vaccine formulation. (II) Adjuvants induce in situ immune activation together with tumor antigens produced by immunogenic cell death resulting from cancer treatment (radiotherapy, chemotherapy, and oncolytic virus treatment). Adapt with permission from ref. 16, Mount Sinai Health System. | ||
A variety of material platforms with adjuvant effects have been exploited to trigger anti-tumor immune responses. In terms of the mechanism of action, the role of the adjuvant material is to mimic pathogen invasion or cell damage, which will transmit a danger signal to the innate immune system; this signal is then recognized by pattern recognition receptors (PRRs) as pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs).16–18 PAMPs reflect the invasion of the body by pathogens, causing microbe-derived components (e.g., lipopolysaccharides and flagellin derived from bacteria) to activate PPRs on innate immune cells to alert the immune system and elicit pathogen clearance and immune memory. DAMPs, which are self-derived substances (e.g., calreticulin, high mobility group box 1, and uric acid) released due to tissue injury or cell death, can also be recognized by PRRs and activate innate immune cells.3,19 There are five families of PRRs: toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), RIG-I-like receptors (RLRs), and cytoplasmic DNA sensors (CDs).14,20,21 PRRs dimerize upon binding their cognate ligands (Fig. 2), leading to conformational changes that allow the subsequent recruitment of adaptor molecules [e.g., toll-interleukin-1 receptor (TIR) domain-containing adaptor protein; TIR-domain-containing adapter-inducing interferon-β; myeloid differentiation primary response 88 (MyD88); and TRIF-related adaptor molecule] and the upregulation of genes related to inflammatory response. Taking the most widely studied TLR as an example, once it is activated by adjuvants such as lipopolysaccharides (LPS) and flagellin, TLR stimulates the downstream signal through the MyD88 pathway.3 MyD88 first interacts and forms a complex with interleukin-1 receptor-associated kinase 4 (IRAK-4). The MyD88/IRAK-4 complex recruits IRAK-1 and IRAK-2, resulting in the phosphorylation of IRAKs. The IRAKs then mediate the translocation of TRAF6 from the membrane to the cytosol and activate transforming growth factor β-activated kinase 1 (TAK-1) and TGF-beta-activated kinase 1 binding protein 2/3 (TAB2/3). Consequently, IkappaB (IκB) and mitogen-activated protein kinase (MAPK) are activated, which finally results in the translocation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein-1 (AP-1) and the production of cytokines and interferons.16
 | ||
| Fig. 2 Molecular targets and downstream signaling pathways of adjuvants. The production of proinflammatory cytokines and type I interferon (IFN-I) is upregulated when PRRs are recognized by respective ligands. The downstream signaling pathway is then stimulated. IRAK: interleukin (IL)-1 receptor-associated kinase; TRAF: tumor necrosis factor receptor-associated factor; TAK: transforming growth factor-β-activated kinase; TAB: transforming growth factor-β-activated kinase 1-binding protein; NFκB: nuclear factor-κB; and MAPK: mitogen-activated protein kinase. Adapted with permission from ref. 16, Mount Sinai Health System. | ||
Despite the great achievements in vaccines against infectious diseases, the application of therapeutic vaccines in cancer treatment is far from mature. To date, only the dendritic cell vaccine, sipuleucel-T, has been approved for cancer immunotherapy.10 To overcome the challenges resulting from poor antigen availability, researchers have attempted to leverage adjuvant systems to evoke effective anti-tumor immunity in the existing antigens.10 The concept of adjuvants has evolved along with our in-depth understanding of the human immune system and the development of vaccine technology. It is clear now that classic licensed adjuvants can no longer meet the requirements of current cancer immunotherapy due to their low efficiency and limited applicability to cancer antigens, especially nucleic acid antigens.3,22 Recently, molecular PRR agonists, self-adjuvanting polymer/lipid materials, inorganic materials designed to have specific physiochemical properties, and bio-derived materials have been widely explored for the development of cancer vaccine adjuvants and exhibit superior therapeutic performance over traditional adjuvants. However, the clinical application of adjuvant materials for therapeutic purposes is still restricted. Here, we define four key challenges in the development and application of cancer vaccine adjuvants: (1) improving the delivery of PRR agonists to APCs to induce effective anti-tumor immune responses and minimize potential toxic side effects; (2) obtaining synthetic adjuvant systems that effectively deliver antigens and simplify the vaccine manufacturing process; (3) determining how the physicochemical features of adjuvant materials affect their immune activity and how to modulate these characteristics to manipulate the immunogenicity; and (4) developing bio-derived materials through genetic or chemical engineering approaches to make safe and potent adjuvants for next-generation cancer vaccines.
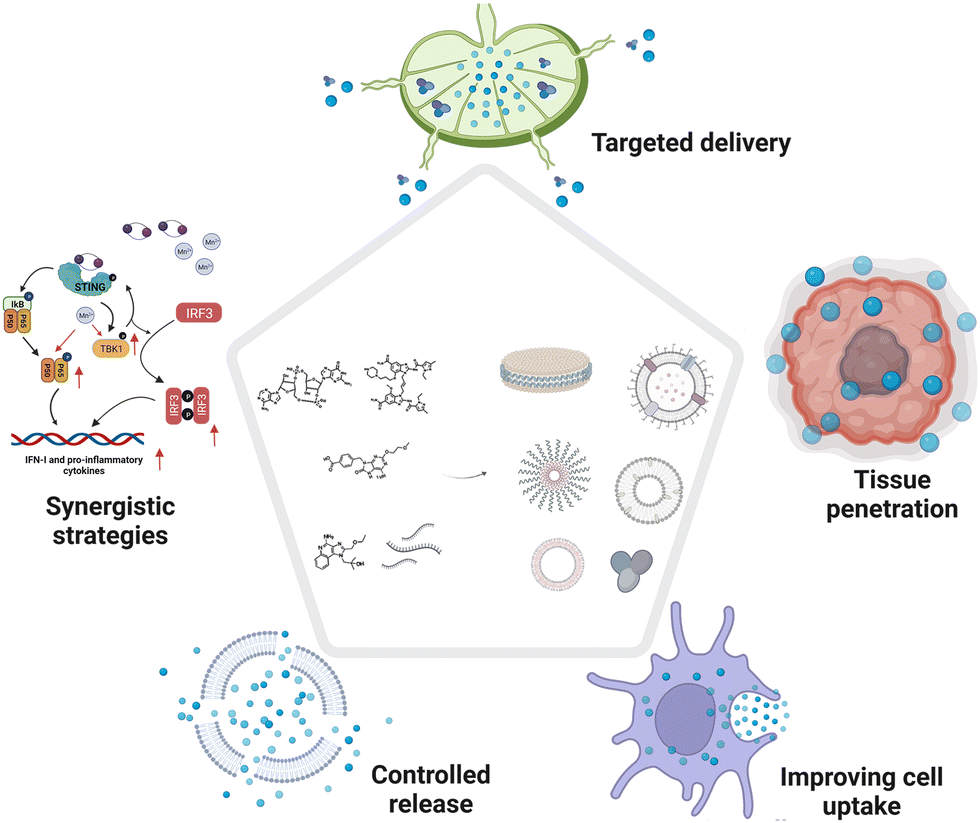
In this tutorial review, we (1) summarize innovations in materials engineering techniques to improve the delivery and augment the potency of cancer vaccine adjuvants, and (2) deliberate on how delivery approaches, molecular mechanisms, topography effects, multivalent interactions, and biomimetic technologies will be involved in adjuvant design (Fig. 3). Furthermore, we discuss the safety challenges, opportunities, and clinical translation potential of these novel cancer vaccine adjuvants.
 | ||
| Fig. 3 Overview of materials engineering strategies in cancer vaccine adjuvant development. This review describes (1) engineered molecular immune agonists, (2) synthetic self-adjuvanting materials, (3) organic adjuvants with versatile physicochemical functions, and (4) genetically or chemically engineered bio-derived adjuvants. | ||
2. Physicochemically engineered molecular immune agonists
Adjuvants in traditional vaccines are mostly complex mixtures, and their molecular mechanisms are not fully understood; this remains the major obstacle hindering the rational optimization and development of new vaccine adjuvants.12,23 In preclinical studies, a variety of molecular PRR agonists have been used to enhance vaccine potency. Due to the widespread distribution of PRR receptors, the systemic administration of molecular PRR receptor agonists usually causes severe systemic immunotoxicity, which restricts the translation of these molecular agonists from the bench to the bedside.3,17 For example, the rapid diffusion of small-molecule TLR7/8 agonists (imiquimod and resiquimod) induced severe systemic inflammatory responses when administered locally.23 In addition, some natural agonists have very limited in vivo applications due to their unfavorable physicochemical features. For example, cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), a natural product of cGAS (cyclic GMP-AMP synthase) in mammalian cells, encounters low cellular internalization and enzymatic instability due to the net negative charge and hydrolysis of the phosphodiester linkage.24 The rational design of PPR agonists in vaccines is thus needed to optimize their pharmaceutical behavior and amplify the potency of PRR agonists while minimizing side effects.Among the five families of PRRs, molecular agonists that activate TLRs and CDs have been studied extensively in cancer immunotherapy owing to their clearly defined pharmacological mechanisms. Various chemical engineering approaches have been developed to improve the adjuvant activity and therapeutic effects while reducing the side effects associated with systemic inflammatory response to achieve targeted delivery, improved cellular internalization, programmable release, and synergistic therapeutic effects (Fig. 4 and Table 2). It is evident that the precise accumulation of PRR molecular agonists in lymphatic organs or tumor tissues will maximize the activation of local immunity while reducing off-target toxicity.25 Apart from the delivery efficiency, the synergistic effect induced by the co-delivery of multiple PRR agonists is critical to elicit robust anti-tumor immune responses.26–28 Specifically, metal ions have been identified as a new type of molecular adjuvant and are formulated within cancer vaccines to potentiate immune responses.29,30 In this section, we discuss the engineering strategies applied in molecular agonist-based adjuvant design and future prospects for the rational design of adjuvants for cancer immunotherapy.
 | ||
| Fig. 4 Engineering strategies applied in molecular agonist-based adjuvant design. | ||
| PRRs | Subsets | Agonists | Adjuvant designs |
|---|---|---|---|
| TLRs | TLR3 | dsRNA | Assembled into a pH-responsive DNA nanodevice31 |
| TLR4 | MPLA | Formulated in nanodiscs with CpG32 | |
| TLR7 | 1V209 | Cho-1V209 conjugations formulated in liposomes33 | |
| TLR7/8 | Resiquimod (R848) | Lipid-R848 conjugate formulated in liposomes or polymeric nano-suspension34 | |
| Imidazoquinoline | Enzyme-responsive polymer-agonist conjugates assembled in vesicles;20 peptide-agonists self-assemble into nanoparticles (NPs)35 | ||
| TLR9 | CpG | CpG loops assembled into a pH-responsive DNA nanodevice;31 Cho-CpG conjugations formulated in nanodiscs;36 lipid–CpG conjugates;37 EB–CpG conjugations;38 CpG–vitamin E conjugations self-assemble into spherical nucleic acids39 | |
| CDs | cGAMP | Encapsulated into endosomolytic polymersomes40 or cubic PLGA microparticles;41 binding on ultrasound-responsive microbubbles42 | |
| DMXAA | Grafted onto acid-activatable polymers and encapsulated in micellar NPs43 | ||
| ADU-S100 (CDA) | Coordinating with Mn2+ and self-assemble into lipid polymers;44 conjugated to PEGylated lipids and incorporated into nanodiscs24 | ||
| NLRs | NOD2 | Mifamurtide | Intercalated into the phospholipid bilayer of a liposomal vector45 |
2.1 Targeted delivery strategies in molecular PRR agonist-based adjuvant design
Lymph nodes (LNs) are composed of lymphoid tissue and lymphatic sinuses; dendritic cells (DCs) are usually found in the medulla, while T cells and B cells are mainly located in the lymphatic sinuses.46 LNs provide a structural support and contact field for DCs, macrophages, and lymphocytes for tumor antigen processing and humoral or cellular immune activation.37 Therefore, the preferential accumulation of PRR agonists in draining LNs can effectively activate the anti-tumor immune responses of immune cells in the lymph nodes.46 In the past decades, two main strategies have emerged for the rational design of adjuvants for targeted LN delivery: (1) programming the properties (e.g., size and shape) of adjuvants to passively target LNs; and (2) chemically modifying PRR agonists to hitchhike on natural LN-targeting molecules (e.g., albumin) to enable active LN-targeted delivery.Due to their filter-like structures, LNs can specifically intercept particles with certain sizes, which enables passive targeting.25 Generally, particles with sizes ranging from 5 to 200 nm can effectively migrate through the tissue fluid and lymphatic fluid to accumulate in the LNs (Fig. 5). Specifically, NPs with sizes of 5–15 nm prefer to localize in follicular dendritic cells and are quickly cleared within 48 h, while particles with sizes of 50–100 nm can remain in the LNs for over five weeks.47 Moreover, when the particle size is above 500 nm, the administered NPs will be trapped locally in the subcutaneous tissues and will only reach the LNs when taken up by phagocytic cells.25 Based on the size-dependent passive targeting principle, PRR agonists have been engineered into versatile nanoformulations for LN-targeted delivery. For example, some lipids or phosphate groups with intrinsic properties that allow them to bind to endogenous biomolecules (IgG, albumin, etc.) have been conjugated to PRR agonists to drive the self-formation of liposomes, lipoproteins, and aluminum particles via hydrophobic or electrostatic interactions. This chemical engineering strategy is an effective approach to obtain LN-targeting adjuvants without adverse effects.23,33,36 In addition to particle size, other physicochemical properties such as surface charge and particle stiffness also affect the passive targeting of LNs.25 Positively charged lipid nanoparticles (LNPs) have been reported to adsorb easily on local tissues, and modification with polyethylene glycol (PEG) can improve the mobility of the LNPs and prolong their retardation in lymphoid tissues for efficacious cancer immunotherapy.25
 | ||
| Fig. 5 Schematic of the LN structure and design principle of vaccines for passive targeting. NPs larger than 500 nm will be trapped locally in the subcutaneous tissues. NPs ranging from 5 to 200 nm in size can effectively accumulate in the LNs. Albumin-hitchhiking molecules or NPs can passively migrate and accumulate in the LNs.25 | ||
Albumin hitchhiking is another delivery strategy to obtain PRR agonists that passively target LNs.37 Albumin, which accounts for nearly 50–60% of total proteins in the blood and 30% of total tissue fluid proteins, usually has a long half-life in circulation.48 The high albumin concentration in blood and the large pressure difference between blood and interstitial fluid will force the migration of albumin from the interstitial fluid into lymphoid tissue.38 In addition, albumins are inclined to be drained to LNs due to their specific size (66 kDa), which exceeds the cutoff of dissemination from interstitial fluid. When PRR agonist molecules bind with albumins, whether in a covalent or non-covalent manner, they will acquire the natural properties of albumins, providing an attractive alternative for LN delivery.38 Structurally, albumin has multiple hydrophobic binding domains for interacting with lipophilic or amphiphilic molecules, including some hydrophobic molecules (paclitaxel),49 lipophilic dyes (indocyanine green),50 and fatty acids with long aliphatic chains (diacyl lipid).37 These non-covalent interactions can be tailored to the effective transport of PRR agonists to LNs. Irvine et al. first proposed the concept of “albumin hitchhiking” and applied it to the delivery of unmethylated cytosine–guanine dinucleotide (CpG) oligonucleotide adjuvants. In their study, CpG oligonucleotides were chemically conjugated with cholesterol, monoacyl lipid, and diacyl lipid. They found that when modified with the diacyl lipid, the lipid–CpG conjugates (Lipo–CpG) formed an amphiphilic structure with adequate endogenous albumin binding affinity. The subcutaneous administration of these Lipo–CpG conjugates resulted in remarkable LN accumulation compared with free CpG. The authors also demonstrated that the increased accumulation was largely a result of albumin binding rather than the self-assembled Lipo–CpG conjugates. It is worth noting that the Lipo–CpG conjugates dramatically outperformed free CpG in cancer immunotherapy, resulting in a 30-fold enhancement in T-cell priming response while diminishing systemic toxicity.37
Evans Blue (EB) is a typical albumin-binding dye.51 Inspired by its high binding capacity, our group developed a series of EB-based drug delivery platforms, including small-molecule drugs, therapeutic peptides or nucleic acids, and radionuclides for diagnostic imaging and radionuclide therapy.38,52,53 By coupling the CpG oligonucleotide with a truncated EB molecule, we developed a nanovaccine platform termed AlbiCpG. By controlling the length of the PEG linker between the CpG and EB molecules, a mini library of EB–CpG conjugates was established with discernible differences in albumin binding affinity and adjuvant potency. Both in vivo PET pharmacoimaging and ex vivo flow cytometry revealed the efficient LN delivery of the resulting AlbiCpG nanocomplexes (Fig. 6A). The co-administration of AlbiCpG together with EB-conjugated antigen peptides (termed Albivax) resulted in a nearly 100-fold increase in LN and LN-derived APC internalization compared to the traditional incomplete Freund's adjuvant, resulting in potent and durable antigen-specific anti-tumor T-cell responses with minimal inflammation-related side effects. Furthermore, the therapeutic outcome in multiple mouse tumor models verified that Albivax provides a platform for personalized neoantigen-based cancer immunotherapy.38
 | ||
| Fig. 6 Design principles of molecular PRR agonist-based adjuvants for targeted delivery, tumor penetration and enhanced APC uptake. (A) Chemical structure of MEB and schematic of the structure of albumin/MEB nanocomplexes (left panel) and working mechanism of albumin/AlbiVax nanocomplexes for LN targeting (right panel). Reproduced with permission from ref. 38, Copyright 2017, The Author(s). (B) STING agonist delivery by tumor-penetrating PEG-lipid nanodiscs: (a) chemical structure of the CDN-PEG-Lipid; (b) components and structure of LNDs; (c and d) results of molecular dynamics simulations of LNDs and liposomes passing through biological barriers with different pore sizes; (e) confocal microscope images of fluorescent LNDs and liposomes penetrating into tumor spheroids; and (f) LND and liposome penetration represented in tumor tissue sections harvested from mice treated with fluorescent LNDs or liposomes. Reproduced with permission from ref. 24, Copyright 2022, The Author(s). (C) Schematic of polymeric TLR-7/8 agonists with low and high conjugation densities and their aggregation states. (D) Preparation and structure of ncMBs and the process of cGAMP in ncMBs delivered into the cytosol of the APCs induced by sonoporation. | ||
2.2 Tumor tissue penetration in molecular PRR agonist-based adjuvant design
Nanomedicine has been validated to play a critical role in treating human diseases, especially cancers. However, the delivery of nanodrugs to tumors is hampered by several biological barriers, including abnormal tumor vasculature, interstitial fluid pressure (IFP), and dense extracellular matrix (ECM).54 The abnormal tumor microvasculature drastically diminishes the supply of oxygen and nutrients, especially in the core regions of solid tumors. In addition, the IFP increases when moving from the margins to the center regions, restricting the diffusion of NPs to the central tumor areas. Moreover, the ECM also blocks the access of NPs to solid tumor tissues.54 To improve the penetration of NPs, many engineering strategies have been investigated for NP development, including modulating the topographies of NPs and regulating the TME by, for example, using hyaluronidase to destroy hyaluronic acid and regulate the tumor ECM. Nevertheless, the further application of these approaches has been restricted by the tedious manufacturing processes of delivery systems and the complicated and dynamic TME.54Leveraging adjuvants that activate PRRs locally in the tumor immune microenvironment will provide a new way to potentiate effective antitumor immunity. However, enhancing the tumor penetration of adjuvants to achieve therapeutic doses remains a challenge. Recently, properly designed nanotechnologies have been exploited to improve adjuvant penetration and distribution to tumor tissues, with lipid nanodiscs and nanoscale coordination polymers developed as delivery vehicles.24,55 Dane et al. incorporated stimulator of interferon genes (STING)-activating cyclic dinucleotides (CDNs) into lipid nanodiscs (LNDs) by conjugating the PEGylated lipid with CDNs using a cleavable dialanine peptide linker (Fig. 6B). Molecular dynamics simulations demonstrated the superior tumor permeation ability of the LNDs compared with state-of-the-art PEGylated liposomes, which can likely be attributed to the suitable elasticity and deformation properties of the LNDs. Correspondingly, pharmacokinetic profiling demonstrated that the LNDs outperformed liposomes in tumor penetration in both a tumor spheroid model and in vivo animal tumor models; a single LND treatment also induced robust T-cell activation to effectively eliminate the established tumors.24 Yang et al. developed a tumor-targeting nanosystem using zinc cyclic di-AMP nanoparticles (ZnCDA), consisting of non-toxic zinc phosphate and a PEG-conjugated phospholipid with cyclic dimeric adenosine monophosphate (CDA) loaded in the hydrophilic core. The administration of ZnCDA prolonged the circulation and tumor accumulation of CDNs by disrupting the tumor vasculature. In two immunologically “cold” cancer models, ZnCDA elicited effective anti-tumor immunity. These features of ZnCDA make it a promising nanoplatform for the combination immunotherapy of intractable human cancers.55
2.3 Improving cell uptake in molecular PRR agonist-based adjuvant design
Some PRRs, including TLRs 1, 2, 4, 5, and 6, and CLRs are known to locate on the outer cell membrane, while more PRRs that sense intracellular changes are located inside the cell cytoplasm (Fig. 2). For example, TLRs 3, 7, 8, and 9 are present in endosomes. Other PRRs detected inside the cytoplasm include cGAS-STING, NLRs, and RLRs. For these cytosolic PRRs, agonists can work only after their internalization by target cells.3 Given that the cell internalization of molecular agonists usually relies on a simple diffusion mechanism, which is less efficient and can be easily influenced by the lipophilic properties of the agonist molecules, improving the cellular uptake of the cytosolic or endosomal targeting agonists (e.g., CpG and cGAMP) remains a challenge for the rational design of adjuvants. For most hydrophobic agonists, various nanoplatforms have emerged with satisfactory loading and delivery efficiency, including polymer-, liposome-, and lipid-based nanoparticles.33,36,56 Seder's group has been devoted to TLR-7/8 agonist (TLR-7/8a) research and development.34,35 Strikingly, they developed an adjuvant (Poly-7/8a) platform by conjugating the classic TLR-7/8a, imidazoquinoline, on a biocompatible and hydrophilic N-(2-hydroxypropyl) methacrylamide (HPMA) polymer-based scaffold (Fig. 6C). When evaluating the effects of the physicochemical properties of TLR-7/8a and its derivatives on in vivo immune activation, the authors found that increasing the hydrophobicity of the TLR agonist led to structural changes of the polymers.35 Generally, the polymers remained individual swollen coils at low levels of TLR agonist conjugation; upon increasing the TLR agonist density, the polymer chains underwent self-aggregation to form submicron-sized polymer particles. Notably, compared with the individual polymer chains, the polymer particles had impressively higher uptake in LNs, thereby inducing a large amount of antibody production and strong T-cell priming. The researchers further investigated the difference in cell uptake between the polymer particles and individual polymer chains, which might result from the distinctive endocytosis mechanisms of APCs. They found that individual polymer chains were more likely to be internalized through pinocytosis, while the assembled polymer particles may trigger the more efficient phagocytosis in APCs.35Hydrophilic nucleic acids/nucleotide derivatives including CpG oligonucleotides and cyclic dinucleotide-based STING agonists represent another important class of agonists.57 However, these agonists are hindered by poor cellular uptake.58 While cationic carriers are effective tools for the intracellular delivery of nucleic acid agonists, their application is typically limited by cellular toxicity or immune response-related adverse events.58 Recently, a carrier-free spherical nucleic acid (SNA) platform was developed for effective in vivo nucleic acid delivery. Chen et al. designed self-assembled nanoparticles by linking CpG with a poly(vitamin E) segment. The poly(vitamin E) tail provided a hydrophobic driving force and synergistically amplified the immune response of CpG owing to the inherent adjuvant effect of vitamin E.59 Interestingly, although no carrier is involved in this system, the self-assembled SNA achieved rapid cellular uptake into endocytic compartments, and CpG was finally delivered through caveolae/lipid raft-dependent endocytosis with an approximately 23-fold increase in efficiency.26,39
Moreover, some clinically approved techniques such as ultrasound have been applied to increase the uptake of PRR agonists. When microbubbles are bound to the APC surface and exposed to ultrasound, they create transient pores on the APC membrane for cargo delivery. Accordingly, Li et al. designed nanocomplex-conjugated microbubbles (ncMBs) and decorated their surfaces with biocompatible spermine-modified dextran (SpeDex) and anti-CD11b antibodies (Fig. 6D). cGAMP could efficiently bind on the ncMBs through electrostatic adsorption mediated by positively charged SpeDex. Modification with anti-CD11b endowed the ncMBs with APC targeting ability. In vitro studies showed that the ncMBs could accurately bind with CD11b+ THP-1 human macrophages, while CD11b− EO771 breast cancer cells exhibited weak binding ability. Under sonoporation at 1 MHz, the amount of CDNs internalized by ncMB-treated macrophages was four times higher than that in the free CDN-treated group. In murine breast cancer models, the ncMBs delivered seven times as many CDNs to tumor-associated CD11b+ cells compared with non-targeted IgG-ncMB, while the non-specific uptake of CDNs in CD11b− cells was negligible. These findings reveal that this ultrasound-guided delivery platform provides a robust strategy to overcome the limitation of inefficient CDN cytosolic entry.42
2.4 Controlled release strategies in molecular PRR agonist-based adjuvant design
The undesired release of immunotherapeutic agents into normal tissues may lead to severe side effects.60 In contrast, the insufficient release of agonists due to entrapment in carriers or uncleavable conjugation may also decrease the adjuvant efficacy. The on-demand release of these therapeutic adjuvants into the target tissues and maintaining a high concentration of adjuvants are critical to achieving efficient antitumor immunity while minimizing side effects. Variations in physiological conditions provide the possibility to design bio-responsive materials for the controlled release of PRR agonists at the desired time and location. Programmable degradable materials that are less sensitive to the physiological environment have been used for the controlled release of PRR agonists to maintain the immune response in tumors or LNs at a certain level. Typical classes of biomaterial scaffolds including implantable scaffolds,61,62 injectable hydrogels,63,64 microneedle patches,65 and supramolecular scaffolds66 have been used for controlled release. Here, we focus on some nanoparticle-based strategies for controlled release.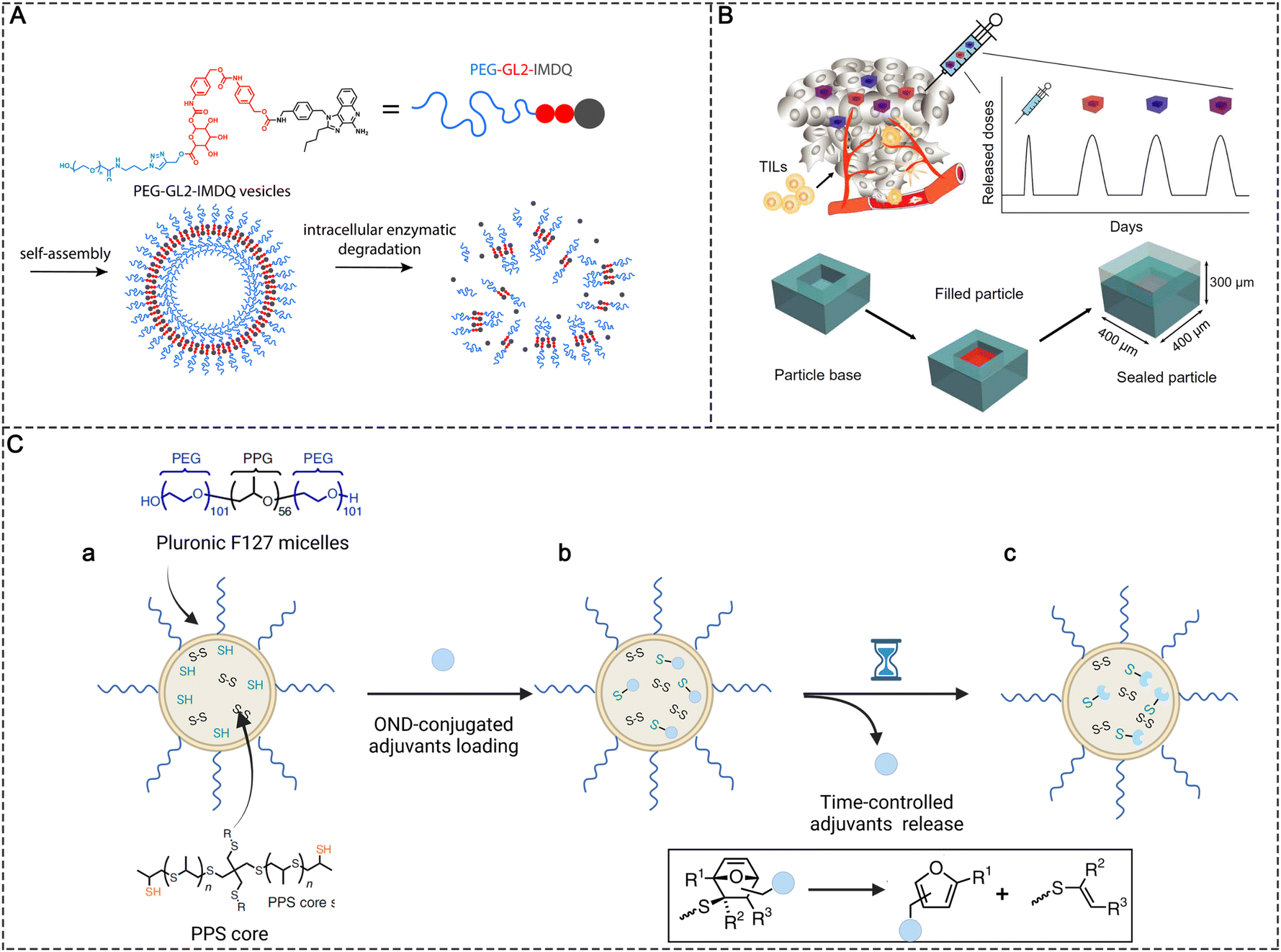 | ||
| Fig. 7 Controlled release strategies in molecular PRR agonist-based adjuvant design. (A) Chemical structure of PEG-GL2-IMDQ and schematic of enzymatically induced drug release by PEG-GL2-IMDQ self-assembled vesicular nanoparticles. Reproduced with permission from ref. 20, Copyright 2020, American Chemical Society. (B) Schematic of pulsed release from a single intratumoral injection containing PLGA microparticles with different release rates. Reproduced with permission from ref. 41, Copyright 2020, the American Association for the Advancement of Science. (C) Schematic representations of (a) the preparation of thiolated PPS NPs, (b) adjuvants conjugated with PPS NPs via thiol-reactive OND linkers, and (c) time-controlled release of loaded adjuvants based on the first-order retro-Diels–Alder mechanism. | ||
NPs in the size ranging from 5 to 200 nm can passively target LNs; however, the reticular network of LNs restricts the access of these particles to the cortical parenchyma, which also prevents the agonists from reaching more lymphocyte subpopulations.46 To improve LN penetration, the sufficient release of molecular adjuvants in the LNs is important. Nanoparticles with stable release kinetics make it possible to continuously release agonists after they are accumulated in the LNs. For example, Schudel et al. synthesized a two-stage delivery system using thiolated poly(propylene sulfide) (PPS) NPs (Fig. 7C). The adjuvant cargos were conjugated with thiol-reactive oxanorbornadiene (OND) linkers and further loaded in PPS NPs by linking OND with excess thiol in the PPS NPs. The two-stage nanoplatform accumulated in LNs effectively after peripheral administration. Meanwhile, the adjuvant cargos were released from the PPS NPs with different kinetics according to a first-order retro-Diels–Alder mechanism, with the half-life spanning from hours to days. The half-life of release kinetics determined the intra-lymphatic delivery efficiency. A delivery system with a half-life of ∼10 h was found to efficiently traffic adjuvants to specific LN structures such as the cortex and paracortex, where T and B cells and some DCs are located, significantly improving the immunotherapeutic effects compared with other pharmacokinetic profiles (e.g., half-lives of 17 min and 29 h).46
2.5 Synergistic strategies in molecular PRR agonist-based adjuvant design
Live attenuated viral vaccines can induce strong cellular immune responses because the virus can replicate to produce abundant antigens. Moreover, live virus particles can activate multiple PRRs.3,21 For example, the live-attenuated yellow fever 17D (YF-17D) virus in YF vaccine can induce the activation of TLR2, 3, and 7–9 and MDA5 of RLRs. However, the immune activation of a single PRR signaling pathway, even at large doses, has limited effects, is poorly tolerated, and is prone to safety issues.3 The co-delivery of multiple PRR agonists by nanomaterials can mimic microbial invasion and elicit more effective cellular immunity.21 For example, Zheng et al. designed a synthetic adjuvant vector (BMV) including monophosphoryl lipid A (MPLA), mifamurtide (MFT), recombinant flagellin (rFljB), and single-strand DNA containing CpG motifs to cooperatively trigger the activation of multiple PRRs (Fig. 8A). BMVs containing multiple agonists separately activated the TLR4, NOD2, TLR5, and TLR9 pathways. To mimic the bacterial components and structure, these components were rationally engineered and used to modify the liposome membranes (MPLA, MFT, and rFljB) or encapsulated inside the liposome core (CpG motifs). In vivo experiments showed that four agonist-loaded adjuvant vectors (BMVs) activated both the interferon regulatory factor 3 (IRF3) and NF-κB pathways and improved anti-tumor activity compared with any single agonist-based adjuvant vector. More importantly, in contrast to the standard Freund's adjuvant (FA), this multi-component adjuvant vector induced long-lasting immunological memory while reducing treatment-related toxicity.45 Similarly, our group developed a bi-adjuvant neoantigen nanovaccine (banNV) through the programmable self-assembly of amphiphilic nucleic acid-polymer conjugates and cationic polypeptide; banNV is composed of a colorectal cancer-specific neoantigen peptide, a small-molecule TLR7/8 agonist (R848), and a TLR9 agonist (CpG).67 The synergistic effect of the two agonists greatly enhanced the immunogenicity of the neoantigen while reducing acute systemic toxicity. Combination therapy with banNV and aPD-1 resulted in the complete regression of 70% of neoantigen-specific tumors without recurrence, demonstrating the strong potential of this co-delivery nanosystem in tumor immunotherapy (Fig. 8B).67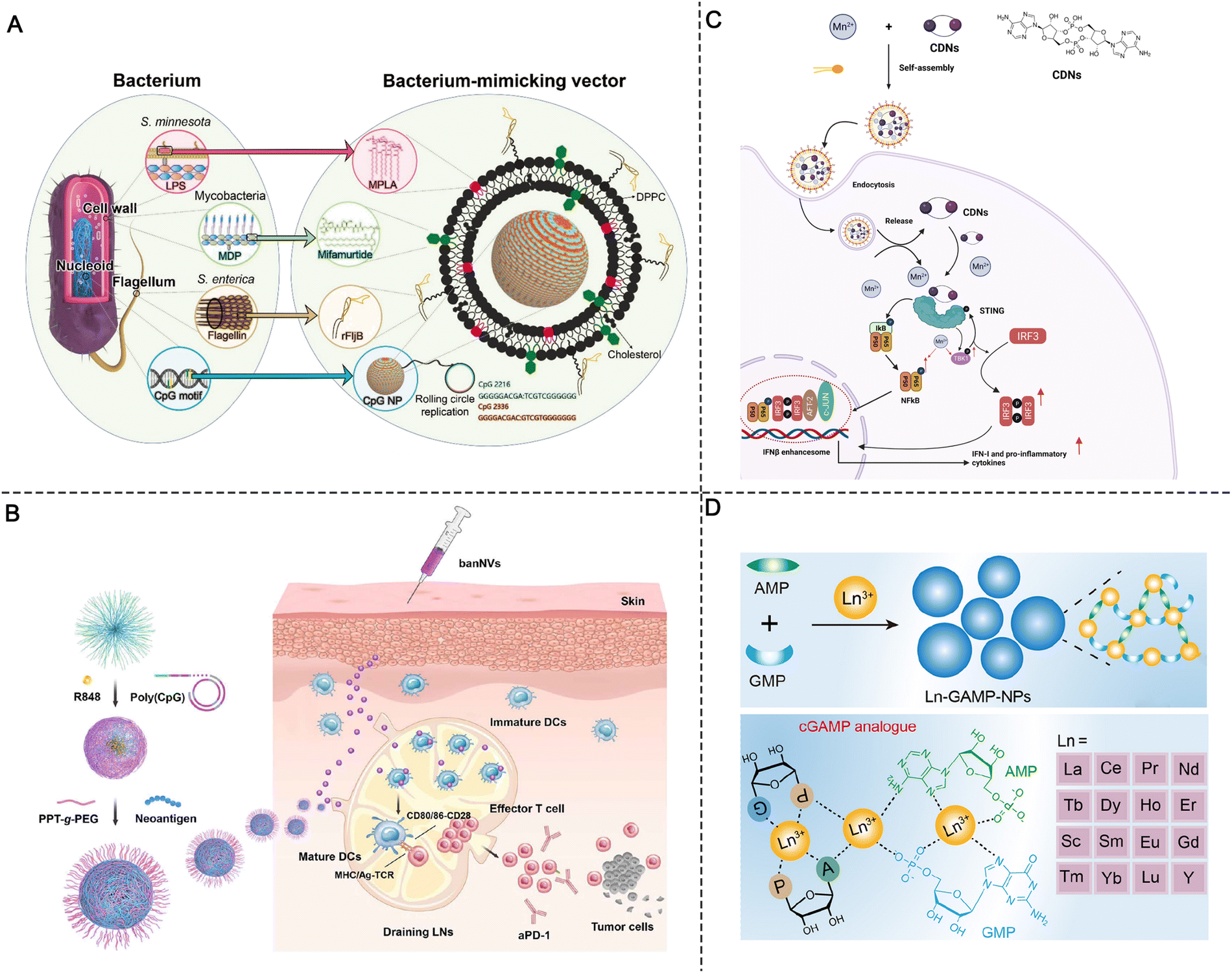 | ||
| Fig. 8 Synergistic strategies in molecular PRR agonist-based adjuvant design. (A) Schematic of an engineered liposomal vector comprising multiple adjuvants that mimic the bacterium. Reproduced with permission from ref. 45, Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (B) Schematic of the construction of banNVs for potentiation of neoantigen immunogenicity. Reproduced with permission from ref. 67, Copyright 2022, The Author(s). (C) Schematic of the preparation of Mn2+- and CDN-coordinated NPs for amplified STING activation and the synergistic activation mechanism. (D) Design of lanthanide-based coordination NPs for STING activation. Reproduced with permission from ref. 74, Copyright 2022, American Chemical Society. | ||
It is important to note that the use of PRR agonists to induce immune activation may lead to exhausted immune cells which often do not achieve optimal immunotherapy results. When using multiple molecular adjuvants to synergistically activate innate immunity, the phenomenon of immune cell exhaustion can be overcome by adjusting the release kinetics of different adjuvant molecules, thereby improving the effect of synergistic therapy.68 For example, Jin et al. designed a liposomal nanoadjuvant that can deliver TLR3 agonists (polyinosinic:polycytidylic acid, polyI:C) and TLR7/8 agonists (R848) simultaneously. Among them, TLR3 agonists can be rapidly released after being ingested, thus forming the first wave of immune stimulation. Meanwhile, TLR7/8 agonists and cholesterol in liposomes are covalently coupled through disulfide bonds and released in response to γ-interferon-inducible lysosomal thiol reductase (GILT) in endosomes, thus forming a second wave of immune stimulation. Due to the characteristics of TLR7/8 agonist-responsive release, there is a 4 h time window for the two waves of immune stimulation; it is the presence of this time window that allows the two agonists to have a significant synergistic effect, which resulted in greater upregulation of co-stimulatory molecules and secretion of various cytokines in bone marrow-derived dendritic cell (BMDC) cell models. Moreover, this design can achieve the long-term and continuous secretion of pro-inflammatory cytokines to prevent DC exhaustion. In contrast, when the cells were first treated with R848 followed by polyI:C stimulation, the synergistic effect and inhibition of DC exhaustion were not observed.69
Metal ions have been shown to have multiple important roles in fundamental life processes, including as enzyme cofactors and mediators of electron transport.70 Recently, various immune processes have been reported to be manipulated by metal ions. For example, Ca2+ was found to regulate T-cell receptor activation by modulating the charges of cell membrane lipids.71 Potassium released from tumors suppressed the T-cell activity, and increasing the potassium efflux capacity of tumor-specific T cells enhanced the anticancer activity of T cells.72 In particular, advances in the use of metal ions to drive cGAS-STING-associated immune responses are providing mechanistic insights into the development of new cancer vaccines. For example, Wang et al. found that Mn2+ could enhance the binding affinity of cGAS-STING to enhance STING activity.73 Moreover, Mn2+ could also enhance the sensitivity of cGAS to dsDNA and enhance the production of messenger cGAMP.73
According to the finding that Mn2+ can sensitize cGAS-STING, Sun et al. designed co-assembled nano-adjuvants by coordinating a cyclic dinucleotide (CDN) with Mn2+ for cancer metalloimmunotherapy. After screening from various metal ions, Mn2+ was found to significantly enhance the ability of STING agonists to induce IFN-I for cancer immunotherapy. Upon local or systemic administration, the coordinated CDN-Mn2+ particles (CMP) reversed the immunosuppression of the TME and elicited robust antitumor immunity.44 Mechanistically, instead of enhancing the binding affinity of STING agonists to the receptors, Mn2+ induced TANK-binding kinase 1 (TBK1) and p65 phosphorylation without the involvement of STING, while the subsequent augmentation of the phosphorylation of IRF3 was associated with STING agonists, finally amplifying the cascade STING signaling and enhancing IFN-I production (Fig. 8C).44
Due to the complicated synthetic procedure and unsatisfactory systemic delivery of metal ions, Luo et al. developed a series of hybrid NPs (termed Ln-GAMP-NPs) via the coordination of guanosine monophosphate (GMP), adenosine monophosphate (AMP), and lanthanides (Fig. 8D). The hybrid Ln-GAMP-NPs simplified the general small-molecule synthetic process of cGAMP, in which GMP and AMP are covalently conjugated to form a cyclic dimer via two phosphate bonds. Instead, the Ln-GAMP-NPs were prepared by the coordination-driven self-assembly of AMP/GMP with lanthanide ions. Despite the structural changes of GAMP, the Ln-GAMP-NPs efficiently promoted DC maturation and antigen presentation. Molecular docking revealed a high binding affinity of Ln-GAMP-NPs for STING originating from both mice and humans. Markedly, in the B16F10-OVA tumor model, the Ln-GAMP-NPs eliminated tumor growth and prolonged survival compared with natural 2’3’-cGAMP, suggesting that metal ions can potentiate STING agonists and improve immune priming.74
3. Synthetic self-adjuvanting materials
Tumor antigens can be classified into (1) synthetic proteins or peptides, (2) nucleic acids (DNA and mRNA), and (3) cell fragments from lysed tumor tissue based on the antigen source.75 Among them, protein or peptide antigens are often captured by APCs through endocytosis and trapped in lysosomes, generally resulting in the induction of relatively weak major histocompatibility class (MHC) I-restricted CD8+ T-cell responses. Delivery vectors are usually required to improve the cross-presentation of antigens and prime the antigen-specific T-cell responses. DNA and mRNA antigens are more suitable to deliver antigens for MHC I presentation.10 Delivery vehicles are necessary for mRNA antigens to protect them from degradation and facilitate delivery to lymphatic organs. Compared with mRNA, DNA antigens require more steps for antigen processing because the cytomembrane and nuclear membrane serve as barriers to penetration. Virus vectors or electroporation are commonly used for DNA antigen transfection.11 Moreover, tumor cell-derived antigens have low immunogenicity due to the accompanying trashy proteins and nucleotides; thus, numerous adjuvants are usually required to assist in immune stimulation.4 To meet the requirements for the in vivo delivery of different antigens, the properties of the antigens and their various immunologic mechanisms should be fully considered in the design of suitable delivery carriers and adjuvants for vaccine development.76 As mentioned earlier, while molecular PRR agonists can be chemically programmed to enhance their adjuvant potency, the variability in physicochemical properties between antigens and agonists presents challenges for cancer vaccine manufacturing. In addition, new formulations of antigens and adjuvants, including polymersomes, liposomes, and PLGA NPs, are usually empirical and rely on proprietary techniques from individual labs with the varying manufacturing technologies in different antigens and adjuvants.34 Therefore, the development of a general vaccine platform for different tumor antigens in an optimal format is desired. Moreover, apart from conventional vaccine components, the diverse physicochemical properties of antigens and adjuvants also raises technical concerns over the sterilization method, bioavailability, and stability.77Recently, some synthetic polymer- and lipid-based nanomaterials have emerged as vaccine adjuvants that exhibit self-adjuvant properties for cancer immunotherapy. These self-adjuvanting materials exhibit immune activity to effectively trigger innate immune responses. Moreover, compared with conventional adjuvants, they are able to activate some specific innate immune signaling pathways (e.g., STING, TLR, and NLR).78 Another notable feature is that these self-adjuvanting materials have a relatively simple chemical structure, which conceptually improves the reproducibility for adjuvant manufacturing and quality control. Additionally, the physicochemical superiority of these synthetic materials could simplify the manufacturing procedure of vaccine formulations (Fig. 9).
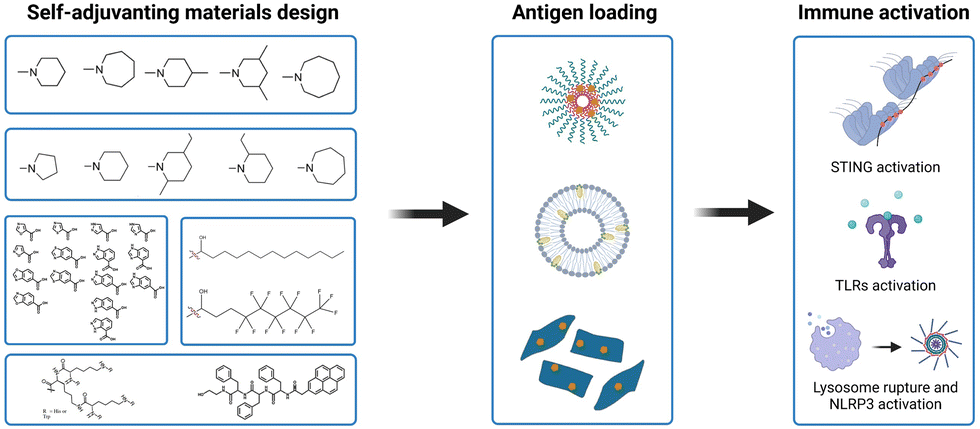 | ||
| Fig. 9 Synthetic self-adjuvanting materials design, simplified vaccine manufacturing and immune activation mechanism. | ||
3.1 Self-adjuvanting materials for STING signaling pathway activation
The STING signaling pathway is indispensable for managing the transcription of multiple defense genes when abnormal cytoplasmic DNA species are recognized.79,80 This pathway is also an alert system that fights against the progression of cancer by promoting anti-tumor immune responses.68 As a cytosolic DNA sensor, cGAS can be activated by direct binding to DNA species. Once activated, cGAS will convert GTP and ATP into the second messenger, cGAMP. After binding to STING, cGAMP then initiates downstream signaling pathways. STING is a transmembrane protein that predominantly resides in the endoplasmic reticulum (ER) and consists of four transmembrane helices, a C-terminal tail (CTT), and a dimeric cytosolic ligand-binding domain (LBD). The binding of cGAMP to LBD will lead to conformational changes of STING molecules. These STING molecules then construct a complex with higher-order oligomerization. The entire complex translocates from the ER to the Golgi apparatus, where TBK1 is recruited. TBK1 molecules gather on oligomeric STING complexes and phosphorylate adjacent TBK1 and neighboring STING dimers along with the recruited interferon regulatory factor 3 (IRF3), thus initiating the transcription of IFN-β (Fig. 10).40 Overall, the assembly of STING oligomers into oligomeric conformations is a key process in the downstream activation of TBK1 and IRF3, which is also an important step in the initiation of STING-mediated anti-tumor immunity.68 | ||
| Fig. 10 cGAMP-STING-TBK1 complex. cGAMP binding to STING induces the lid-closing and oligomerization of the STING dimer to form a higher-ordered scaffold. The scaffold then translocates to the Golgi apparatus. On the Golgi membrane, STING residues are bound and phosphorylated by TBK1, which also phosphorylates other TBK1 molecules. IRF3 is recruited, phosphorylated, and finally translocated to the nucleus followed by the induction of transcription of IFN-I. | ||
Synthetic self-adjuvanting materials for STING activation have also been investigated. Different from natural cGAMP, these self-adjuvanting materials typically encompass unique immune mechanisms for STING pathway activation.81 For example, Gao et al. reported a class of ultra-pH-sensitive PEG block copolymers with cyclic amines. Among them, a seven-membered amine ring (PC7A) was found to induce significant innate immune response through the formation of a PC7A-STING condensate (Fig. 12A).82 In multiple animal models, the synthetic PC7A NPs generated antitumor immunity by stimulating the STING pathway and inducing CD8+ T-cell activity.82 Compared with the natural STING ligand cGAMP, the PC7A polymer was found to bind to a non-competitive STING surface site on the α5 helix of STING, which is distinct from the cGAMP binding pocket.78 Moreover, the formation of PC7A-STING condensates enabled sustained STING activity over 6–48 h, whereas STING-mediated immune response was induced by cGAMP peaks at around 6 h and then declines rapidly (Fig. 12A).
The underlying biophysical mechanism of the PC7A polymer in STING activation is thought to rely on the multivalent condensation effect of PC7A-STING.78 The valency of biomolecules such as nucleic acids and proteins is determined as the quantity of independent binding units with the same or similar target-binding structure. Compared to monovalent interactions, multivalent interactions are more powerful since the target can interact with multiple binding units simultaneously (Fig. 11A). Therefore, synthetic multivalent ligands generally regulate biological processes more efficiently than monovalent ligands. The PC7A polymer can act as a multivalent supramolecular scaffold and bind with STING molecules to promote STING multimerization for immune activation (Fig. 11B).78 It is worth mentioning that the binding efficiency of STING-PC7A depends on the number of repeating units in the polymer. The PC7A polymer with 70 repeating units exhibited the maximum STING activation. Further increasing the number of repeating units beyond 70 resulted in excessive cross-linking and weak molecular dynamics, which led to an inferior signaling capacity and weaker STING activity (Fig. 12A).68
 | ||
| Fig. 11 Multivalent effect in STING activation. (A) Schematic of monovalent and multivalent ligand–receptor interactions. (B) STING oligomerization and condensation driven by PC7A through multivalent interactions. Reproduced with permission from ref. 78, Copyright 2021, The Author(s). | ||
 | ||
| Fig. 12 Synthetic self-adjuvanting materials. (A) STING activation of the PC7A polymer: (a) polymer structures with cyclic amines can generate STING activation; (b) PC7A polymer induced prolonged STING activation compared with cGAMP, as indicated by the Ifnb1 and Cxcl10 mRNA levels; and (c) STING activation was correlated with the PC7A valency. Reproduced with permission from ref. 78, Copyright 2021, The Author(s). (B) Structures of lipids with heterocyclic amines that induce STING activation. (C) Structures of azole-modified PEI molecules that induce STING activation. Reproduced with permission from ref. 81, Copyright 2022, Wiley-VCH GmbH. (D) Structure of F-PEI adjuvants that induce TLR4 activation and the preparation process of F-PEI/OVA vaccine. (E) Synthesis of cationic lipid-like compounds by PAMAM dendrimers for mRNA delivery. Reproduced with permission from ref. 96, Copyright 2021, The Author(s). (F) Structure of T34 (LR+) and T62 (LR−), which induced controllable NLRP3 inflammasome activation. Reproduced with permission from ref. 94, Copyright 2018, The Author(s). (G) Structure of polymer–PDP conjugate adjuvants; the acidic environment (pH 5.6) leads to the release of PDP, which re-assembles into the nanosheet structure. | ||
With the advances in nucleic acid nanotechnology, mRNA vaccines have emerged as a promising platform for cancer immunotherapy. However, effective mRNA vaccination requires both efficient antigen expression and appropriate immune activation.76 Lipid-based nanoparticles are currently the most researched and clinically advanced mRNA delivery vehicles.83 A class of lipids with STING activity and great potential in cancer vaccine-coded mRNA delivery was recently reported. Miao and co-workers have screened over 1000 ionizable lipids for mRNA cancer vaccine delivery.76 They found that the top-performing lipids share a common structure with cyclic amine head groups (Fig. 12B). LNPs with cyclic amine head groups provide both efficient antigen translation and potent immune activation in vivo. This specific class of LNPs induced APC maturation by activating the STING pathway, as opposed to activating TLRs.76 Heterocyclic azole molecules were also identified as another class of self-adjuvanting materials with STNG activity. Zhao et al. developed a series of azole molecule end-capped PEI (PEI-M); over 60% of these PEI-M polymers were found to possess innate immune stimulating activity through the STING pathway (Fig. 12C). Based on these findings, the authors proposed a minimalist nanovaccine fabricated by simply mixing model ovalbumin antigens with PEI-M. In vivo study indicated that PEI-4BImi, one derivative of the most efficient PEI-M, elicited robust antitumor immune responses when administered subcutaneously. This platform also enabled the fabrication of personalized binary nanovaccines by mixing autologous tumor cell membranes with PEI-4BImi. When combined with immune checkpoint inhibitors, a 60% postoperative cure rate was observed.81
3.2 Self-adjuvanting materials for TLR signaling pathway activation
Recently, cationic polymers have been applied in cancer vaccines as self-adjuvant TLR activators. Cationic materials such as polyethylenimine (PEI), which has a high charge density, have long been used for cancer vaccine delivery.84–86 Due to its intrinsic properties, PEI can easily form complexes with proteins or peptides through electrostatic interactions. After being engulfed by cells through endocytosis, the “proton sponge effect” of PEI results in endosome rupture, which allows the payload to escape from the endosome.87 In addition to cytosolic delivery, PEI can also increase the immunogenicity of antigens. Therefore, PEI has been widely applied as a vaccine adjuvant. Specifically, together with glycoprotein antigens, PEI has been incorporated into mesoporous silica nanoparticles to achieve effective mucosal delivery for personalized cancer immunotherapy.87 Nevertheless, the weak immunogenicity and cellular toxicity hamper the clinical translation of PEI.81,88,89 Emerging evidence suggests that the chemical modification of PEI usually decreases its toxicity and endows it with novel properties.As a striking example, grafting cationic polymers with fluorocarbon chains can enhance their transmembrane delivery owing to the lipophobicity and low surface energy of fluoroalkane. Xu et al. synthesized fluoroalkane-grafted PEI (F-PEI) via an amine–epoxide reaction (Fig. 12D). The conjugation of fluorocarbon chains significantly reduced the cytotoxicity of PEI. In addition, owing to the efficient cytosolic antigen delivery, F-PEI NPs loaded with tumor antigens (OVA) promoted high levels of antigen cross-presentation. F-PEI can also prompt DC maturation through the TLR4-mediated signaling pathway. Furthermore, personalized nanovaccines acquired by mixing F-PEI with cell membranes from resected autologous primary tumors can effectively prevent post-operative tumor recurrence and tumor metastases.90
Self-adjuvanting materials have also shown great potential in the intracellular delivery of mRNA-based cancer vaccines.76 Zhang et al. developed a library of lipid nanoparticles in which poly(amidoamine) (PAMAM) dendrimers (Fig. 12E) were linked with different lipid tails at different grafting ratios. After screening the lipid nanoparticle library, the authors found that PAMAM with a 12-carbon lipid tail (PAMAM-12) could effectively deliver mRNA to antigen-presenting cells. With ovalbumin (OVA) mRNA as a research model, a minimalist nanovaccine was prepared by mixing 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG 2000), PAMAM-12, and OVA mRNA to form the lipid nanoparticles. The minimalist PAMAM-12 nanovaccine facilitated efficient mRNA translation and elicited robust T-cell responses both in vitro and in vivo. Unexpectedly, PAMAM-12 itself was found to induce the production of proinflammatory cytokines such as IL-1β, IL-6, IFN-α1, and IFN-α4. PAMAM-12 activated the DCs mainly through the TLR4 signaling pathway.22
In addition to polymer-based self-adjuvanting materials, self-assembling small molecules have emerged as another efficient class of TLR activators. Small molecules with specific chemical structures can spontaneously self-assemble and form large structures through noncovalent interactions, which provides a robust material construction method. Shuyu Ji et al. reported a library of self-assembling small molecules that serve as adjuvants. After optimization, they discovered that the self-assemblies of cholicamide can enter cells and stimulate the innate immune response via TLR7. Notably, the small molecule of cholicamide is distinct from nucleoside deoxycholate derivatives such as imidazoquinolines, but the cholicamide assemblies can bind to the RNA-binding pocket of TLR7.91 In a subsequent study, the researchers investigated the application of self-assembled cholicamide as an antigen carrier. They found that the assemblies of antigen-cholicamide conjugates with cholicamide could induce high levels of MHC II presentation of antigen peptides and cytokine release.92
3.3 Self-adjuvanting materials for NLR signaling pathway activation
NLRs, which belong to cytosolic PRRs, can recognize intracellular PAMPs (such as microbial toxins) and DAMPs (such as ATP, uric acid, and K+).93 NLRs regulate inflammation and apoptotic response by cooperating with TLRs. There are multiple subfamilies of NLRs, including NODs, NLRPs, and IPAF. NLR stimulation can activate the NF-κB pathway and induce cytokine production to enhance immune activity. NLRs also play a critical role in the functioning of inflammasomes, which control the production of caspase-1 and induce the production of inflammatory cytokines such as IL-1β and IL-18. Within the NLR family, NLRP3 inflammasome has been extensively studied. Currently, numerous vaccine adjuvants are found to be involved in NLRP3 inflammasome activation, including alum and QS-21.3 The concept of synthesizing self-adjuvanting materials with the ability to activate NLRP3 inflammasomes has also been proposed.Although inflammasomes are important in the inflammatory responses of vaccines, there are two major challenges in engineering inflammasome activators as feasible adjuvant materials: (1) the precise conditions under which inflammasomes can be activated are still unknown; and (2) the degree of inflammasome activation cannot be controlled with the current activators. To overcome these issues, Manna et al. developed two categories of dendritic polymers (denoted T34/LR+ and T62/LR−) with multiple lysine-based dendrons conjugated to an l-lysine-dicysteine backbone. The polymer backbones were further modified by different percentages (34% and 62%) of tetraethylene glycol (TEG) chains to modulate the space between adjacent dendrons (Fig. 12F).94 The different TEG percentages resulted in different lysosome rupture abilities, which led to significant differences in inflammasome activation. The polymer entered the cells through endocytosis. Endosomal acidification led to the protonation of the histidine residues of polymers, which changed the osmotic pressure of intracellular lysosomes and caused their rupture. The authors demonstrated that the cationic charge of the tryptophan residues facilitated cellular uptake and lysosomal membrane lysis, leading to the escape of polymers from lysosomes and rapid diffusion into the cytosol. A greater amount of dendrons and more protonation sites for the protonation of T34/LR+ induced remarkable changes in osmolarity and stronger lysosome rupture.94
It appears that peptides re-assembled in lysosomes can induce lysosome rupture followed by NALP3-activation, and this phenomenon has been applied in adjuvant design. Accordingly, Gong et al. designed a proton-driven nanotransformer from a pyrene-conjugated d-peptide (PDP) (Fig. 12G).95 The nanotransformer could load antigen peptide and assemble into 100-nm spherical nanotransformer vaccine (NTV) at pH 7.4. After entering the acidic endosomal environment, NTV dramatically reassembled into 5–8-μm nanosheets. The morphological changes disrupted the endosomal membrane and released the antigenic peptide into the cytoplasm, thereby boosting the anti-tumor immunity by activating the NLRP3 inflammasome pathway. When combined with anti-PD-L1 antibodies, the NTV induced complete tumor regression in 50% of B16F10 tumor mice.95
4. Inorganic adjuvants with versatile physicochemical features
In the past decades, adjuvant design has focused more on identifying the chemical compositions that stimulate PRRs.27 However, recent efforts have demonstrated that the topological features of adjuvants are also involved in modulating the innate immune activity.97 Generally, the morphological characteristics of adjuvant materials, such as particle size and shape, are considered to be important features that impact the delivery and potency of adjuvants. Currently, the topological features of adjuvant materials are shown to modulate the in vivo immunological properties of adjuvants.98 For example, uric acid, the product of purine nucleotide degradation, is normally produced by the body and dissolved in the blood. However, high concentrations of uric acid promote the formation of monosodium urate crystals, leading to acute inflammation at the joint.99 Inspired by the strong correlation between immunological responses and physicochemical features, a novel design principle was been proposed for adjuvant development. In this section, we discuss some recent advances related to the influence of physicochemical features, including shape, size, surface topology, and chirality, on adjuvant immunogenicity (Fig. 13). | ||
| Fig. 13 Overview of structure-based physically activated adjuvant materials design. Shape and size (a), surface topology (b), and chirality (c) influence the activation of the NLRP3 inflammasome pathway to promote DCs maturation. | ||
4.1 Shape- and size-dependent adjuvant activation
The morphological features (e.g., size and shape) of particles have been demonstrated to be correlated with cellular uptake and delivery efficiency.100,101 NPs with larger surface-to-volume ratios or higher surface charges are favorable for cellular uptake, leading to drug-loaded nanoformulations with superior therapeutic efficacy.101 In addition, nanoparticle size and shape have been found to be important in immunomodulation (Table 3). In a recent study, smaller hydroxyapatite (HA) particles (sizes of 0.1–20 μm) with needle-shaped morphologies significantly enhanced IL-1β secretion compared with larger particles (size >30 μm) (Fig. 14A). The different immunomodulatory effects were mainly related to the cellular uptake of the HA particles. The needle-shaped HA particles possessed high aspect ratios, and small particles with high aspect ratios are readily phagocytosed by macrophages, thereby inducing the secretion of IL-1β.102 However, these rules do not apply to all materials. For example, mesoporous silica (MS) particles with diameters of 2.5 μm induced higher CD86 expression and IL-12p70 production in DCs than MS particles with diameters of 270 nm; the small MS particles (270 nm) were prone to becoming trapped within the endosomes, whereas the large MS particles could escape endosomal entrapment.103| Composition | Morphological features | Immunogenic activity |
|---|---|---|
| AlOOH NPs | Long-rod-shaped vs. short-rod-shaped | Long-rod-shaped AlOOH NPs could induce higher CD86/CD80 expression and IL-1β/IL-6 on secretion BMDCs.105 |
| MS particles | Particle size 30–50 nm vs. 100–200 nm | Particles with size of 110–200 nm induced higher GM-CSF secretion in macrophages.106 |
| NPs (270 nm) vs. microparticles (2.5 μm) | Microparticles (2.5 μm) induced higher CD86 expression and IL-12p70 production in monocyte-derived DCs.103 | |
| HA particles | Particle size (0.1 μm, 5 μm, 20 μm, 100 μm); needle-shaped/spherical | Needle-shaped HA with size of 0.1–20 μm generated prolonged inflammatory response102 |
| Gold NPs | Spherical (20 and 40 nm vs. rod (40 × 10 nm) vs. cubic (40 × 40 × 40 nm) | Rod gold NPs mainly induced secretion of IL-1β/IL-18 in BMDCs; cubic and 40 nm spherical gold NPs induced higher secretion of inflammatory cytokine (TNF-α, IL-6, IL-12, and GM-CSF).107 |
 | ||
| Fig. 14 Physicochemical feature-dependent adjuvant activation. (A) HA with various shapes and sizes and the corresponding immunogenicity. Reproduced with permission from ref. 102, Copyright 2017, The Author(s). (B) AlOOH NPs with different morphologies and the corresponding immunogenicity. Reproduced with permission from ref. 105, Copyright 2013, American Chemical Society. (C) Schematic of spiky particles and their immune activation mechanism. (D) Preparation of mesoporous silica NPs (MSNs) and the induced cross-presentation effects. Reproduced with permission from ref. 109, Copyright 2020, The Author(s). (E) Chiral NPs induced immune activation (preparation of chiral gold NPs, characterization of circular dichroism and g-factor, and the distribution of interaction force between BMDCs and NPs). Reproduced with permission from ref. 110, Copyright 2022 Wiley-VCH GmbH. | ||
Nanoparticles with different shapes and sizes can act through different biophysical mechanisms to activate immune responses, including Th1 and Th2 responses mediated by T helper 1/2 cells (Th1/Th2 cells). In general, Th1 cells mainly produce interleukin (IL)-12 and IFN-γ to prompt CD8+ T cell-based cellular immunity. Th2 cells generally induce humoral immunity by secreting cytokines including IL-4 and IL-5 and activating B cells to produce antibodies.6 In previous studies, immunization with polylactide microparticles with sizes of approximately 200–600 nm tended to induce the Th1 immune response, whereas particles with sizes of 2–8 μm tended to induce the Th2 immune response.104 Sun et al. showed that long-rod-shaped aluminum oxyhydroxide nanoparticles (AlOOH NPs) improved IL-1β production and oxidative stress in THP-1 cells compared with short-rod-shaped AlOOH NPs, and the effect was mediated by the NLRP3 pathway (Fig. 14B). The long-rod-shaped AlOH NPs also improved mouse BMDC maturation (CD86/CD80 expression on CD11c+ cells, etc.).105
4.2 Surface topology-dependent adjuvant activation
Many natural pathogens (such as the influenza virus) have spiky nanostructures on their surfaces, which have been shown to be important for adsorption and infection.108 The spike-like nanostructures in turn alarm the immune system of the approaching intruders and trigger the adaptive immune response. To explore the correlation between immunogenicity and particle surface topology, Wang et al. synthesized TiO2 microparticles modified with nanospikes (spiky particles) through a two-step hydrothermal method. During phagocytosis by macrophages and DCs, the spiky particles exerted more mechanical stress on the cells compared with smooth-surfaced TiO2 microparticles, leading to potassium efflux and inflammasome activation (Fig. 14C). The injectable spiky particles upregulated the expression of the co-stimulatory molecules CD40 on DCs in an inflammasome-dependent manner. CD40 is an important co-stimulatory molecule on which antigen cross-presentation by DCs is dependent. The upregulation of this signal molecule is important for initiating CD8+ T cells for cancer cell killing. Moreover, by mixing with MPL A (a TLR4 agonist), the spiky particles evoked intensive antigen-specific cellular and humoral immune responses in vivo and substantially increased tumor clearance.108Mesoporous materials have been widely applied in nanomedicine and drug delivery due to their unique structures for hosting, protecting, and transporting cargos. Recent investigations have proposed mesoporous materials as important self-adjuvants. An extraordinary finding is that the surface topological features of mesoporous particles, including the pore size, remarkably affect the immunomodulatory effects. Hong et al. fabricated mesoporous silica nanoparticles (MSNs) with different pore sizes (7.8, 10.3, and 12.9 nm) by adjusting the concentration of tetraethyl orthosilicate (TEOS) (Fig. 14D).109 All of the silica nanoparticle sizes were around 80 nm. Taking OVA as the model antigen, a silica nanoparticle-based vaccine (OVA@MSNs) was fabricated by loading OVA protein in MSNs. Three kinds of OVA@MSNs with different pore sizes showed comparable delivery efficiency to LNs due to the consistent particle size. Furthermore, no significant difference is observed in the internalization of DCs or their maturation performance among the three types of OVA@MSNs tested. All three types of OVA@MSNs induced significantly increased expression of costimulatory factors CD80 and CD86. However, MSNs with large pore size (12.9 nm) showed a five-fold higher cross-presentation efficiency of OVA. The ROS generated by the MSNs destroyed the endosomal/lysosomal membrane, which expedited OVA escape from the lysosomes for MHC I-restricted presentation. The pore size was also correlated with the antigen release rate and cytotoxicity. MSNs with large pore sizes released antigens faster and were less cytotoxic, resulting in more efficient antigen cross-presentation.109
4.3 Chirality-dependent adjuvant activation
Chirality is widely prevalent in nature and endows biomaterials with unique and specific biological and chemical properties. The two enantiomeric forms in nature can be defined separately as the D-type and L-type.97 In contrast to molecular chirality, supramolecular chirality and its potential role in biology have not been fully explained. Chiral inorganic nanostructures are considered valuable due to their potential biomedicine applications. To determine the effects of chiral nanostructures on complex immune networks, Xu and coworkers developed chiral gold NPs in the presence of chiral L- and D-cysteine-phenylalanine (CYP) under circularly polarized light (CPL) illumination. The gold NPs displayed distinct chiral shapes with a g-factor of 0.44, resulting in significant differences in the ability to induce in vitro and in vivo immune responses.97 Compared with the D-P-NPs, the L-P-NPs induced higher expressions of co-stimulatory biochemical markers such as CD40, CD80, and CD86 in mouse BMDCs. Moreover, the L-P-NPs facilitated the secretion of a larger amount of pro-inflammatory cytokines such as IL-12 and TNF-α. The researchers further investigated the underlying mechanisms by which the chiral NPs induce immune responses. Specifically, the results showed that CD97 and epidermal growth factor-like module receptor 1 (EMR1) had higher binding affinities for L-P-NPs compared with D-P-NPs. By blocking the receptors of CD97 and EMR1 in mouse BMDCs, the uptake of the chiral NPs was almost completely suppressed, indicating that the CD97 and EMR1 receptors mediate the endocytosis of the chiral NPs. Furthermore, endocytosed NPs activated the downstream NLRP3 inflammasome pathway to enhance immune response.97Furthermore, in a study by Wang et al., the immunological effects induced by chiral gold nanomaterials were investigated, revealing that chiral NPs with a g factor of 0.44 can significantly enhance innate and adaptive immunity against tumor growth compared to achiral NPs (Fig. 14E).110 The researchers found that chiral NPs stimulate DCs, leading to the activation of CD8+ T cells and natural killer cells (CD69+ NK cells), and that L-type chiral NPs outperformed D-type chiral NPs in activating CD8+ T and CD69+ NK cells in tumor tissues.110 Furthermore, L-type chiral NPs induced tumor cell apoptosis and prolonged the survival of mice in an EG7.OVA tumor model. Mechanistic studies demonstrated that chiral NPs reinforce the activation of NK cells and CD8+ T cells by exerting mechanical force on BMDCs and promoting cytokine production. Moreover, L-type NPs showed a higher interaction force against target cells compared to D-type NPs, further enhancing their intratumor tissue infiltration.110
5. Engineered bio-derived adjuvants
Microorganisms such as bacteria and viruses can induce strong immune responses.3 Moreover, endogenous immune cells and their derivatives such as exosomes also carry a large number of immune-activating molecules.111 Through rational design, bio-derived adjuvant materials can mimic the natural properties of invading pathogen and biological processes. Traditional bio-derived adjuvants (e.g., the LPS derived from the outer membrane of Gram-negative bacteria, which has a strong immune-activating effect) have played important roles in vaccine development. However, the toxicity of LPS limits its application. MPL obtained via the chemical modification of the LPS has a significantly lower toxicity than the LPS while retaining the adjuvant activity.3 In recent years, many new bio-derived adjuvants have been developed through biomimicry and biomimetic techniques; these adjuvants are more suitable for the rapid development of tumor-personalized vaccines than traditional bioderived adjuvants.112 For example, hybrid biomimetic vaccines are prepared by extracting tumor cell membranes and hybridizing them with bacterial membranes, coating oncolytic virus with tumor cell membranes to induce anti-tumor immunity, and using DC vesicles to directly present tumor antigens (Fig. 15). These bio-derived adjuvant materials show great promise for generating highly potent antitumor vaccines. Furthermore, the development of bio-derived adjuvants using bioinspired or biomimetic technologies can enhance the antigenic activity and spatiotemporal controllability for on-demand immune activation, thus offering great potential to address existing limitations and expedite the clinical translation of cancer vaccines.111,113 In this section, we discuss recent advances in materials engineering strategies for the rational design of bio-derived adjuvants.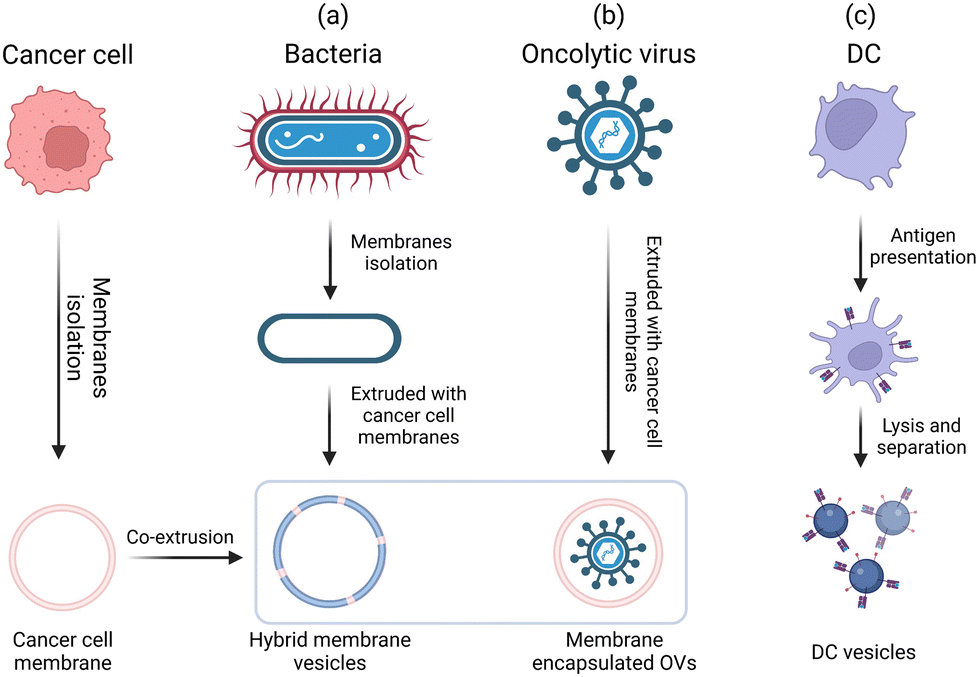 | ||
| Fig. 15 General engineering approaches for bio-derived adjuvants. | ||
5.1 Bio-derived adjuvants from bacteria/viruses
Although synthetic adjuvants are being actively developed, they may still not be sufficient to fully recapitulate the complexity of living biological systems. Bacteria-based natural or synthetic PAMPs have been widely used as adjuvants in cancer vaccine development.3,12 However, single ingredients are considered to be less effective than the counterparts of whole pathogens, which contain multiple types of PAMPs to synergistically activate a broad repertoire of TLRs.45 However, whole-pathogen adjuvants will likely raise safety concerns such as cytokine storm and sepsis. Therefore, engineering bacteria to minimize the pathogenicity while maintaining the adjuvant potency will facilitate the development of cancer vaccines. Chen et al. proposed a bacteria engineering strategy in which the cytoplasmic membrane is separated from the bacterial cell wall to produce vaccine adjuvants. By discarding the toxic cell wall components, adjuvants composed of cytoplasmic membranes can reduce the strength of the danger signals, making them safer and avoiding drastic immune side effects. Using this strategy, the authors developed a hybrid membrane vesicle composed of engineered Escherichia coli cytoplasmic membrane (EM) adjuvants and tumor cell membranes (TMs) from resected autologous tumor tissues (Fig. 16A).113 The integration of the EM into hybrid membrane nanoparticle vaccines (HM-NPs) induced DC maturation followed by the activation of tumor-specific T cells. In immunogenic CT26 colon and 4T1 breast tumor models, the HM-NPs extended postoperative animal survival and conferred long-term (up to three months) protection against tumor rechallenge.113 | ||
| Fig. 16 Engineered bio-derived adjuvants. (A) Preparation of HM-NPs. Reproduced with permission from ref. 113, Copyright 2021, The American Association for the Advancement of Science. (B) Schematic of ExtraCRAd production. Reproduced with permission from ref. 114, Copyright 2019, The Author(s). (C) Generation of DCNVs and schematic showing the specific activation and proliferation of CD8+ T cells by DCNV-rAd-Ag.112 | ||
Oncolytic viruses (OVs) are native adjuvants that provoke antitumor immunity. In recent efforts, OVs selectively eradicated tumor cells and released TAAs in situ, demonstrating great potential for comprehensive tumor treatment.10 Although several types of OVs have been tested in Phase III clinical trials, one major challenge is that oncolytic virus therapy mostly elicits immunity against viruses rather than tumors due to the high immunogenicity.114 Therefore, it is important to direct the adjuvant activity of OVs from antivirus to antitumor applications. An emerging concept is that encapsulating OVs within tumor cell membranes can decrease the fast clearance of OVs while equipping them with cancer targeting ability in vivo.115 Based on this biomimetic cell-membrane technique, Fusciello et al. developed OV-containing nanoparticles (extra-conditionally replicating adenoviruses, ExtraCRAd) by encapsulating OVs together with an antigen pool inside of fusion cancer cell membranes derived from well-characterized cancer cell lines (Fig. 16B). ExtraCRAd exerted strong antitumor immunity and significantly inhibited tumor growth in multiple tumor models.114 It is widely accepted that the extracellular environment of cancer cells is acidic (pH in the range of 6.4–7.0), and the low-pH environment can be used to improve the fusogenic activity of OVs. Huang et al. designed a vesicular stomatitis virus glycoprotein (VSVG)-engineered tumor cell membrane. VSVG is a viral fusion protein that is folded in neutral environments and can be converted into an unfolded fusion state in acidic environments, thereby promoting fusion between cell membranes. A carrier membrane was decorated with VSVG by vesicular stomatitis virus infection and then coated on OVs (V-M@OV). After systemic administration, V-M@OV efficiently accumulated inside tumor tissues. The low pH of the TME caused VSVG to transform into an unfolded fusion state, which triggered membrane fusion between V-M@OV and tumor cells and led to sequential OV release and duplication.115
5.2 Adjuvants that originate from or mimic DCs and other cells
Antigen processing and presentation are key processes in adaptive immunity. To allow the recognition of CD4+ or CD8+ T cells, antigens must be presented to immune cells by MHC I or II molecules on the surfaces of APCs.116 Manipulating the antigen presentation process is crucial to effective cancer immunotherapy. Conventional adjuvants are designed to assist antigens in eliciting immune responses. Although they can promote the expression of MHC molecules on APCs, they have limited effects on antigen processing and presentation. Thus, it is necessary to design T cell-targeted delivery platforms to enable the direct manipulation of T cells.112 Generally, both antigen presentation and co-stimulation signaling pathways are involved in the full activation of tumor-specific CTLs. The first signal is provided by the interactions between the T-cell receptors (TCRs) on the lymphocytes with the MHC/antigen complexes present on the APCs. The interactions between CD28 on the lymphocytes and CD80/86 on the APCs provide the necessary co-stimulatory second signal. Without this second signal, the lymphocytes would become apoptotic or anergic and be unable to effectively respond to the presented antigens.116 Although the DC-based immunotherapy sipuleucel-T achieved clinical success in prostate cancer, the complexity of production and short storage life significantly restricted its large-scale application.112Emerging evidence suggests that DCs play a crucial role in regulating the initiation of adaptive immunity through the secretion of exosomes, which are nano-sized membrane structures that serve as a type of extracellular vesicle.117 These vesicles are a key means of intercellular communication and, as secretions of living cells, carry the identity of their parent cells. Exosomes have been found to possess good biocompatibility. Mature DCs that have taken up antigens secrete exosomes that express key components of the immune system, including MHC-I/II and the co-stimulatory molecules CD80 and CD86. These exosomes can directly stimulate T-cell responses in CD4 and CD8 cells that are reactive to the antigens.118 However, the use of DC exosomes as tumor vaccines has not yet produced satisfactory results, which may be due to the suppressive tumor immune microenvironment. In light of this, Phung et al. modified exosomes from OVA-treated mice BMDCs with an anti-CTLA-4 monoclonal antibody (EXO-OVA-mAb). Compared to unmodified exosomes, EXO-OVA-mAb greatly improved T-cell targeting, induced a strong tumor-specific T-cell response, and increased the ratio of intratumoral effector T-cells to regulatory T-cells, ultimately leading to a significant inhibition of tumor growth. This study highlights the potential of modifying exosomes to improve the efficacy of DC-based immunotherapy.119
Moreover, engineered DC membrane nanovesicles containing MHC and CD80/86 molecules are capable of cognate T-cell activation. Recent works have rationally engineered DC nanovesicles to endow them with desirable functions such as enhanced T-cell priming and immunosuppression reverse. Through biomimetic synthesis, Liu et al. designed a genetically engineered DC membrane nanovesicle platform (ASPIRE) that integrates antigen self-presentation and immune tolerance blockade (Fig. 16C).112 The ASPIRE nanovaccine was derived from DCs, and the antigen-MHC I complex, B7 co-stimulatory molecules, and anti-PD1 antibody were programmably anchored on.112 ASPIRE markedly enhanced the immune checkpoint blockade and stimulated powerful CTL responses against established tumors.112 Similarly, Cheng et al. used cell membrane coating nanotechnology to develop DC-like nanoparticles (“mini DC”) by coating DC membranes on IL-2-loaded PLGA NPs. The DC membranes were extracted from cancer cell lysate-treated DCs. MiniDC retained the functional surface membrane proteins (including MHC, CD86, and CD40) for T-cell activation, and the release of IL-2 was not restricted by immunosuppressive TAMs. MiniDC elicited robust T-cell response both in vitro and in vivo and inhibited tumor progression in mouse ovarian cancer models. Furthermore, miniDC suppressed the metastasis of ovarian cancer in the abdomen. This platform thus provides a robust and safe strategy for cancer immunotherapy.120
Given that almost all cells express MHC-I, which can be monitored by the immune system in the event of infection, cancer cells have been engineered to elude immune detection by manipulating MHC-I expression. Relying on the antigen presentation machinery, another idea is that cancer cells could be employed as natural vaccines to interact directly with T cells. Zhang's group engineered a model cancer cell line (B16F10 melanoma cell line) to express the co-stimulatory marker CD80. The cancer cell membrane was extracted and coated on PLGA NPs to fabricate a biomimetic nanoformulation capable of presenting its own antigens directly to cancer-specific T cells in an immunostimulatory context.116
6. Conclusions and future perspectives
Success in the clinical translation of cancer vaccines has sparked great interest in improving the vaccine adjuvants. However, for conventional approved adjuvants, challenges remain related to their low immunogenicity and unclear molecular mechanisms. Seeking to overcome these limitations, materials engineering techniques have emerged as attractive approaches for cancer vaccine adjuvant development to improve both adjuvant potency and safety. Selective examples include (1) engineered molecular adjuvants with the ability to precisely target LNs, penetrate tissues, and achieve controlled release to elicit robust immune responses and minimize toxic side effects; (2) synthetic self-adjuvanting materials that simplify the vaccine manufacturing process; (3) materials with adjustable physicochemical features (e.g., topological characteristics) for improved immunogenicity; and (4) genetically and chemically engineered bio-derived materials as safe and potent vaccine adjuvants in cancer immunotherapy. This Review describes current progress in materials engineering strategies for the rational design of well-defined adjuvants as well as the associated mechanisms.Despite their great promise, these engineering strategies for adjuvant materials face challenges in terms of both mechanistic elucidation and clinical translation. Above all, although vaccine adjuvants enhance the quality and longevity of anticancer immune response, strong and sustained immune stimulation is not the only pursuit of adjuvant material design. Inappropriate immune stimulation not only causes toxic side effects such as granulomas, it may also lead to immune tolerance and the deterioration of T-cell function (defined as T-cell exhaustion), severely restricting tumor immunotherapeutic efficacy. In view of the discontinuity theory of immunity, our immune system would respond to the sudden changes in stimuli rather than the emergence of the stimulus itself; in other words, slow or continuous stimulation will induce immune system tolerance.121 Therefore, adjuvant design should consider the frequency of immune stimulation. To avoid immune tolerance, the dosing frequency or release kinetics should be further optimized according to longer-term experimental results. Although few attempts have been made, chemical synthesis and biomedical engineering strategies are powerful and practical approaches for modulating the pharmaceutical profiles of adjuvants.
At present, most of the current cancer vaccine adjuvants in clinical trials are molecular agonists. Compared with conventional adjuvants, these molecular agonists (e.g., Resiquimod, cGAMP, and diamidobenzimidazole) tend to be chemically well defined, stimulate a stronger immune response, have a clear mechanism of action, and be suitable for large-scale industrial production in terms of quality control and safety evaluation. Although engineered adjuvant materials have overcome some limitations of existing adjuvants and demonstrated better performance in cancer immunotherapy in terms of their biocompatibility, safety, and potency, their clinical translation is still hindered by the following factors. (1) Differences between animal and human immune systems. Although animal experiments provide valuable guidance, subtle differences between the immune systems of the different species may lead to failed clinical translation. A typical example is the failure of DMXAA (5,6-dimethylxanthenone-4-acetic acid, a STING agonist) in a Phase III clinical trial because DMXAA can selectively bind to mouse STING but not human STING. Although these two species share high sequence and structural similarity, the inability of DMXAA to recognize human STING diminished its therapeutic efficacy.18 The success of engineered adjuvant materials in animal models does not translate into success in humans. Therefore, species differences should be considered when evaluating vaccine adjuvants in the future. (2) Heterogeneity in tumor and immunity among individuals.10 Heterogeneity in the TME as well as in system immunity has long been thought to limit the efficacy of therapeutic drugs. Therefore, the design of adjuvant materials that provide precise and individualized treatment for wide ranges of patients is critical for successful clinical translation. Recent advances in high-throughput synthesis and screening techniques hold great promise to revolutionize adjuvant design for cancer vaccines.122 These tools provide more options for designing new and personalized adjuvant formulations that target different signaling pathways with improved selectivity, safety, and efficacy; they also improve our understanding of how exactly the variations in adjuvants (e.g., differences in their physicochemical properties, structures, and pharmaceutical activities) influence how they induce immune responses. In addition, there is an urgent need for new potential pre-clinical models that could be translated to human immunity.
In this Review, we have summarized advances in the development of cancer vaccine adjuvants, including chemically engineered molecular agonists, versatile organic/inorganic self-adjuvanting materials, and genetically engineered bio-derived materials, along with our understanding of their mechanisms of immune activation. Although the widespread implementation of materials engineering approaches in vaccine adjuvant development remains challenging due to safety, manufacturing, and socioeconomic issues, advances in chemical synthesis and biomedical engineering will help revolutionize the development of new cancer vaccine adjuvants. These efforts are expected to accelerate in the future to speed the clinical translation of material-based cancer immunotherapy.
Abbreviations
| APCs | Antigen-presenting cells |
| BMDCs | Bone marrow-derived dendritic cells |
| CpG | Cytosine phosphoguanine |
| CDNs | Cyclic dinucleotides |
| CDs | Cytoplasmic DNA sensors |
| cGAMP | Cyclic guanosine monophosphate-adenosine monophosphate |
| cGAS | Cyclic GMP-AMP synthase |
| CTLs | Cytotoxic T lymphocytes |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| MHC I/II | Major histocompatibility class I/II |
| MPL | Monophosphoryl lipid |
| IFN-α1/α4/β/γ | Interferon-α1/α4/β/γ |
| IFN-I | Type I interferon |
| IL-1β/2/6/12/12p70/18 | Interleukin-1β/2/6/12/12p70/18 |
| IRF3 | Interferon regulatory factor 3 |
| LNPs | Lipid nanoparticles |
| LNs | Lymph nodes |
| MAPK | Mitogen-activated protein kinase |
| MyD88 | Myeloid differentiation primary response 88 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PAMPs | Pathogen-associated molecular patterns |
| PRRs | Pattern recognition receptors |
| STING | Stimulator of interferon genes |
| TAAs | Tumor-associated antigens |
| TBK1 | TANK-binding kinase 1 |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor-α |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National University of Singapore Start-up Grant (NUHSRO/2020/133/Startup/08, NUHSRO/2022/005/Startup/02), The Ministry of Education (MOE) Academic Research Fund (AcRF) Tier 1 (NUHSRO/2022/068/T1/Seed-Mar/04), and the NUS School of Medicine Nanomedicine Translational Research Programme (NUHSRO/2021/034/TRP/09/Nanomedicine), Open Fund Young Individual Research Grant (OFYIRG22jul-0019), and National Research Foundation (NRF) Competitive Research Programme (CRP) (CRP28-2022RS-0001). XZ thanks China Scholarship Council (CSC) for a scholarship allowing him to study in Singapore.Notes and references
- L. Kraehenbuehl, C. H. Weng, S. Eghbali, J. D. Wolchok and T. Merghoub, Nat. Rev. Clin. Oncol., 2022, 19, 37–50 CrossRef CAS.
- K. D. Miller, L. Nogueira, T. Devasia, A. B. Mariotto, K. R. Yabroff, A. Jemal, J. Kramer and R. L. Siegel, Ca-Cancer J. Clin., 2022, 72, 409–436 CrossRef.
- B. S. Pulendran, P. Arunachalam and D. T. O'Hagan, Nat. Rev. Drug Discovery, 2021, 20, 454–475 CrossRef CAS.
- T. Ye, F. Li, G. Ma and W. Wei, Adv. Drug Delivery Rev., 2021, 177, 113927 CrossRef CAS.
- G. Del Giudice, R. Rappuoli and A. M. Didierlaurent, Semin. Immunol., 2018, 39, 14–21 CrossRef CAS.
- C. Pifferi, R. Fuentes and A. Fernandez-Tejada, Nat. Rev. Chem., 2021, 5, 197–216 CrossRef CAS.
- M. L. Bookstaver, S. J. Tsai, J. S. Bromberg and C. M. Jewell, Trends Immunol., 2018, 39, 135–150 CrossRef.
- S. G. Reed, S. Bertholet, R. N. Coler and M. Friede, Trends Immunol., 2009, 30, 23–32 CrossRef CAS.
- E. Blass and P. A. Ott, Nat. Rev. Clin. Oncol., 2021, 18, 215–229 CrossRef.
- M. Saxena, S. H. van der Burg, C. J. M. Melief and N. Bhardwaj, Nat. Rev. Cancer, 2021, 21, 360–378 CrossRef CAS PubMed.
- U. Sahin and O. Tureci, Science, 2018, 359, 1355–1360 CrossRef CAS PubMed.
- T. W. Dubensky, Jr. and S. G. Reed, Semin. Immunol., 2010, 22, 155–161 CrossRef PubMed.
- F. Lang, B. Schrors, M. Lower, O. Tureci and U. Sahin, Nat. Rev. Drug Discovery, 2022, 21, 261–282 CrossRef CAS PubMed.
- O. Demaria, S. Cornen, M. Daeron, Y. Morel, R. Medzhitov and E. Vivier, Nature, 2019, 574, 45–56 CrossRef CAS PubMed.
- S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Chem. Soc. Rev., 2022, 51, 1212–1233 RSC.
- M. Aleynick, J. Svensson-Arvelund, C. R. Flowers, A. Marabelle and J. D. Brody, Clin. Cancer Res., 2019, 25, 6283–6294 CrossRef CAS PubMed.
- S. Bhagchandani, J. A. Johnson and D. J. Irvine, Adv. Drug Delivery Rev., 2021, 175, 113803 CrossRef CAS PubMed.
- S. Van Herck, B. Feng and L. Tang, Adv. Drug Delivery Rev., 2021, 179, 114020 CrossRef CAS PubMed.
- X. Cao, Nat. Rev. Immunol., 2016, 16, 35–50 CrossRef CAS PubMed.
- B. Wang, S. Van Herck, Y. Chen, X. Bai, Z. Zhong, K. Deswarte, B. N. Lambrecht, N. N. Sanders, S. Lienenklaus, H. W. Scheeren, S. A. David, F. Kiessling, T. Lammers, B. G. De Geest and Y. Shi, J. Am. Chem. Soc., 2020, 142, 12133–12139 CrossRef CAS PubMed.
- S. P. Kasturi, I. Skountzou, R. A. Albrecht, D. Koutsonanos, T. Hua, H. I. Nakaya, R. Ravindran, S. Stewart, M. Alam, M. Kwissa, F. Villinger, N. Murthy, J. Steel, J. Jacob, R. J. Hogan, A. Garcia-Sastre, R. Compans and B. Pulendran, Nature, 2011, 470, 543–547 CrossRef CAS PubMed.
- H. Zhang, X. You, X. Wang, L. Cui, Z. Wang, F. Xu, M. Li, Z. Yang, J. Liu, P. Huang, Y. Kang, J. Wu and X. Xia, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2005191118 CrossRef CAS PubMed.
- T. Y. Wu, M. Singh, A. T. Miller, E. De Gregorio, F. Doro, U. D'Oro, D. A. Skibinski, M. L. Mbow, S. Bufali, A. E. Herman, A. Cortez, Y. Li, B. P. Nayak, E. Tritto, C. M. Filippi, G. R. Otten, L. A. Brito, E. Monaci, C. Li, S. Aprea, S. Valentini, S. Calabromicron, D. Laera, B. Brunelli, E. Caproni, P. Malyala, R. G. Panchal, T. K. Warren, S. Bavari, D. T. O'Hagan, M. P. Cooke and N. M. Valiante, Sci. Transl. Med., 2014, 6, 263ra160 Search PubMed.
- E. L. Dane, A. Belessiotis-Richards, C. Backlund, J. Wang, K. Hidaka, L. E. Milling, S. Bhagchandani, M. B. Melo, S. Wu, N. Li, N. Donahue, K. Ni, L. Ma, M. Okaniwa, M. M. Stevens, A. Alexander-Katz and D. J. Irvine, Nat. Mater., 2022, 21, 710–720 CrossRef CAS PubMed.
- Y. Chen, S. De Koker and B. G. De Geest, Acc. Chem. Res., 2020, 53, 2055–2067 CrossRef CAS PubMed.
- C. H. Choi, L. Hao, S. P. Narayan, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7625–7630 CrossRef CAS PubMed.
- M. H. Teplensky, M. E. Distler, C. D. Kusmierz, M. Evangelopoulos, H. Gula, D. Elli, A. Tomatsidou, V. Nicolaescu, I. Gelarden, A. Yeldandi, D. Batlle, D. Missiakas and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119093119 CrossRef CAS PubMed.
- P. Kumthekar, C. H. Ko, T. Paunesku, K. Dixit, A. M. Sonabend, O. Bloch, M. Tate, M. Schwartz, L. Zuckerman, R. Lezon, R. V. Lukas, B. Jovanovic, K. McCortney, H. Colman, S. Chen, B. Lai, O. Antipova, J. Deng, L. Li, S. Tommasini-Ghelfi, L. A. Hurley, D. Unruh, N. V. Sharma, M. Kandpal, F. M. Kouri, R. V. Davuluri, D. J. Brat, M. Muzzio, M. Glass, V. Vijayakumar, J. Heidel, F. J. Giles, A. K. Adams, C. D. James, G. E. Woloschak, C. Horbinski and A. H. Stegh, Sci. Transl. Med., 2021, 13, eabb3945 CrossRef CAS PubMed.
- J. Li, H. Ren and Y. Zhang, Coord. Chem. Rev., 2022, 455, 214345 CrossRef CAS.
- J. Lotscher, I. L. A. A. Marti, N. Kirchhammer, E. Cribioli, G. M. P. Giordano Attianese, M. P. Trefny, M. Lenz, S. I. Rothschild, P. Strati, M. Kunzli, C. Lotter, S. H. Schenk, P. Dehio, J. Loliger, L. Litzler, D. Schreiner, V. Koch, N. Page, D. Lee, J. Grahlert, D. Kuzmin, A. V. Burgener, D. Merkler, M. Pless, M. L. Balmer, W. Reith, J. Huwyler, M. Irving, C. G. King, A. Zippelius and C. Hess, Cell, 2022, 185, 585–602.e529 CrossRef CAS PubMed.
- S. Liu, Q. Jiang, X. Zhao, R. Zhao, Y. Wang, Y. Wang, J. Liu, Y. Shang, S. Zhao, T. Wu, Y. Zhang, G. Nie and B. Ding, Nat. Mater., 2020, 20, 421–430 CrossRef PubMed.
- R. Kuai, X. Sun, W. Yuan, L. J. Ochyl, Y. Xu, A. Hassani Najafabadi, L. Scheetz, M. Z. Yu, I. Balwani, A. Schwendeman and J. J. Moon, J. Controlled Release, 2018, 282, 131–139 CrossRef CAS PubMed.
- D. Wan, H. Que, L. Chen, T. Lan, W. Hong, C. He, J. Yang, Y. Wei and X. Wei, Nano Lett., 2021, 21, 7960–7969 CrossRef CAS PubMed.
- G. M. Lynn, C. Sedlik, F. Baharom, Y. Zhu, R. A. Ramirez-Valdez, V. L. Coble, K. Tobin, S. R. Nichols, Y. Itzkowitz, N. Zaidi, J. M. Gammon, N. J. Blobel, J. Denizeau, P. de la Rochere, B. J. Francica, B. Decker, M. Maciejewski, J. Cheung, H. Yamane, M. G. Smelkinson, J. R. Francica, R. Laga, J. D. Bernstock, L. W. Seymour, C. G. Drake, C. M. Jewell, O. Lantz, E. Piaggio, A. S. Ishizuka and R. A. Seder, Nat. Biotechnol., 2020, 38, 320–332 CrossRef CAS PubMed.
- G. M. Lynn, R. Laga, P. A. Darrah, A. S. Ishizuka, A. J. Balaci, A. E. Dulcey, M. Pechar, R. Pola, M. Y. Gerner, A. Yamamoto, C. R. Buechler, K. M. Quinn, M. G. Smelkinson, O. Vanek, R. Cawood, T. Hills, O. Vasalatiy, K. Kastenmuller, J. R. Francica, L. Stutts, J. K. Tom, K. A. Ryu, A. P. Esser-Kahn, T. Etrych, K. D. Fisher, L. W. Seymour and R. A. Seder, Nat. Biotechnol., 2015, 33, 1201–1210 CrossRef CAS PubMed.
- R. Kuai, L. J. Ochyl, K. S. Bahjat, A. Schwendeman and J. J. Moon, Nat. Mater., 2017, 16, 489–496 CrossRef CAS PubMed.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. Van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS PubMed.
- G. Zhu, G. M. Lynn, O. Jacobson, K. Chen, Y. Liu, H. Zhang, Y. Ma, F. Zhang, R. Tian, Q. Ni, S. Cheng, Z. Wang, N. Lu, B. C. Yung, Z. Wang, L. Lang, X. Fu, A. Jin, I. D. Weiss, H. Vishwasrao, G. Niu, H. Shroff, D. M. Klinman, R. A. Seder and X. Chen, Nat. Commun., 2017, 8, 1954 CrossRef PubMed.
- P. Chen, D. Wang, Y. Wang, L. Zhang, Q. Wang, L. Liu, J. Li, X. Sun, M. Ren, R. Wang, Y. Fang, J. J. Zhao and K. Zhang, Nano Lett., 2022, 22, 4058–4066 CrossRef CAS PubMed.
- D. Shae, K. W. Becker, P. Christov, D. S. Yun, A. K. R. Lytton-Jean, S. Sevimli, M. Ascano, M. Kelley, D. B. Johnson, J. M. Balko and J. T. Wilson, Nat. Nanotechnol., 2019, 14, 269–278 CrossRef CAS PubMed.
- X. Lu, L. Miao, W. Gao, Z. Chen, K. J. McHugh, Y. Sun, Z. Tochka, S. Tomasic, K. Sadtler, A. Hyacinthe, Y. Huang, T. Graf, Q. Hu, M. Sarmadi, R. Langer, D. G. Anderson and A. Jaklenec, Sci. Transl. Med., 2020, 12, eaaz6606 CrossRef CAS PubMed.
- X. Li, S. Khorsandi, Y. Wang, J. Santelli, K. Huntoon, N. Nguyen, M. Yang, D. Lee, Y. Lu, R. Gao, B. Y. S. Kim, C. de Gracia Lux, R. F. Mattrey, W. Jiang and J. Lux, Nat. Nanotechnol., 2022, 17, 891–899 CrossRef CAS PubMed.
- L. Zhou, B. Hou, D. Wang, F. Sun, R. Song, Q. Shao, H. Wang, H. Yu and Y. Li, Nano Lett., 2020, 20, 4393–4402 CrossRef CAS PubMed.
- X. Sun, Y. Zhang, J. Li, K. S. Park, K. Han, X. Zhou, Y. Xu, J. Nam, J. Xu, X. Shi, L. Wei, Y. L. Lei and J. J. Moon, Nat. Nanotechnol., 2021, 16, 1260–1270 CrossRef CAS PubMed.
- B. Zheng, J. Xu, G. Chen, S. Zhang, Z. Xiao and W. Lu, Adv. Funct. Mater., 2019, 29, 1901437 CrossRef.
- A. Schudel, A. P. Chapman, M. K. Yau, C. J. Higginson, D. M. Francis, M. P. Manspeaker, A. R. C. Avecilla, N. A. Rohner, M. G. Finn and S. N. Thomas, Nat. Nanotechnol., 2020, 15, 491–499 CrossRef CAS PubMed.
- Y. N. Zhang, J. Lazarovits, W. Poon, B. Ouyang, L. N. M. Nguyen, B. R. Kingston and W. C. W. Chan, Nano Lett., 2019, 19, 7226–7235 CrossRef CAS PubMed.
- W. Wei, C. Luo, J. Yang, B. Sun, D. Zhao, Y. Liu, Y. Wang, W. Yang, Q. Kan, J. Sun and Z. He, J. Controlled Release, 2018, 285, 187–199 CrossRef CAS PubMed.
- F. Kratz, J. Controlled Release, 2014, 190, 331–336 CrossRef CAS PubMed.
- J. Pan, Y. Wang, C. Zhang, X. Wang, H. Wang, J. Wang, Y. Yuan, X. Wang, X. Zhang, C. Yu, S. K. Sun and X. P. Yan, Adv. Mater., 2018, 30, 1704408 CrossRef PubMed.
- J. Lau, O. Jacobson, G. Niu, K. S. Lin, F. Benard and X. Chen, Bioconjugate Chem., 2019, 30, 487–502 CrossRef CAS PubMed.
- F. Zhang, G. Zhu, O. Jacobson, Y. Liu, K. Chen, G. Yu, Q. Ni, J. Fan, Z. Yang, F. Xu, X. Fu, Z. Wang, Y. Ma, G. Niu, X. Zhao and X. Chen, ACS Nano, 2017, 11, 8838–8848 CrossRef CAS PubMed.
- Z. Zhou, C. Du, Q. Zhang, G. Yu, F. Zhang and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 21033–21039 CrossRef CAS PubMed.
- M. U. Munir, Cancers, 2022, 14, 2904 CrossRef CAS PubMed.
- K. Yang, W. Han, X. Jiang, A. Piffko, J. Bugno, C. Han, S. Li, H. Liang, Z. Xu, W. Zheng, L. Wang, J. Wang, X. Huang, J. P. Y. Ting, Y. X. Fu, W. Lin and R. R. Weichselbaum, Nat. Nanotechnol., 2022, 17, 1322–1331 CrossRef CAS PubMed.
- E. L. Dane, A. Belessiotis-Richards, C. Backlund, J. Wang, K. Hidaka, L. E. Milling, S. Bhagchandani, M. B. Melo, S. Wu, N. Li, N. Donahue, K. Ni, L. Ma, M. Okaniwa, M. M. Stevens, A. Alexander-Katz and D. J. Irvine, Nat. Mater., 2022, 21, 710–720 CrossRef CAS PubMed.
- C. C. de Oliveira Mann and V. Hornung, Eur. J. Immunol., 2021, 51, 1897–1910 CrossRef CAS PubMed.
- A. I. S. van den Berg, C. O. Yun, R. M. Schiffelers and W. E. Hennink, J. Controlled Release, 2021, 331, 121–141 CrossRef CAS PubMed.
- A. Franchini, S. Bertuzzi, C. Tosarelli and G. Manfreda, Poult. Sci., 1995, 74, 666–671 CrossRef CAS PubMed.
- L. Li, Z. Yang and X. Chen, Acc. Chem. Res., 2020, 53, 2044–2054 CrossRef CAS PubMed.
- H. Phuengkham, C. Song, S. H. Um and Y. T. Lim, Adv. Mater., 2018, 30, e1706719 CrossRef PubMed.
- H. Phuengkham, C. Song and Y. T. Lim, Adv. Mater., 2019, 31, e1903242 CrossRef PubMed.
- Z. Luo, Q. Wu, C. Yang, H. Wang, T. He, Y. Wang, Z. Wang, H. Chen, X. Li, C. Gong and Z. Yang, Adv. Mater., 2017, 29, 1601776 CrossRef PubMed.
- G. A. Roth, E. C. Gale, M. Alcantara-Hernandez, W. Luo, E. Axpe, R. Verma, Q. Yin, A. C. Yu, H. Lopez Hernandez, C. L. Maikawa, A. A. A. Smith, M. M. Davis, B. Pulendran, J. Idoyaga and E. A. Appel, ACS Cent. Sci., 2020, 6, 1800–1812 CrossRef CAS PubMed.
- P. Makvandi, R. Jamaledin, G. Chen, Z. Baghbantaraghdari, E. N. Zare, C. Di Natale, V. Onesto, R. Vecchione, J. Lee, F. R. Tay, P. Netti, V. Mattoli, A. Jaklenec, Z. Gu and R. Langer, Mater. Today, 2021, 47, 206–222 CrossRef CAS PubMed.
- J.-J. Wu, F.-Y. Chen, B.-B. Han, H.-Q. Zhang, L. Zhao, Z.-R. Zhang, J.-J. Li, B.-D. Zhang, Y.-N. Zhang, Y.-X. Yue, H.-G. Hu, W.-H. Li, B. Zhang, Y.-X. Chen, D.-S. Guo and Y.-M. Li, CCS Chem., 2022, 0, 1–17 Search PubMed.
- Q. Ni, F. Zhang, Y. Liu, Z. Wang, G. Yu, B. Liang, G. Niu, T. Su, G. Zhu, G. Lu, L. Zhang and X. Chen, Sci. Adv., 2020, 6, eaaw6071 CrossRef CAS PubMed.
- Z. T. Bennett, S. Li, B. D. Sumer and J. Gao, Semin. Immunol., 2021, 56, 101580 CrossRef CAS PubMed.
- S. M. Jin, Y. J. Yoo, H. S. Shin, S. Kim, S. N. Lee, C. H. Lee, H. Kim, J. E. Kim, Y. S. Bae, J. Hong, Y. W. Noh and Y. T. Lim, Nat. Nanotechnol., 2023 DOI:10.1038/s41565-022-01296-w.
- Q. Pena, A. Wang, O. Zaremba, Y. Shi, H. W. Scheeren, J. M. Metselaar, F. Kiessling, R. M. Pallares, S. Wuttke and T. Lammers, Chem. Soc. Rev., 2022, 51, 2544–2582 RSC.
- X. Shi, Y. Bi, W. Yang, X. Guo, Y. Jiang, C. Wan, L. Li, Y. Bai, J. Guo, Y. Wang, X. Chen, B. Wu, H. Sun, W. Liu, J. Wang and C. Xu, Nature, 2013, 493, 111–115 CrossRef PubMed.
- K. G. Chandy and R. S. Norton, Nature, 2016, 537, 497–499 CrossRef CAS PubMed.
- C. Wang, Y. Guan, M. Lv, R. Zhang, Z. Guo, X. Wei, X. Du, J. Yang, T. Li, Y. Wan, X. Su, X. Huang and Z. Jiang, Immunity, 2018, 48, 675–687.e677 CrossRef CAS PubMed.
- Z. Luo, X. Liang, T. He, X. Qin, X. Li, Y. Li, L. Li, X. J. Loh, C. Gong and X. Liu, J. Am. Chem. Soc., 2022, 144, 16366–16377 CrossRef CAS PubMed.
- O. J. Finn, N. Engl. J. Med., 2008, 358, 2704–2715 CrossRef CAS PubMed.
- L. Miao, L. Li, Y. Huang, D. Delcassian, J. Chahal, J. Han, Y. Shi, K. Sadtler, W. Gao, J. Lin, J. C. Doloff, R. Langer and D. G. Anderson, Nat. Biotechnol., 2019, 37, 1174–1185 CrossRef CAS PubMed.
- H. G. Hu and Y. M. Li, Front. Chem., 2020, 8, 601 CrossRef CAS PubMed.
- S. Li, M. Luo, Z. Wang, Q. Feng, J. Wilhelm, X. Wang, W. Li, J. Wang, A. Cholka, Y. X. Fu, B. D. Sumer, H. Yu and J. Gao, Nat. Biomed. Eng., 2021, 5, 455–466 CrossRef CAS PubMed.
- G. N. Barber, Nat. Rev. Immunol., 2015, 15, 760–770 CrossRef CAS PubMed.
- S. L. Ergun, D. Fernandez, T. M. Weiss and L. Li, Cell, 2019, 178, 290–301.e210 CrossRef CAS PubMed.
- J. Zhao, Y. Xu, S. Ma, Y. Wang, Z. Huang, H. Qu, H. Yao, Y. Zhang, G. Wu, L. Huang, W. Song, Z. Tang and X. Chen, Adv. Mater., 2022, 34, e2109254 CrossRef PubMed.
- M. Luo, H. Wang, Z. Wang, H. Cai, Z. Lu, Y. Li, M. Du, G. Huang, C. Wang, X. Chen, M. R. Porembka, J. Lea, A. E. Frankel, Y. X. Fu, Z. J. Chen and J. Gao, Nat. Nanotechnol., 2017, 12, 648–654 CrossRef CAS PubMed.
- X. Huang, N. Kong, X. Zhang, Y. Cao, R. Langer and W. Tao, Nat. Med., 2022, 28, 2273–2287 CrossRef CAS PubMed.
- Y. Zhao, W. Wang, S. Guo, Y. Wang, L. Miao, Y. Xiong and L. Huang, Nat. Commun., 2016, 7, 11822 CrossRef PubMed.
- J. Li, W. Wang, Y. He, Y. Li, E. Z. Yan, K. Zhang, D. J. Irvine and P. T. Hammond, ACS Nano, 2017, 11, 2531–2544 CrossRef CAS PubMed.
- S. Duan, W. Yuan, F. Wu and T. Jin, Angew. Chem., Int. Ed., 2012, 51, 7938–7941 CrossRef CAS PubMed.
- A. W. Li, M. C. Sobral, S. Badrinath, Y. Choi, A. Graveline, A. G. Stafford, J. C. Weaver, M. O. Dellacherie, T. Y. Shih, O. A. Ali, J. Kim, K. W. Wucherpfennig and D. J. Mooney, Nat. Mater., 2018, 17, 528–534 CrossRef CAS PubMed.
- H. Chang, J. Lv, X. Gao, X. Wang, H. Wang, H. Chen, X. He, L. Li and Y. Cheng, Nano Lett., 2017, 17, 1678–1684 CrossRef CAS PubMed.
- C. Yu, E. Tan, Y. Xu, J. Lv and Y. Cheng, Bioconjugate Chem., 2019, 30, 413–417 CrossRef CAS PubMed.
- J. Xu, J. Lv, Q. Zhuang, Z. Yang, Z. Cao, L. Xu, P. Pei, C. Wang, H. Wu, Z. Dong, Y. Chao, C. Wang, K. Yang, R. Peng, Y. Cheng and Z. Liu, Nat. Nanotechnol., 2020, 15, 1043–1052 CrossRef CAS PubMed.
- S. Jin, H. T. Vu, K. Hioki, N. Noda, H. Yoshida, T. Shimane, S. Ishizuka, I. Takashima, Y. Mizuhata, K. Beverly Pe, T. Ogawa, N. Nishimura, D. Packwood, N. Tokitoh, H. Kurata, S. Yamasaki, K. J. Ishii and M. Uesugi, Angew. Chem., Int. Ed., 2021, 60, 961–969 CrossRef CAS PubMed.
- S. Jin, S. H. Zhuo, Y. Takemoto, Y. M. Li and M. Uesugi, Chem. Commun., 2022, 58, 12228–12231 RSC.
- W. Song, M. Das, Y. Xu, X. Si, Y. Zhang, Z. Tang and X. Chen, Mater. Today Nano, 2019, 5, 100029 CrossRef.
- S. Manna, W. J. Howitz, N. J. Oldenhuis, A. C. Eldredge, J. Shen, F. N. Nihesh, M. B. Lodoen, Z. Guan and A. P. Esser-Kahn, ACS Cent. Sci., 2018, 4, 982–995 CrossRef CAS PubMed.
- N. Gong, Y. Zhang, X. Teng, Y. Wang, S. Huo, G. Qing, Q. Ni, X. Li, J. Wang, X. Ye, T. Zhang, S. Chen, Y. Wang, J. Yu, P. C. Wang, Y. Gan, J. Zhang, M. J. Mitchell, J. Li and X. J. Liang, Nat. Nanotechnol., 2020, 15, 1053–1064 CrossRef CAS PubMed.
- H. Zhang, X. You, X. Wang, L. Cui, Z. Wang, F. Xu, M. Li, Z. Yang, J. Liu, P. Huang, Y. Kang, J. Wu and X. Xia, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2005191118 CrossRef CAS PubMed.
- L. Xu, X. Wang, W. Wang, M. Sun, W. J. Choi, J.-Y. Kim, C. Hao, S. Li, A. Qu, M. Lu, X. Wu, F. M. Colombari, W. R. Gomes, A. L. Blanco, A. F. de Moura, X. Guo, H. Kuang, N. A. Kotov and C. Xu, Nature, 2022, 601, 366–373 CrossRef CAS PubMed.
- V. Hornung, F. Bauernfeind, A. Halle, E. O. Samstad, H. Kono, K. L. Rock, K. A. Fitzgerald and E. Latz, Nat. Immunol., 2008, 9, 847–856 CrossRef CAS PubMed.
- N. Dalbeth, A. L. Gosling, A. Gaffo and A. Abhishek, Lancet, 2021, 397, 1843–1855 CrossRef CAS PubMed.
- S. Kumar, A. C. Anselmo, A. Banerjee, M. Zakrewsky and S. Mitragotri, J. Controlled Release, 2015, 220, 141–148 CrossRef CAS PubMed.
- D. Zhang, L. Wei, M. Zhong, L. Xiao, H. W. Li and J. Wang, Chem. Sci., 2018, 9, 5260–5269 RSC.
- F. Lebre, R. Sridharan, M. J. Sawkins, D. J. Kelly, F. J. O'Brien and E. C. Lavelle, Sci. Rep., 2017, 7, 2922 CrossRef PubMed.
- H. Vallhov, S. Gabrielsson, M. Stromme, A. Scheynius and A. E. Garcia-Bennett, Nano Lett., 2007, 7, 3576–3582 CrossRef CAS PubMed.
- V. Kanchan and A. K. Panda, Biomaterials, 2007, 28, 5344–5357 CrossRef CAS PubMed.
- B. Sun, Z. Ji, Y. P. Liao, M. Wang, X. Wang, J. Dong, C. H. Chang, R. Li, H. Zhang, A. E. Nel and T. Xia, ACS Nano, 2013, 7, 10834–10849 CrossRef CAS PubMed.
- X. Wang, X. Li, A. Ito, Y. Sogo and T. Ohno, Acta Biomater., 2013, 9, 7480–7489 CrossRef CAS PubMed.
- K. Niikura, T. Matsunaga, T. Suzuki, S. Kobayashi, H. Yamaguchi, Y. Orba, A. Kawaguchi, H. Hasegawa, K. Kajino, T. Ninomiya, K. Ijiro and H. Sawa, ACS Nano, 2013, 7, 3926–3938 CrossRef CAS PubMed.
- J. Wang, H. J. Chen, T. Hang, Y. Yu, G. Liu, G. He, S. Xiao, B. R. Yang, C. Yang, F. Liu, J. Tao, M. X. Wu and X. Xie, Nat. Nanotechnol., 2018, 13, 1078–1086 CrossRef CAS PubMed.
- X. Hong, X. Zhong, G. Du, Y. Hou, Y. Zhang, Z. Zhang, T. Gong, L. Zhang and X. Sun, Sci. Adv., 2020, 6, eaaz4462 CrossRef CAS PubMed.
- W. Wang, J. Zhao, C. Hao, S. Hu, C. Chen, Y. Cao, Z. Xu, J. Guo, L. Xu, M. Sun, C. Xu and H. Kuang, Adv. Mater., 2022, 34, e2109354 CrossRef PubMed.
- M. Li, H. Zhou, W. Jiang, C. Yang, H. Miao and Y. Wang, Nano Today, 2020, 35, 101007 CrossRef CAS.
- C. Liu, X. Liu, X. Xiang, X. Pang, S. Chen, Y. Zhang, E. Ren, L. Zhang, X. Liu, P. Lv, X. Wang, W. Luo, N. Xia, X. Chen and G. Liu, Nat. Nanotechnol., 2022, 17, 531–540 CrossRef CAS PubMed.
- L. Chen, H. Qin, R. Zhao, X. Zhao, L. Lin, Y. Chen, Y. Lin, Y. Li, Y. Qin, Y. Li, S. Liu, K. Cheng, H. Chen, J. Shi, G. J. Anderson, Y. Wu, Y. Zhao and G. Nie, Sci. Transl. Med., 2021, 13, eabc2816 CrossRef CAS PubMed.
- M. Fusciello, F. Fontana, S. Tahtinen, C. Capasso, S. Feola, B. Martins, J. Chiaro, K. Peltonen, L. Ylosmaki, E. Ylosmaki, F. Hamdan, O. K. Kari, J. Ndika, H. Alenius, A. Urtti, J. T. Hirvonen, H. A. Santos and V. Cerullo, Nat. Commun., 2019, 10, 5747 CrossRef CAS PubMed.
- L. L. Huang, W. Wang, Z. Wang, H. Zhang, H. Liu, G. Wu, W. Nie and H. Y. Xie, Adv. Funct. Mater., 2022, 33, 202209056 Search PubMed.
- Y. Jiang, N. Krishnan, J. Zhou, S. Chekuri, X. Wei, A. V. Kroll, C. L. Yu, Y. Duan, W. Gao, R. H. Fang and L. Zhang, Adv. Mater., 2020, 32, e2001808 CrossRef PubMed.
- Y. Zhao, L. Liu, R. Sun, G. Cui, S. Guo, S. Han, Z. Li, T. Bai and L. Teng, Asian J. Pharm. Sci., 2022, 17, 193–205 CrossRef PubMed.
- Z. Xu, S. Zeng, Z. Gong and Y. Yan, Mol. Cancer, 2020, 19, 160 CrossRef CAS PubMed.
- C. D. Phung, T. T. Pham, H. T. Nguyen, T. T. Nguyen, W. Ou, J. H. Jeong, H. G. Choi, S. K. Ku, C. S. Yong and J. O. Kim, Acta Biomater., 2020, 115, 371–382 CrossRef CAS PubMed.
- S. Cheng, C. Xu, Y. Jin, Y. Li, C. Zhong, J. Ma, J. Yang, N. Zhang, Y. Li, C. Wang, Z. Yang and Y. Wang, Adv. Sci., 2020, 7, 1903301 CrossRef CAS PubMed.
- T. Pradeu and E. Vivier, Sci. Immunol., 2016, 1, aag0479 Search PubMed.
- G. Yamankurt, E. J. Berns, A. Xue, A. Lee, N. Bagheri, M. Mrksich and C. A. Mirkin, Nat. Biomed. Eng., 2019, 3, 318–327 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |