DOI:
10.1039/D2VA00034B
(Communication)
Environ. Sci.: Adv., 2022,
1, 438-442
Room-temperature coupling of methane with singlet oxygen†
Received
22nd February 2022
, Accepted 30th June 2022
First published on 3rd August 2022
Abstract
Owing to emission of methane (CH4) causing global warming and waste of resources, conversion of CH4 to value-added chemicals can mitigate environmental sustainability and energy concerns. Direct room-temperature coupling of CH4 to form ethane (CH3CH3) challenges chemists owing to the strong C–H bonds requiring high temperature (>700 °C) for dehydrogenation of CH4. Oxidative coupling is a promising approach for CH4 conversion to C2H6 using solar energy at room temperature. To achieve high efficiency of C2H6 formation, using an appropriate oxidant is a potential strategy to avoid overoxidation during the CH4 coupling process. Singlet oxygen (1O2) has typically manifested a mild redox capacity with a high selectivity to attack organic substrate CH4. Here, we report a synergistic photocatalytic-oxidative route for direct CH4 coupling. Under solar light irradiation, a high CH3CH3 generation rate of 647 μmol g−1 h−1 is achieved at 25 °C. Our work demonstrates that the solar-oxidative route can result in new and useful C1-based catalytic behaviors.
Environmental significance
Owing to emission of methane (CH4) causing global warming and waste of resources, conversion of CH4 to value-added chemicals can mitigate environmental and energy concerns. Direct room-temperature coupling of CH4 to form ethane (CH3CH3) challenges chemists owing to the strong C–H bonds requiring high temperature (>700 °C) for dehydrogenation of CH4. Oxidative coupling is a promising approach for CH4 conversion to C2H6 using solar energy at room temperature. To achieve high efficiency of C2H6 formation, using an appropriate oxidant is a potential strategy to avoid overoxidation during the CH4 coupling process. Singlet oxygen (1O2) has typically manifested a mild redox capacity with a high selectivity to attack organic substrate CH4. Here, we report a synergistic photocatalytic-oxidative route for direct CH4 coupling. Under solar light irradiation, a high CH3CH3 generation rate of 647 μmol g−1 h−1 is achieved at 25 °C. Our work demonstrates that the solar-oxidative route can result in new and useful C1-based catalytic behaviors.
|
Taking into account the environmental pollution and global warming caused by the use of traditional fossil energy and the shortage of its reserves, increasing the use of natural gas mainly composed of methane (CH4) is an inevitable trend,1 since methane has the advantage of being abundant and relatively inexpensive and clean. However, methane itself is also a greenhouse gas whose greenhouse effect is about 25 times that of carbon dioxide of the same mass.2 Thus, methane emissions contribute to global warming. On the other hand, the direct use of natural gas as a fuel will also cause environmental pollution and waste of resources, since methane storage and transportation are difficult and it is prone to leakage. These factors have made scientists invest a lot of energy in the research and development of simple and feasible technologies for converting methane into value-added chemical raw materials.
However, a high temperature (>700 °C) is required for thermodynamic dehydrogenation of CH4 due to the strong C–H bonds (434 kJ mol−1), leading to energy consumption and low selectivity of CH4 conversion.3 Photocatalytic methane conversion is a safe, low-energy and environmentally friendly strategy for the direct conversion of methane, since the dissociation of methane at room temperature can be achieved by means of photocatalytic methods using the light energy of sunlight and a suitable photocatalyst. Photocatalytic oxidation is also a promising approach for coupling of CH4 to form C2H6 using solar energy at room temperature.4 Nevertheless, the major challenge of CH4 coupling via the photocatalytic route is insufficient yield of target CH3CH3 and large production of by-products, e.g., HCOOH, CO, and CO2. Furthermore, noble metal co-catalysts, such as Au, Pd, and Pt, were generally used for promoting the efficiency of coupling of CH4.5 Developing oxidative-coupling and noble-metal-free catalyst systems, thus, is highly desirable for photocatalytic CH4 coupling at room temperature.
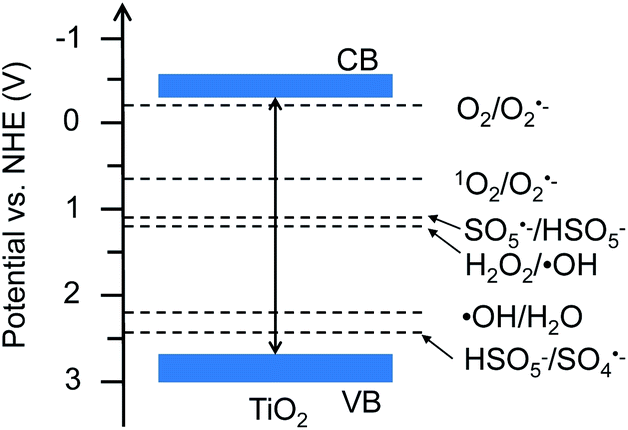
Traditionally, chemical oxidants, including O2, H2O2, and CO2, have been proven to be important in oxidative activation of CH4 to hydrocarbons, such as methanol.6 Actually, these oxidants potentially generate reactive oxygen species (ROS), such as the superoxide radical anion (O2˙−), hydroxyl radical (˙OH), sulfate radical (SO4˙−) and singlet oxygen (1O2), which are crucial in activation of CH4.7 In particular, the 1O2-based system typically manifested a mild redox capacity (2.2 V) with a high selectivity to attack organic substrates, compared to other free radicals such as SO4˙− (2.5–3.1 V) and ˙OH (2.7 V) (see Fig. 1).8
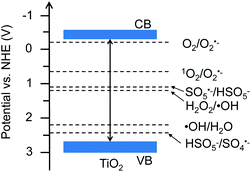 |
| | Fig. 1 Band structure of TiO2 (Degussa P25) and redox potentials of reactive oxygen species. | |
Peroxymonosulfate (P) as an excellent alternative oxidant has been confirmed to be the main source of HSO5− which can produce 1O2, ˙OH, and SO4˙− radicals.9 In particular, P can be utilized for selective oxidation of organic substances during which 1O2 is generated and serves as a mild oxidant with distinct reactivity towards different substrates.10 Importantly, the HSO5− molecule has a higher oxidizing potential (1.82 V) than H2O2 (1.76 V), and is thus more promising for activation of CH4.7 Therefore, P is often applied as an electron acceptor in photocatalytic degradation of organic pollutants.11 Nevertheless, P has never been studied for selective activation of CH4.
Herein, we develop a TiO2–1O2 system for the photocatalytic-oxidative route for CH4 coupling to form CH3CH3 with solar light at room temperature. Other oxidants, including O2, H2O2, and CO2, have been investigated to illustrate the important role of HSO5− in selectively controlling the coupling of CH4 to form C2H6. Further, an 1O2 involving radical-mediated pathway is proposed to explain the high activity of C2H6 formation from CH4. This work provides an alternative new approach for effective coupling of CH4 to form C2H6 at room temperature.
The XRD patterns of TiO2 in Fig. S1a† show the typical anatase and rutile diffraction peaks. The particle morphology with the size range of 10–30 nm and crystalline structure have been clearly indicated by the TEM and HRTEM images of TiO2, respectively (Fig. S1b and c†). Fig. 1 shows the band structure of TiO2 and the redox potentials of H2O2/˙OH, H2O/˙OH, O2/O2˙−, 1O2/O2˙−, SO5˙−/HSO5−, and HSO5−/SO4˙−.12,13 Based on these band and redox positions, the TiO2 material is expected to present enhanced performance for radical generation and activation of CH4.
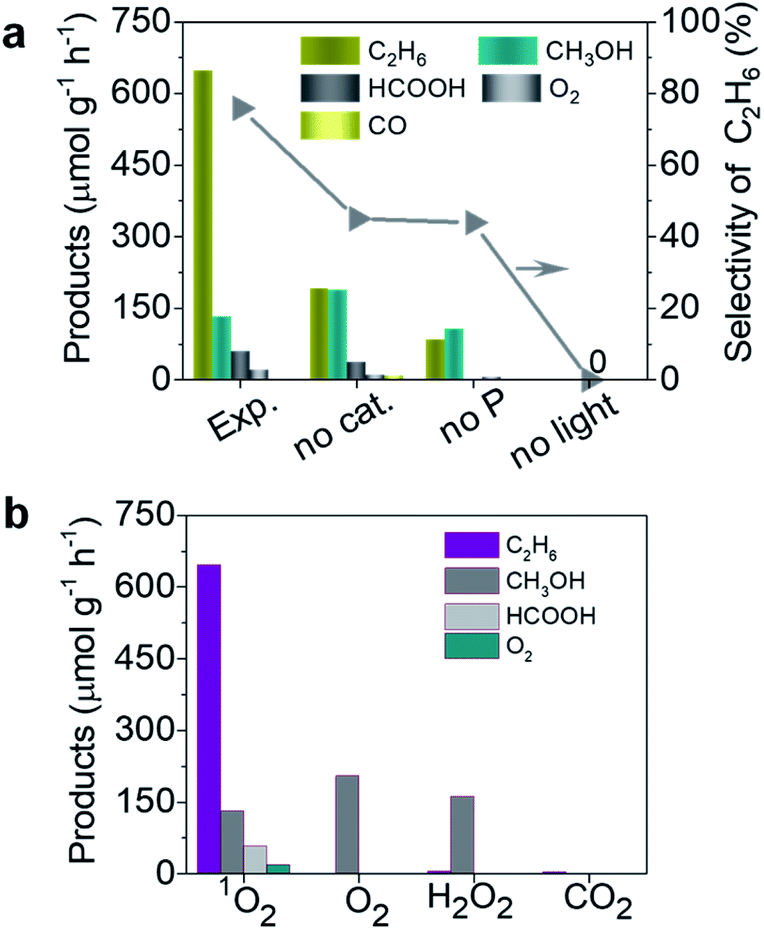
In order to reveal the photocatalytic performance of the TiO2–1O2 system, we first made a comparison of control experiments based on different reaction conditions, including light, catalyst, and HSO5− (see Fig. 2a). Under solar light irradiation, TiO2 with HSO5− as oxidant gave rise to an excellent performance for selective generation of C2H6, with a rate of 647 μmol g−1 h−1, much higher than the 180 and 89 μmol g−1 h−1 for the two by-products CH3OH and HCOOH, respectively, leading to a calculated C2H6 selectivity up to 75%. Notably, HSO5− can be independently activated by solar light with the corresponding reaction: HSO5− → SO4˙− + ˙OH.14 The ˙OH radical enables activation of CH4 to produce ˙CH3 species which are essential for C2H6 and CH3OH generation. Under these conditions only a little C2H6, CH3OH, HCOOH, and CO were detected, as displayed in Fig. 2a. For TiO2 as catalyst, the photo-generated carriers reacting with HSO5− generate more 1O2 which activates CH4 to generate ˙CH3 species, thus accelerating the coupling of ˙CH3 to form C2H6.
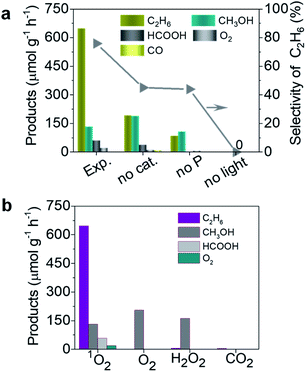 |
| | Fig. 2 Photocatalytic performance of the TiO2–1O2 system under solar light irradiation: (a) comparison of this work (Exp.) and control experiments by varying conditions (no light, no P, and no cat.); (b) products obtained with different oxidants (1O2, O2, H2O2, and CO2). | |
To reveal the crucial role of 1O2 in selective conversion of CH4 to C2H6, a control experiment was conducted using different oxidants for the conversion of CH4. Fig. 2b summarizes the results of CH4 oxidation with various oxidants (1O2, H2O2, O2, and CO2) under solar light irradiation. Apart from a little bit of CH3OH, trace amounts of C2H6 were found for H2O2, O2, and CO2 as oxidants, as shown in Fig. 2b. In contrast, the reaction with 1O2 as oxidant remarkably promotes the conversion of CH4 and selective generation of C2H6. Therefore, we conclude that 1O2 possesses superiority in view of the photocatalytic activity and selectivity for C2H6 generation. This is probably attributed to the specific band structure of TiO2 and more positive redox potential of HSO5−/SO4˙−, thus favouring generation of 1O2, as shown in Fig. 1. Additionally, to further understand the ability of 1O2, we controlled the amount of P which is the source of 1O2 (Fig. S2a†). As the amount of P was increased from 0 to 0.10 mmol, more C2H6 was selectively produced in addition to two other products CH3OH and HCOOH. Meanwhile, much more over-oxidation by-products (HCOOH, CO, and CO2) were generated as it increased to 0.4 mmol, as displayed in Fig. 3a and b. This is presumably owing to the over-oxidation of CH4. It is possible that excessive P may undergo a photoreaction (HSO5− → SO4˙− + ˙OH) and produce ˙OH, leading to the formation of CH3OH and subsequent over-oxidization to HCOOH.
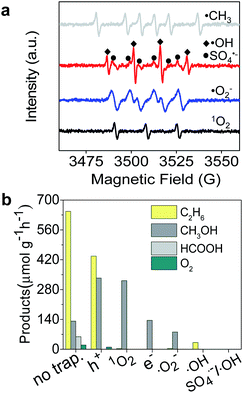 |
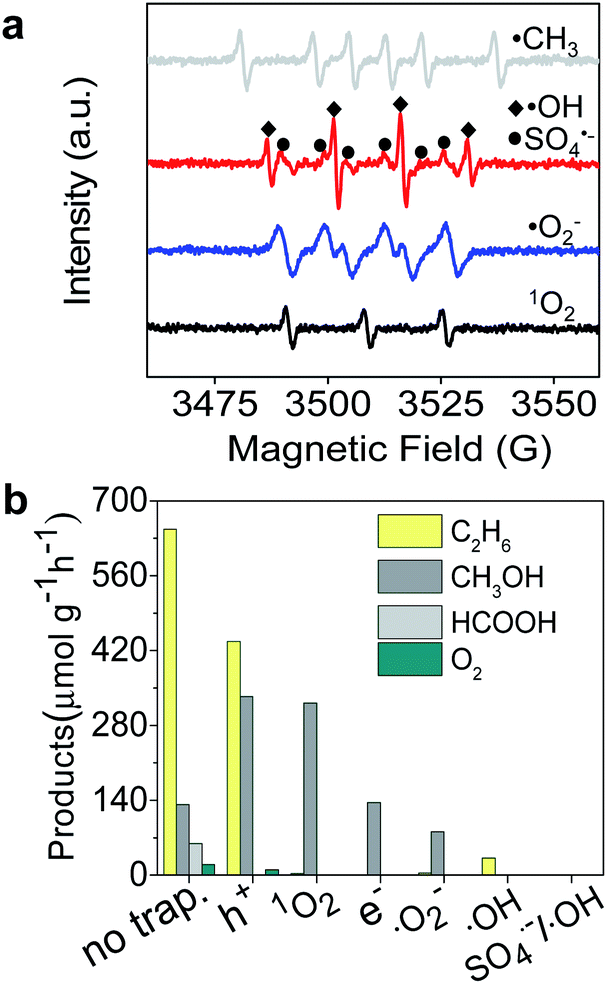
| | Fig. 3 (a) ESR spectra of ˙CH3, ˙OH, SO4˙−, and ˙O2− radicals produced after photocatalytic reaction for 10 min. (b) Products of CH4 conversion after adding scavengers para-quinone, K2Cr2O7, Na2C2O4, and salicylic acid in the reaction system for trapping ˙O2−, e−, h+, and ˙OH, respectively. | |
Therefore, excessive P normally results in formation of other by-products, leading to less C2H6. This result further suggests that an appropriate amount of P contributes to selective conversion of CH4 to C2H6. Importantly, such a noble-metal free catalyst system presents remarkable coupling of CH4 to form CH3CH3 compared to the various reported noble metal-based catalysts (see Table 1).
Table 1 Comparison of photocatalytic conversion of CH4 to CH3CH3 over reported noble-metal-based catalysts
| Catalysts |
Light source |
Temperature (°C) |
C2H6 (μmol g−1 h−1) |
Ref. |
| 6.0 wt% Ag–HPW/TiO2 |
Xe lamp 400 W (200 < λ < 1000 nm) |
30 |
20.3 |
15
|
| 1.0 wt% Pt/HGTS |
Xe lamp 300 W |
60 |
0.63 |
16
|
| 11.7 wt% Au/m-ZnO |
Xe lamp 300 W (solar light) |
30 |
11.3 |
17
|
| AuPd/ZnO (Pd, 1.0 wt%) |
Xe lamp 300 W (solar light) |
30 |
17.7 |
18
|
| 0.5 wt% Pd/Ga2O3 |
Xe lamp 300 W (λ = 254 nm) |
45 |
0.28 |
19
|
| WO3, H2O2 (2 mM) |
Mercury-vapor lamp (UVC-visible light) |
55 |
3.40 |
20
|
| HBEA |
Hg lamp 450 W |
70 |
14.3 |
21
|
| Cu0.1Pt0.5/PC-50 |
LED 40 W (λ = 365 nm) |
40 |
68.0 |
22
|
| Au–ZnO/TiO2 |
Xenon lamp 300 W (300 < λ < 500 nm) |
26 |
188 |
23
|
| TiO2, 1O2 |
Xe lamp 300 W (solar light) |
25 |
647 |
This work |
To gain a better understanding of the mechanism of the photocatalytic process for selective conversion of CH4 to C2H6, we used electron spin resonance (ESR) characterization and performed trapping experiments of active species, where 5-dimethyl-1-pyrroline-N-oxide (DMPO) was used as a trapping reagent to detect ˙CH3, SO4˙−, ˙OH, and O2˙− species. 2,2,6,6-Tetramethylpiperidine (TEMP) was used for detection of 1O2. As shown in Fig. 3a, the typical ˙CH3, SO4˙−, ˙OH, O2˙−, and 1O2 radical species were obviously formed during the photocatalytic process. To verify the distinctive roles of these species in selective conversion of CH4, we carried out trapping experiments using the corresponding scavengers, as displayed in Fig. 2a. The detailed reactions between scavenger reagents and active species are described in the ESI.† In Fig. 3b, the generation of C2H6 was significantly suppressed after trapping the 1O2 in Fig. 2b, while CH3OH production was slightly promoted during this process. This result suggests that 1O2 remarkably facilitates the formation of C2H6 and the ˙OH radical prefers to activate CH4 for generation of CH3OH. However, a fraction of C2H6 was still detectable even after trapping ˙OH in the reactive system, which is probably attributed to the remaining 1O2 radicals. Apart from a certain amount of CH3OH, C2H6 was never found in the absence of O2˙− active species (see Fig. 3b). This indicates that O2˙− also remarkably determined the selective formation of C2H6 which is related to the formation of 1O2. The e− was also essential for the selective conversion as it initiated the 1O2 generation through chain reactions, which was proven by the absence of C2H6 in products after elimination of photogenerated electrons. On the other hand, h+ only partially controlled the formation of C2H6 based on an h+ trapping experiment.
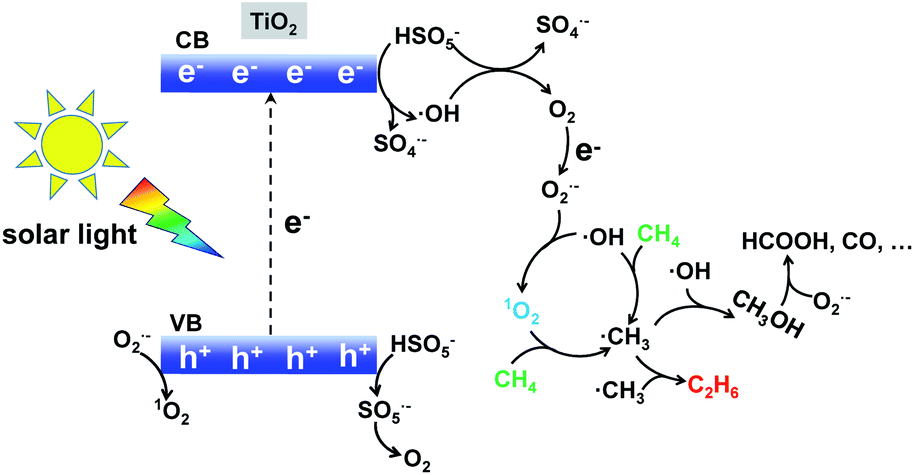
Based on the above experimental analysis, we proposed a plausible mechanism. As displayed in Scheme 1, photo-induced electrons reacted with HSO5− and generated SO4˙− and ˙OH radicals (eqn (1) and (2)).24 Meanwhile, direct light irradiation accelerated the generation of SO4˙− and ˙OH radicals (eqn (3)).7 O2 was formed based on the reactions described by eqn (4)–(6),7 which agrees well with the trace amount of O2 when 1O2 is used as oxidant in Fig. 2b. This O2 further generated the O2˙− radical according to the reactions described by eqn (7). Consequently, 1O2 was finally produced as a result of the presence of the O2˙− radical (see eqn (8) and (9)).25 The synthesized 1O2 was able to selectively dehydrogenize CH4 and generate the ˙CH3 radical which further underwent coupling, hence producing CH3CH3 (eqn (10)–(13)). It is noted that an increasing amount of ˙CH3 prefers to form C2H6,26 which is competitive with the CH3OH generation (˙CH3 + ˙OH → CH3OH).27,28 Therefore, when more ˙OH or O2˙− was present, CH3OH could be generally produced. This well indicates that the dominant product was CH3OH when radicals H2O2 and O2 were selected as oxidants in Fig. 2b. Taken together, 1O2 favoured selective production of CH3CH3, in comparison with H2O2 or O2-based systems. Apart from the products CH3CH3 and CH3OH, over-oxidation by-products such as HCOOH, CO, and even CO2 could also be formed (see eqn (14) and (15)) in the presence of the O2˙− radical.29
| | | HSO5− + e− → OH− + SO4˙− | (1) |
| | | HSO5− + e− → ˙OH + SO42− | (2) |
| | | 2HSO5− + 2˙OH → 2SO4˙− + 2H2O + O2 | (4) |
| | | HSO5− + h+ → H+ + SO5˙− | (5) |
| | | SO5˙− + SO5˙− → 2SO4˙− + O2 | (6) |
| | | ˙OH + O2˙− → 1O2 + OH− | (9) |
| | | CH4 + ˙OH → ˙CH3 + H2O | (11) |
| | | HCOOH + O2˙− → CO + CO2 | (15) |
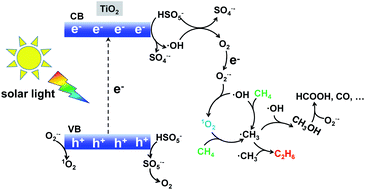 |
| | Scheme 1 Singlet oxygen involving radical-pathway mechanism for conversion of methane to ethane with the TiO2–1O2 system. | |
Conclusions
Solar-light driven selective conversion of methane to ethane has been achieved through a photocatalytic reaction at room temperature. By introducing HSO5− into a TiO2-based photocatalytic system, enhanced yields and selectivity of CH3CH3 are obtained largely due to the presence of 1O2 provided by HSO5−. Solar light stimulates the TiO2 catalyst to produce charge carriers (excited electrons and holes) which further activate HSO5− to generate 1O2. Detection and trapping experiments of active species further prove that the photocatalytic TiO2–1O2 system involves the 1O2 radical pathway mechanism. This report opens up a new possibility for efficient conversion of methane to ethane with solar energy at ambient temperature.
Author contributions
Anhua Huang and Jingsheng Wang prepared the samples, carried out the experiments, analysed the data and prepared the paper; Xingyang Wu and Hangchen Liu assisted with the characterization and photocatalytic tests; Jun Cai and Guo Qin Xu reviewed and edited the manuscript; Song Ling Wang supervised this work and reviewed/edited the manuscript; all authors discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was sponsored by the Shanghai Pujiang Talent Program (No. 19PJ1405200) and the Startup Fund for Youngman Research at SJTU (SFYR at SJTU, No. WF220516003).
Notes and references
- H. Herzog, B. Eliasson and O. Kaarstad, Capturing greenhouse gases, Sci. Am., 2000, 282, 72–79 CrossRef CAS PubMed.
- H. Song, X. Meng, Z.-j. Wang, H. Liu and J. Ye, Solar-energy-mediated methane conversion, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- Y. Zeng, H. C. Liu, J. S. Wang, X. Y. Wu and S. L. Wang, Synergistic photocatalysis–Fenton reaction for selective conversion of methane to methanol at room temperature, Catal. Sci. Technol., 2020, 10, 2329–2332 RSC.
- L. Yuliati and H. Yoshida, Photocatalytic conversion of methane, Chem. Soc. Rev., 2008, 37, 1592–1602 RSC.
- L. Yu, Y. Shao and D. Li, Direct combination of hydrogen evolution from water and methane conversion in a photocatalytic system over Pt/TiO2, Appl. Catal., B, 2017, 204, 216–223 CrossRef CAS.
- Y. Kang, Z. Li, X. Lv, W. Song, Y. Wei, X. Zhang, J. Liu and Z. Zhao, Active oxygen promoted electrochemical conversion of methane on two-dimensional carbide (MXenes): from stability, reactivity and selectivity, J. Catal., 2020, 393, 20–29 CrossRef.
- F. Ghanbari and M. Moradi, Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- S. Zhu, X. Li, J. Kang, X. Duan and S. Wang, Nonradical oxidation in persulfate activation by graphene-like nanosheets (GNS): differentiating the contributions of singlet oxygen (1O2) and sorption-dependent electron transfer, Environ. Sci. Technol., 2019, 53, 307–315 CrossRef CAS PubMed.
- D. Dai, Z. Yang, Y. Yao, L. Chen, G. Jia and L. Luo, Persulfate activation on crystallographic manganese oxides: mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants, Catal. Sci. Technol., 2017, 7, 934–942 RSC.
- Y. Zhou, J. Jiang, Y. Gao, J. Ma, S.-Y. Pang, J. Li, X.-T. Lu and L.-P. Yuan, Activation of peroxymonosulfate by benzoquinone: a novel nonradical oxidation process, Environ. Sci. Technol., 2015, 49, 12941–12950 CrossRef CAS PubMed.
- M. Ahmadi, F. Ghanbari and M. Moradi, Photocatalysis assisted by peroxymonosulfate and persulfate for benzotriazole degradation: effect of pH on sulfate and hydroxyl radicals, Water Sci. Technol., 2015, 72, 2095–2102 CrossRef CAS PubMed.
- Y. Shiraishi, Y. Ueda, A. Soramoto, S. Hinokuma and T. Hirai, Photocatalytic hydrogen peroxide splitting on metal-free powders assisted by phosphoric acid as a stabilizer, Nat. Commun., 2020, 11, 3386 CrossRef CAS PubMed.
- J. Lim, Y. Yang and M. Hoffmann, Activation of Peroxymonosulfate by Oxygen Vacancies-Enriched Cobalt-Doped Black TiO2 Nanotubes for the Removal of Organic Pollutants, Environ. Sci. Technol., 2019, 53, 6972–6980 CrossRef CAS PubMed.
- M. G. Antoniou, A. A. de la Cruz and D. D. Dionysiou, Degradation of microcystin-LR using sulfate radicals generated through photolysis, thermolysis and e-transfer mechanisms, Appl. Catal., B, 2010, 96, 290–298 CrossRef CAS.
- X. Yu, V. L. Zholobenko, S. Moldovan, D. Hu, D. Wu, V. V. Ordomsky and A. Y. Khodakov, Stoichiometric methane conversion to ethane using photochemical looping at ambient temperature, Nat. Energy, 2020, 5, 511–519 CrossRef CAS.
- S. Wu, X. Tan, J. Lei, H. Chen, L. Wang and J. Zhang, Ga-doped and Pt-loaded porous TiO2–SiO2 for photocatalytic nonoxidative coupling of methane, J. Am. Chem. Soc., 2019, 141, 6592–6600 CrossRef CAS PubMed.
- L. Meng, Z. Chen, Z. Ma, S. He, Y. Hou, H.-H. Li, R. Yuan, X.-H. Huang, X. Wang and X. Wang, Gold plasmon-induced photocatalytic dehydrogenative coupling of methane to ethane on polar oxide surfaces, Energy Environ. Sci., 2018, 11, 294–298 RSC.
- W. Jiang, J. Low, K. Mao, D. Duan, S. Chen, W. Liu, C.-W. Pao, J. Ma, S. Sang and C. Shu, Pd-Modified ZnO–Au Enabling Alkoxy Intermediates Formation and Dehydrogenation for Photocatalytic Conversion of Methane to Ethylene, J. Am. Chem. Soc., 2020, 143, 269–278 CrossRef PubMed.
- S. P. Singh, A. Anzai, S. Kawaharasaki, A. Yamamoto and H. Yoshida, Non-oxidative coupling of methane over Pd-loaded gallium oxide photocatalysts in a flow reactor, Catal. Today, 2020, 375, 264–272 CrossRef.
- K. Villa, S. Murcia-López, T. Andreu and J. R. Morante, Mesoporous WO3 photocatalyst for the partial oxidation of methane to methanol using electron scavengers, Appl. Catal., B, 2015, 163, 150–155 CrossRef CAS.
- S. Murcia-López, M. C. Bacariza, K. Villa, J. M. Lopes, C. Henriques, J. R. Morante and T. Andreu, Controlled photocatalytic oxidation of methane to methanol through surface modification of beta zeolites, ACS Catal., 2017, 7, 2878–2885 CrossRef.
- X. Li, J. Xie, H. Rao, C. Wang and J. Tang, Platinum-and CuOx-Decorated TiO2 Photocatalyst for Oxidative Coupling of Methane to C2 Hydrocarbons in a Flow Reactor, Angew. Chem., Int. Ed., 2020, 59, 19702–19707 CrossRef CAS PubMed.
- S. Song, H. Song, L. Li, S. Wang, W. Chu, K. Peng, X. Meng, Q. Wang, B. Deng, Q. Liu, Z. Wang, Y. Weng, H. Hu, H. Lin, T. Kako and J. Ye, A selective Au-ZnO/TiO2 hybrid photocatalyst for oxidative coupling of methane to ethane with dioxygen, Nat. Catal., 2021, 4, 1032–1042 CrossRef CAS.
- X. Chen, W. Wang, H. Xiao, C. Hong, F. Zhu, Y. Yao and Z. Xue, Accelerated TiO2 photocatalytic degradation of Acid Orange 7 under visible light mediated by peroxymonosulfate, Chem. Eng. J., 2012, 193, 290–295 CrossRef.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Excellent photo-Fenton catalysts of Fe–Co Prussian blue analogues and their reaction mechanism study, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, Partial oxidation of methane to methanol using bismuth-based photocatalysts, ACS Catal., 2014, 4, 3013–3019 CrossRef.
- K. Sahel, L. Elsellami, I. Mirali, F. Dappozze, M. Bouhent and C. Guillard, Hydrogen peroxide and photocatalysis, Appl. Catal., B, 2016, 188, 106–112 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Superoxide ion: generation and chemical implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar and D. Ma, J. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species, Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c and
Song Ling
Wang
c and
Song Ling
Wang