
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the influence of counterions on a hysteretic spin-transition in isomorphous iron(II) complex salts†‡
Thomas D.
Roberts
a,
Christopher M.
Pask
a,
Izar
Capel Berdiell
b,
Floriana
Tuna
 c and
Malcolm A.
Halcrow
*a
c and
Malcolm A.
Halcrow
*a
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: m.a.halcrow@leeds.ac.uk
bCenter for Material Science and Nanomaterials (SMN), University of Oslo, Sem Sælands 26, 0371 Oslo, Norway
cSchool of Chemistry and Photon Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 11th October 2022
Abstract
[FeL2]X2·2H2O (L = 2,6-bis{5-methyl-1H-pyrazol-3-yl}pyridine; X− = BF4− or ClO4−) are readily dehydrated upon mild heating. Anhydrous [FeL2][BF4]2 exhibits an abrupt spin-transition at T1/2 = 205 K, with a 65 K thermal hysteresis loop which narrows upon repeated scanning. The isomorphous ClO4− salt remains high-spin on cooling, however, which is investigated further in this study. Unlike the iron complex, [ZnL2][ClO4]2·2H2O undergoes single-crystal-to-single-crystal dehydration; the tetragonal anhydrous crystals transform to a new triclinic phase upon cooling. The phase change is apparently sluggish and transition temperatures between 268 K and <200 K were measured by different techniques, implying it depends on the measurement conditions or sample history. Powder diffraction shows the zinc complex is a good model for the structural chemistry of [FeL2][ClO4]2. The spin states of mixed-anion salts of the iron complex [FeL2][BF4]z[ClO4]2−z (z = 1.5 and 1.0) are also investigated. Their spin-transitions evolve more slowly on repeated scanning, as z decreases, and efficient thermally induced kinetic trapping is observed below 120 K when z = 1.0. Taken together, these data imply structural rearrangements in the anhydrous materials during thermal cycling occur more slowly in the presence of the larger ClO4− ion. Hence, rather than reflecting any structural differences with the SCO-active BF4− salt, the high-spin nature of [FeL2][ClO4]2 is probably caused by kinetic inhibition of its putative spin-transition.
Introduction
The structural chemistry of cooperative, first order spin-crossover (SCO) transitions in solid materials is of continuing interest.1–5 Materials applications of SCO compounds5–9 as components in macroscopic devices10–15 and in nano-electronics16–18 are being continuously developed. Exploitation of those applications requires materials with bespoke switching properties, which usually (but not always) operate at room temperature. Producing new SCO materials to meet those specifications is still a challenging goal of molecular design and crystal engineering.19–21 More fundamentally, SCO materials are useful mechanistic models of phase transitions in molecular crystals, at the macroscopic22 and atomistic levels.23,24SCO transitions exhibiting wide thermal hysteresis have particular interest.25 Hysteretic spin-state switches are bistable at temperatures inside the hysteresis loop, which is a pre-requisite for applications requiring reversible switching at a specific temperature.18,26,27 However, hysteresis is associated with large structural changes between the spin states,28–36 which can lead to crystal decomposition during the transition.37–40 The structural basis underpinning hysteretic SCO can thus be difficult to elucidate.37–46
We have reported that [FeL2]X2·2H2O (Scheme 1; L = 2,6-bis{5-methyl-1H-pyrazol-3-yl}pyridine; 1a·2H2O, X− = BF4−; 1b·2H2O, X− = ClO4−) undergo facile and reversible dehydration on mild heating.46,47 The anhydrous materials exhibit complex temperature-dependent structural chemistry on recooling by powder diffraction. For 1a, this culminates in an abrupt spin-transition at T1/2 = 205 K with a 65 K thermal hysteresis loop (although the hysteresis gradually narrows when the transition is cycled multiple times).46 In contrast, anhydrous 1b remains high-spin at all temperatures, despite being isomorphous with the BF4− salt by powder diffraction (Fig. S1, ESI‡).47
The structural basis for these behaviors could not be probed crystallographically, because the crystals of 1a·2H2O and 1b·2H2O decompose during the dehydration reaction. However a more recent study of [ZnL2][BF4]2·2H2O (2a·2H2O), whose crystals survive the dehydration process, shed some light on the question.48 Dehydrated crystals of 2a (phase B; tetragonal, P42/n, Z = 2) are isomorphous with anhydrous 1a by powder diffraction. The anhydrous zinc crystal undergoes a phase transition near room temperature to triclinic phase C (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2). Unexpectedly, the structure of phase C evolves continuously on further cooling. That reflects a temperature-dependent molecular structure distortion of the complex cation, involving a rotational displacement of one L ligand around the metal ion as the temperature is lowered. The same temperature dependence is also shown by phase C of 1a, by powder diffraction. The spin-transition in 1a involves a transformation from phase C to a new low-spin phase (phase E), which was not structurally characterised but may involve a tripling of the phase C asymmetric unit.48
, Z = 2). Unexpectedly, the structure of phase C evolves continuously on further cooling. That reflects a temperature-dependent molecular structure distortion of the complex cation, involving a rotational displacement of one L ligand around the metal ion as the temperature is lowered. The same temperature dependence is also shown by phase C of 1a, by powder diffraction. The spin-transition in 1a involves a transformation from phase C to a new low-spin phase (phase E), which was not structurally characterised but may involve a tripling of the phase C asymmetric unit.48
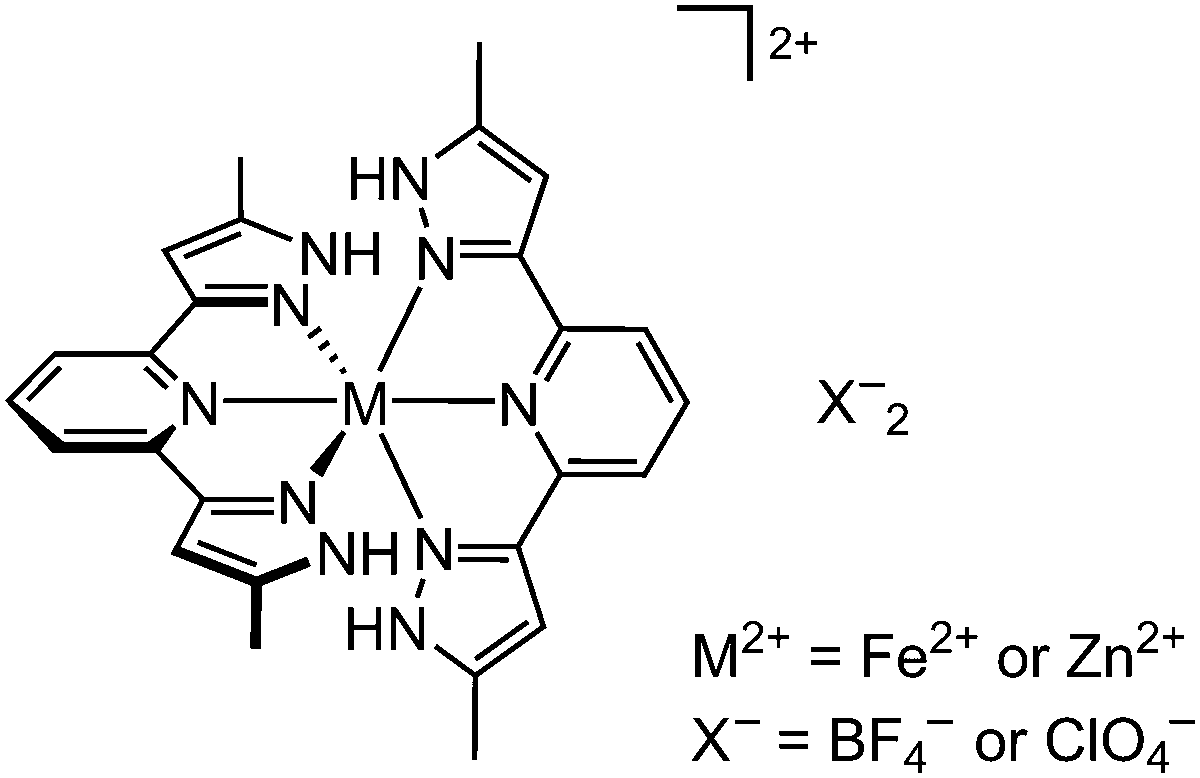 | ||
| Scheme 1 Structures of [FeL2]X2 (1a, X− = BF4−; 1b, X− = ClO4−) and [ZnL2]X2(2a, X = BF4−; 2b, X− = ClO4−). | ||
We now report further investigations of the different behavior of 1a and 1b, through two experiments. First is a structural investigation of [ZnL2][ClO4]2 (2b), as a model for the high-spin iron perchlorate salt 1b. Second, is a series of mixed-anion formulations [FeL2][BF4]z[ClO4]2−z, to probe how SCO in 1a evolves as the anion composition changes in the material.49–51
Results and discussion
As per our previous study,482b was synthesised by treatment of hydrated Zn[ClO4]2 with 2 equiv. L in methanol, which yielded a white microcrystalline solid after the usual work-up. In contrast to the BF4− salt 2a, 2b is too insoluble in water to be recrystallised from that solvent. However, slow recrystallisation from undried nitromethane using diethyl ether vapour as antisolvent yielded small pale yellow crystals of the hydrate phase 2b·2H2O. Six datasets were collected and refined from one of these crystals, under the conditions described below.The structural chemistry of 2b·2H2O resembles 2a·2H2O.48 The as-isolated crystal adopts the tetragonal space group P42/n with Z = 2. A unique one-quarter of a complex molecule lies on a ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) symmetry site, with half-ClO4− and half-water equivalents being disordered within the same lattice cavity near a crystallographic C2 axis. The lattice is a form of “terpyridine embrace”,52 with four-fold interdigitated cation layers in the (001) plane which are separated by hydrogen bonded chains of anions and water molecules (Fig. 1, left). Each pyrazolyl group in the complex forms N–H⋯O hydrogen bond to two anions and two water molecules, in a random distribution reflecting the local four-fold crystallographic symmetry.
symmetry site, with half-ClO4− and half-water equivalents being disordered within the same lattice cavity near a crystallographic C2 axis. The lattice is a form of “terpyridine embrace”,52 with four-fold interdigitated cation layers in the (001) plane which are separated by hydrogen bonded chains of anions and water molecules (Fig. 1, left). Each pyrazolyl group in the complex forms N–H⋯O hydrogen bond to two anions and two water molecules, in a random distribution reflecting the local four-fold crystallographic symmetry.
 | ||
| Fig. 1 Packing diagrams of 2b·2H2O (left), phase B of 2b (centre); and phase C (right), all viewed perpendicular to the (001) plane. Only one orientation of the disordered anions in the tetragonal phases is shown, but both components of the anion disorder about a C2 axis in phase B are included. All atoms have arbitrary radii, with the complex cations de-emphasised for clarity. | ||
In situ dehydration of the 2b·2H2O crystal was achieved by annealing at 350 K on the diffractometer for 30 minutes. The anhydrous crystal exhibits the same crystal symmetry between 350–200 K (tetragonal, P42/n, Z = 2; phase B). Data from phase B were collected at four temperatures within that range, and showed minimal differences in each case.
Allowing for the different measurement temperatures, a is ca. 0.12 Å shorter and c is ca. 0.3 Å longer in phase B compared to the hydrated precursor phase (Table S1, ESI‡), leading to a small decrease in unit cell volume of ca. 22 Å3 following dehydration. The disordered water half-molecule is no-longer apparent in phase B, with the ClO4− ion instead being more disordered to fill the space in the lattice (Fig. 1, centre). Since each anion is equally disordered between two hydrogen bond acceptor groups related by C2 symmetry, on average each cation in phase B only forms two N–H⋯O hydrogen bonds. The reduced hydrogen bonding in phase B might contribute to lengthening of the distal Zn–N bonds from 2.179(3) Å in 2b·2H2O (at 120 K) to 2.226(3) Å in 2b phase B (at 200 K).
The zinc cation structure is unchanged between 350–200 K. However, there is some evidence for reorientation of the disordered ClO4− ions on cooling between 300 and 250 K, which changes the hydrogen bond distribution in the crystal (Fig. S7, ESI‡). The symmetry-imposed anion disorder makes this hard to quantify, but displacement of the anions is an important step in the transformation to phase C described below.
The 200 K dataset of phase B has a high mosaicity, and gave larger refinement residuals than at the higher temperatures. Further cooling the same crystal to 120 K transformed it to a new phase C (triclinic, P, Z = 2). The crystal in phase C exhibited four-fold 90° rotational twinning in the (001) plane, but this was successfully resolved in the. hkl file allowing a full structure refinement. The asymmetric unit of phase C contains one cation and two anions, all on general crystallographic sites with no apparent disorder (Fig. 1, right).
The six-coordinate complex molecule in phase C is significantly distorted from the strict D2d symmetry found in phase B (Fig. 2). That mostly reflects an acute trans-N{pyridine}–Fe–N{pyridine} angle (ϕ) from 180° in phase B to 168.09(9)° in phase C. This distortion positions each ClO4− ion in phase C to accept N–H⋯O hydrogen bonds from two different cations, thus doubling the number of hydrogen bonds in the lattice compared to phase B.
 | ||
| Fig. 2 The [ZnL2]2+ cations in 2b phase B at 200 K (top), and phase C at 120 K (bottom), highlighting the smaller trans-N{pyridine}–Fe–N{pyridine} angle (ϕ) in phase C. Displacement ellipsoids are at the 50% probability level, and C-bound H atoms are omitted. Colour code: C, white; H, pale grey; Fe, green; N, blue. | ||
SCO in high-spin [Fe(bpp)2]2+ (bpp = 2,6-di{pyrazolyl}pyridine) derivatives with reduced values of ϕ requires a significant change in coordination geometry, towards the less distorted geometry with ϕ ≈ 180° preferred by the low-spin state.53 That can lead to cooperative, hysteretic spin-transitions where SCO is observed.36,54 However, it can also kinetically inhibit SCO in a material whose solid lattice is too rigid to accommodate that structure rearrangement.55 Hence, [Fe(bpp)2]2+ complexes with ϕ ≤ 172° are generally less likely to exhibit SCO below room temperature.54 However, at 120–150 K ϕ in phase C of 2a [167.62(7)°]22 and 2b [168.09(9)°] differ by only 4σ, and are essentially equal crystallographically. Hence the molecular structure in phase C cannot, by itself, explain the inactivity of 1b towards SCO.
Rather, the most significant crystallographic difference between the salts 2a and 2b is the temperature of the phase B → C transition. Phase C was observed at 285 K and below in single crystals of 2a,48 but was only achieved at 120 K in 2b. The increased crystal mosaicity of phase B of 2b at 200 K implies that measurement was close to the phase transition temperature. Be that as it may, the phase B → C transition temperature is at least 80–90 K lower in single crystals of the perchlorate salt.
The structural chemistry of 2b was also probed by X-ray powder diffraction. That proved challenging, as inconsistent results were obtained from in situ dehydration of 2b·2H2O inside the sealed capillary sample holders used. Useful data were ultimately achieved from a sample inside an unsealed capillary, which allowed the moisture released by dehydration to escape. The data in Fig. 3 are a good match for crystallographic simulations from the different phases of the material (Fig. S11, ESI‡). There is also excellent agreement between these data and the different phases of 1b,47 confirming 2b is a good structural model for the iron complex (Fig. 4).
 | ||
| Fig. 3 Variable temperature X-ray powder diffraction data for 2b·2H2O. The sample was initially measured at 295 K; annealed at 400 K; then cooled sequentially from there to 150 K. The sample is a mixture of phases B and C at 260 and 250 K. | ||
 | ||
| Fig. 4 Comparison of previously published powder diffraction data for the phases of 1b (black), with simulations based on the crystal structures of 2b (red). The starred peak in phase C implies a fraction of the sample remains in phase B on cooling.48 Data for 1b are taken from ref. 47. | ||
Phase B was generated quantitatively in situ on heating the 2b·2H2O to 400 K. Interestingly, the phase B → C transformation does not happen abruptly under these conditions, but takes place over a 25 ± 5 K temperature range between 270 and 240 K. A small fraction of the sample does not transform to phase C below 240 K, and retains phase B at all temperatures. The same behaviour was shown by 1a, 2a48 and 1b (Fig. 4) in the powder diffractometer. Little change in the powder pattern of phase C was observed on cooling from 240 to 150 K. That is consistent with 1b,47 but contrasts with the BF4− salts 1a and 2a whose phase C structure is very temperature-dependent.48 Evidently the structural plasticity in phase C of 1a and 2a is suppressed in the corresponding perchlorate salts.
A differential scanning calorimetry (DSC) measurement of 2b showed a weak reversible endotherm attributable to the phase B → C transformation, centered at 268 K (Fig. S12, ESI‡). That is consistent with the onset of the transition on cooling in the powder diffraction measurement (Fig. 3). However, there is no evidence in the DSC data for the broadening of the phase transition observed by powder diffraction (Fig. 3). The different characteristics for the phase B → C transition in 2b measured by crystallography, powder diffraction and DSC imply it depends on the measurement conditions and/or the history of the sample. That was not the case for 2a, whose phase change was consistent within experimental error from same three measurement techniques.48
In a second experiment, the effect of replacing BF4− ions with ClO4− on the spin-transition in 1a was probed using mixed-anion samples. Two compositions of [FeL2][BF4]z[ClO4]2−z·2H2O (1c·2H2O, z = 1.50; 1d·2H2O, z = 1.00) were isolated by co-crystallising preformed 1a and 1b in the appropriate ratios from undried nitromethane solution. The formulae of the products derived by C, H, N, Cl microanalysis were a good match for the expected compositions. Both the as-isolated materials were isomorphous with 1a·2H2O by powder diffraction (Fig. S13, ESI‡), and show the same dehydration behaviour on heating by TGA (Fig. S14, ESI‡).
As-isolated 1a·2H2O has a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 high:low-spin population, but becomes fully high-spin around 350 K following in situ dehydration (Fig. 5, left; scan 1).46 Anhydrous 1a exhibits an abrupt spin-transition at T1/2 = 205 K, with ΔT = 65 K hysteresis width on the initial scan (scan 2). The hysteresis narrows as the sample is aged by repeated thermal scanning (scans 3–6), eventually settling at T1/2 = 208 K and ΔT = 37 K (Table 1). There are no further structural changes by powder diffraction during this process, so the hysteresis narrowing should simply reflect an increased number of defects and reduced crystal domain sizes in the aged material.56–58
:1 high:low-spin population, but becomes fully high-spin around 350 K following in situ dehydration (Fig. 5, left; scan 1).46 Anhydrous 1a exhibits an abrupt spin-transition at T1/2 = 205 K, with ΔT = 65 K hysteresis width on the initial scan (scan 2). The hysteresis narrows as the sample is aged by repeated thermal scanning (scans 3–6), eventually settling at T1/2 = 208 K and ΔT = 37 K (Table 1). There are no further structural changes by powder diffraction during this process, so the hysteresis narrowing should simply reflect an increased number of defects and reduced crystal domain sizes in the aged material.56–58
 | ||
| Fig. 5 Variable temperature magnetic data for 1a·2H2O (left), 1c·2H2O (centre); and 1d·2H2O (right), at scan rate 2 K min−1. Top: An initial 290 → 5 → 350 K scan of the as-prepared samples showing their in situ dehydration (black), and the first 350 → 80 → 350 K scan of the dehydrated materials (red). Bottom: Repeated 350 → 80 → 350 K scans of the dehydrated compounds. The same colour coding is used for each material. The curves for 1c·2H2O and 1d·2H2O are shown individually in Fig. S15 and S16 (ESI‡), while the data for 1a·2H2O are replotted from ref. 46. | ||
| Sample | Experiment | T 1/2↓/K | T 1/2↑/K | ΔT1/2/K |
|---|---|---|---|---|
| a Data from ref. 46. b Taken from the 0.5 K min−1 scan in Fig. 6. c The final form of the hysteresis loop was not achieved for 1d. | ||||
| 1a | 1st scan | 172 | 237 | 65 |
| Widest hysteresis | 172 | 237 | 65 | |
| Final hysteresis | 190 | 227 | 37 | |
| 1c | 1st scan | 199 | 233 | 34 |
| Widest hysteresis | 149 | 234 | 85 | |
| Final hysteresis | 177 | 223 | 46 | |
| 1d | 1st scan | 196 | 225 | 29 |
| Widest hysteresisb | 145 | 223 | 78 | |
| Final hysteresisc | — | — | — | |
The sample of 1c·2H2O behaves similarly (Fig. 5, centre), although its structural rearrangement after dehydration appears to occur more slowly than in the parent BF4− salt 1a. That is evidenced by weak additional features in scans 2 and 3, which are not present in scans 4–6; and by the SCO hysteresis, which widens substantially between scans 2 and 3 before contracting again in scans 4–6. The widest thermal hysteresis observed in this spin-transition is ΔT = 85 K (scan 3), which transforms to ΔT = 46 K at the end of the experiment (scan 6; Table 1). Hence, introducing 0.25 equiv. ClO4− into the lattice slightly lowers the temperature of the spin transition, but measurably increases its cooperativity.
Unlike 1a and 1c, freshly prepared 1d·2H2O shows a gradual but incomplete SCO on cooling (Fig. 5, right), with features resembling the structured SCO exhibited by the hydrated perchlorate salt (Fig. S1, ESI‡).47 The first thermal scan following dehydration (scan 2) resembles 1c, although the weak additional features on the main spin transition are more pronounced. The initial hysteretic spin-transition in scans 2–3 slowly transforms into a more cooperative transition during scans 4–6, which also becomes steadily less complete on the cooling branch of the hysteresis loop. That incompleteness reflects thermally induced excited spin state trapping (TIESST) of the sample at low temperature.40,54,60–67 That is, kinetic trapping of a fraction of the sample in its high-spin state below ca. 120 K. The TIESST reflects that the cooling branch of the SCO hysteresis loop extends to a temperature where there is insufficient thermal energy in the lattice to facilitate the structural rearrangements associated with SCO.66
The kinetic nature of the effect is supported by the “reverse-SCO” feature on rewarming the sample above 120 K, where thermal relaxation of the material to its thermodynamic low-spin state can now take place.67 Further warming to 210 K leads to an abrupt low → high-spin transition, which corresponds to the warming branch of the thermodynamic SCO thermal hysteresis loop. The TIESST in 1d becomes progressively more efficient as the sample is cycled through scans 4–6. However, extra thermal cycles beyond scan 6 gave no significant additional changes, implying that represents the final form of the material (Fig. 5).
Notwithstanding the TIESST in 1d, in other respects scan 3 of 1c, and scans 5 and 6 of 1d, closely resemble each other in the 2 K min−1 measurements (Fig. 5). At those stages of the experiment, both materials show a wide hysteretic transition, preceded by a weaker high → low-spin step near 200 K on cooling which is not mirrored in the warming branch of the hysteresis loop. The 200 K feature may be a residual contribution from the initial, narrower hysteresis seen in scan 2, which occurs at the same temperature in cooling mode. Additional scanning of 1c causes its evolution to a symmetrical spin-transition hysteresis loop, whereas 1d remains trapped in that intermediate state under the same conditions.
The behaviour of an aged sample of 1d was clarified by measurement at a slower scan rate of 0.5 K min−1, where the kinetically slow high → low-spin transition proceeds to ca. 90% completeness (Fig. 6).66–72 This revealed a structured spin-transition, occurring in two hysteretic steps of approximately equal height. Allowing for the different measurement conditions,59 the narrow hysteresis component (T1/2↓ = 207, T1/2↑ = 232 K) resembles 1d immediately following dehydration (Fig. 5, step 2), whereas the wider hysteresis step is similar to the wide hysteresis form of 1a and 1c (Table 1). Hence, as described above, the aged sample of 1d appears to be a kinetically frozen mixture of intermediates in the transformation pathway towards its final, wide-hysteresis form.73
 | ||
| Fig. 6 Top: Variable temperature magnetic data from an aged sample of 1d at different scan rates. Bottom: First derivative of the 0.5 K min−1 plot, showing the two distinct hysteretic features. | ||
A sample of 1d, dehydrated in situ on the powder diffractometer, adopts phase B at 353 K and phase C below room temperature (Fig. S17, ESI‡). The powder pattern of phase C in 1d is not temperature-dependent, and has no obvious features to distinguish it from 1a or 1b under the same conditions.46–48 The thermodynamic low-spin phase of 1d was not achieved at 149 K, the lowest temperature achieved on that diffractometer. It is unclear how that how that correlates with the spin state properties of 1d in its initial scans (Fig. 5).
Conclusions
Numerous studies have shown that salts of charged SCO complexes with different counterions often exhibit different spin-state behaviors.1,2,19,74–79 When salts of the same complex are not isomorphous, such differences are a natural consequence of the different crystal packing in those materials, which are not then strictly comparable. Structure:function studies of isomorphous complex salts can be more informative however, as in this work.45,65,80–84The zinc(II) complex 2b has proven to be a useful structural model for its iron(II) analogue 1b (Fig. 4). While 2b·2H2O and 2b are isomorphous with previously reported 2a·2H2O and 2a,48 the analysis of 2b leads to two useful observations. First, although the complex in phase C of 2b deviates significantly from an ideal octahedral geometry (Fig. 2), the degree of distortion at 120 K is almost identical to 2a around the same temperature. Powder diffraction shows the structures of high-spin 1b (Fig. 4) and SCO-active 1a48 closely match the zinc complexes under these conditions. Hence, although such distortions can be responsible for inhibition of SCO in complexes related to [FeL2]2+,54,55 the different spin state behaviours of 1a and 1b are not a reflection of their molecular structure.
The phase B → C transition in 2b also occurs sluggishly, and appears to depend on the measurement conditions or sample history. It was observed over a 25 ± 5 K temperature window on cooling between 270–240 K by powder diffraction (Fig. 3), but was only achieved below 200 K on the single crystal diffractometer. That contrasts with 2a, where the same transformation occurs abruptly at 290–295 K by both techniques.48 The inconsistent behavior shown by 2b suggests the structural rearrangement associated with the phase transition may have a higher kinetic barrier than in the BF4− salt 2a.
The BF4− salt 1a·2H2O transforms cleanly upon dehydration to phase C, which initially exhibits a wide, symmetric spin-transition hysteresis [form (ii); Scheme 2] which slowly narrows on repeated scanning [form (iii)].46 Magnetic measurements on the mixed-anion salts [FeL2][BF4]z[ClO4]2−z·2H2O (1c·2H2O and 1d·2H2O) follow the same sequence but more slowly, and reveal a new intermediate state along that pathway. Freshly dehydrated 1c and 1d (scan 2, Fig. 5) show an initial SCO hysteresis width ΔT1/2ca. 30 K at 2 K min−1 [form (i)] which was not observed for 1a. Further scanning converts this species to form (ii), with ΔT1/2 ≥ 80 K. Forms (i) and (ii) are observed together in scan 3 of 1c, and scans 4–6 of 1d (Fig. 5). Form (iii) was ultimately achieved for 1c by additional thermal scans, but 1d remains trapped as a static mixture of forms (i) and (ii) after ca. 5 thermal cycles. Conversion of 1d to form (iii) was not observed in this study.
 | ||
| Scheme 2 The different stages in the transformation of 1a, 1c and 1d after dehydration, as distinguished by their spin-transition thermal hysteresis. | ||
The slower kinetics of the structure transformations in 1d are also evident in the thermal trapping of the high-spin state (TIESST) of form (ii) below 120 K (Fig. 6). That is not exhibited by 1c form (ii), even though its spin-transition occurs at similar temperatures to 1d at a 2 K min−1 scan rate (Fig. 5). Hence, while 1c and 1d have similar spin state energetics, the internal dynamics of 1d must be intrinsically slower than in 1c, to inhibit its SCO.
Forms (ii) and (iii) of 1a are isomorphous by powder diffraction, and undergo the same sequence of phase B {high-spin} → C {high-spin} → E {low-spin} phase transitions on cooling.48 Therefore the smaller hysteresis loop in form (iii) was attributed to it possessing smaller crystal domains or more microstructural defects, following multiple cycling through its various phase changes.56–58
While more limited characterisation of 1d by powder diffraction is available, we found no evidence for additional crystal phases in 1d above 150 K, beyond the same phases B and C shown by the other materials (Fig. S17, ESI‡). Hence, on this evidence, the form (i) → (ii) → (iii) process (Scheme 2) simply involves a slow annealing of the samples upon repeated cycling across the phase B → C → E → C → B phase transitions.48 While the structure of phase E is still unknown, the phase B → C transition involves both a deformation of the complex, and a substantial rearrangement of the anions in the lattice (Fig. 1 and 2). That rearrangement occurs more slowly for 2b than in 2a, implying the larger ClO4− ions in 2b impede that structural reorganisation.
Taken together, the data in this study imply the structural evolution of [FeL2][BF4]z[ClO4]2−z (1a–1d) following dehydration becomes progressively slower as the fraction of perchlorate ions increases, in the order 1a (z = 2) > 1c (z = 1.5) > 1d (z = 1) > 1b (z = 0). The same pattern of crystal phases is observed in 1a, 1b and 1d within the relevant temperature range; and, the molecular structures of 1a and 1b near the spin-transition temperature are essentially the same. While the materials are extensively hydrogen-bonded, that should not contribute significantly to these differences since BF4− and ClO4− are comparably weak hydrogen bond acceptors.85 Rather, we propose the slower structural rearrangements in the presence of ClO4− reflect its larger size, which impedes its reorientation as the phase changes are cycled.86
We conclude the high-spin nature of 1b reflects its slower lattice dynamics compared with isomorphous, SCO-active 1a, in the presence of ClO4− ions. Effectively, the partial TIESST shown by 1d below 120 K at moderate scan rates (Fig. 5 and 6) becomes complete in 1b, so its SCO is kinetically inhibited.66
Experimental
2,6-Bis(5-methyl-1H-pyrazol-3-yl)pyridine (L),87 [FeL2][BF4]2·2H2O (1a·2H2O)46 and [FeL2][ClO4]2·2H2O (1b·2H2O)47 were prepared by the literature procedures.Synthesis of [ZnL2][ClO4]2·2H2O (2b·2H2O)
A solution of L (0.5 g, 2.1 mmol) and Zn[ClO4]2·4H2O (0.41 g, 1.1 mmol) in MeOH (50 cm3) was stirred until all the solid had dissolved. The colourless solution was filtered, and concentrated to ca. 5 cm3. Addition of excess diethyl ether afforded the complex as a white precipitate. The crude material was recrystallised from undried nitromethane by slow diffusion of diethyl ether vapour, which yielded pale yellow dihydrate crystals suitable for X-ray analysis. Yield 0.58 g, 68%. Found: C, 40.4; H, 3.58; N, 17.9%. Calcd for C26H26Cl2N10O8Zn·2H2O C, 40.1; H, 3.88; N, 18.0%.Synthesis of [FeL2][BF4]z[ClO4]2−z·2H2O (1c·2H2O; z ≈ 1.50)
A mixture of 1a·2H2O (0.10 g, 1.4 mmol) and 1b·2H2O (0.034 g, 0.14 mmol) was dissolved in hot water. After all the solid had dissolved the solution was filtered hot, then cooled slowly to room temperature over a period of 24 h. The product was a brown microcrystalline solid. Yield 0.12 g, 90%. Found: C, 41.8; H, 3.95; N, 18.5; Cl, 2.30%. Calcd for C26H26B1.50Cl0.50F6FeN10O2·2H2O C, 41.6; H, 4.03; N, 18.7; Cl, 2.36%.Synthesis of [FeL2][BF4]z[ClO4]2−z·2H2O (1d·2H2O; z ≈ 1.00)
Method as for 1c·2H2O, using 1a·2H2O (0.068 g, 0.095 mmol) and 1b·2H2O (0.070 g, 0.095 mmol). Yield 0.13 g, 94%. Found: C, 41.4; H, 3.90; N, 18.6; Cl, 4.60%. Calcd for C26H26BClF4FeN10O4·2H2O C, 41.3; H, 4.00; N, 18.5; Cl, 4.69%.Caution
Although we have experienced no problems when using the perchlorate salts in this work, metal–organic perchlorates are potentially explosive and should be handled with care in small quantities.Single crystal structure analyses
All diffraction data for 2b·2H2O and 2b were measured from the same crystal, with an Agilent Supernova dual-source diffractometer using monochromated Cu-Kα (λ = 1.5418 Å) radiation. The diffractometer was fitted with an Oxford Cryostream low-temperature device. The structures were solved by direct methods (SHELXS88), and developed by full least-squares refinement on F2 (SHELXL-201888). Crystallographic figures were generated using XSEED,89 while other publication materials were prepared with OLEX2.90 Experimental details for the structure are listed in Table S1 (ESI‡), while details of the crystallographic refinements are also given in the ESI.‡Other measurements
Elemental microanalyses were performed by the microanalytical service at the University of Leeds School of Chemistry, or London Metropolitan University School of Human Sciences. Thermogravimetric analyses employed a TA Instruments TGA Q50 analyser with a temperature ramp of 10 K min−1 under a stream of nitrogen gas, while differential scanning calorimetry measurements used a TA Instruments DSC Q20 calorimeter, also with a temperature ramp of 10 K min−1. X-Ray powder diffraction data for 2b were obtained using a Bruker D8-A25 diffractometer in transmission capillary geometry, with a Ge(111) Johanssen monochromator and a Lynxeye detector. The diffractometer was fitted with an Oxford Cryostream low-temperature device. Other powder diffraction measurements employed a Bruker D8 Advance A25 diffractometer. Both diffractometers employed Cu-Kα1 radiation (λ = 1.5406 Å). Magnetic susceptibility measurements were performed on a Quantum Design MPMS SQUID magnetometer, in an applied field of 5000 G. A temperature ramp of 2 K min−1 was used for these measurements, unless otherwise stated. A diamagnetic correction for the sample was estimated from Pascal's constants,91 and a previously measured diamagnetic correction for the sample holder, were applied to the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Drs Tim Comyn (University of Leeds) and Laurence Kershaw Cook (University of Leeds, now University of Liverpool) for help with measurements on 1c and 1d. This work was funded by the University of Leeds (Brotherton Scholarship to TDR) and the EPSRC (DTP studentship to TDR). The powder diffraction facility at the University of Oslo is supported by the Norwegian national infrastructure for X-ray diffraction and scattering (RECX), and by the Research Council of Norway via the TomoCAT researcher project (no. 301619).Notes and references
- Spin Crossover in Transition Metal Compounds I–III: Topics in Current Chemistry, ed. P. Gütlich and H. A. Goodwin, Springer, New York, 2004, vol. 233–235 Search PubMed.
- Spin-crossover materials – properties and applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013, p. 568 Search PubMed.
- J. Zarembowitch, F. Varret, A. Hauser, J. A. Real and K. Boukheddaden, C. R. Chim., 2018, 21, 1056–1059 CrossRef CAS.
- Z.-S. Yao, Z. Tang and J. Tao, Chem. Commun., 2020, 56, 2071–2086 RSC.
- K. Senthil Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef CAS.
- S. Hayami, S. M. Holmes and M. A. Halcrow, J. Mater. Chem. C, 2015, 3, 7775–7778 RSC.
- A. Enriquez-Cabrera, A. Rapakousiou, M. Piedrahita Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 CrossRef CAS.
- M. Wang, Z.-Y. Li, R. Ishikawa and M. Yamashita, Coord. Chem. Rev., 2021, 435, 213819 CrossRef CAS.
- Y. Sekine, R. Akiyoshi and S. Hayami, Coord. Chem. Rev., 2022, 469, 214663 CrossRef CAS.
- O. I. Kucheriv, V. V. Oliynyk, V. V. Zagorodnii, V. L. Launets and I. A. Gural'skiy, Sci. Rep., 2016, 6, 38334 CrossRef CAS PubMed.
- M. D. Manrique-Juárez, F. Mathieu, A. Laborde, S. Rat, V. Shalabaeva, P. Demont, O. Thomas, L. Salmon, T. Leichle, L. Nicu, G. Molnár and A. Bousseksou, Adv. Funct. Mater., 2018, 28, 1801970 CrossRef.
- V. Nagy, I. Suleimanov, G. Molnár, L. Salmon, A. Bousseksou and L. Csóka, J. Mater. Chem. C, 2015, 3, 7897–7905 RSC.
- M. Piedrahita-Bello, J. E. Angulo-Cervera, R. Courson, G. Molnár, L. Malaquin, C. Thibault, B. Tondu, L. Salmon and A. Bousseksou, J. Mater. Chem. C, 2020, 8, 6001–6005 RSC.
- M. S. Reis, Coord. Chem. Rev., 2020, 417, 213357 CrossRef CAS.
- K. Ridier, Y. Zhang, M. Piedrahita-Bello, C. M. Quintero, L. Salmon, G. Molnár, C. Bergaud and A. Bousseksou, Adv. Mater., 2020, 32, 2000987 CrossRef CAS PubMed.
- K. Senthil Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef PubMed.
- L. Kipgen, M. Bernien, F. Tuczek and W. Kuch, Adv. Mater., 2021, 33, 2008141 CrossRef CAS PubMed and 2021, 33, 2170354 (correction).
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- P. Guionneau, M. Marchivie and G. Chastanet, Chem. – Eur. J., 2021, 27, 1483–1486 CrossRef CAS PubMed.
- M. A. Halcrow, I. Capel Berdiell, C. M. Pask and R. Kulmaczewski, Inorg. Chem., 2019, 58, 9811–9821 CrossRef CAS PubMed.
- H. Fourati, M. Ndiaye, M. Sy, S. Triki, G. Chastanet, S. Pillet and K. Boukheddaden, Phys. Rev. B, 2022, 105, 174436 CrossRef CAS and references therein.
- M. Chergui and E. Collet, Chem. Rev., 2017, 117, 11025–11065 CrossRef CAS PubMed.
- K. J. Gaffney, Chem. Sci., 2021, 12, 8010–8025 RSC.
- M. A. Halcrow, Chem. Lett., 2014, 43, 1178–1188 CrossRef CAS.
- O. Kahn and C. Jay Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- J.-F. Létard, P. Guionneau and L. Goux-Capes, Top. Curr. Chem., 2004, 235, 221–249 CrossRef.
- J.-F. Létard, P. Guionneau, E. Codjovi, O. Lavastre, G. Bravic, D. Chasseau and O. Kahn, J. Am. Chem. Soc., 1997, 119, 10861–10862 CrossRef.
- S. Dorbes, L. Valade, J. A. Real and C. Faulman, Chem. Commun., 2005, 69–71 RSC.
- G. A. Craig, J. S. Costa, O. Roubeau, S. J. Teat and G. Aromí, Chem. – Eur. J., 2011, 17, 3120–3127 CrossRef CAS PubMed.
- S. Kang, Y. Shiota, A. Kariyazaki, S. Kanegawa, K. Yoshisawa and O. Sato, Chem. – Eur. J., 2016, 22, 532–538 CrossRef CAS PubMed.
- A. Djemel, O. Stefanczyk, M. Marchivie, E. Trzop, E. Collet, C. Desplanches, R. Delimi and G. Chastanet, Chem. – Eur. J., 2018, 24, 14760–14767 CrossRef CAS PubMed.
- M. Grzywa, R. Röβ-Ohlenroth, C. Muschielok, H. Oberhofer, A. Błachowski, J. Żukrowski, D. Vieweg, H.-A. Krug von Nidda and D. Volkmer, Inorg. Chem., 2020, 59, 10501–10511 CrossRef CAS PubMed.
- A. Benchohra, Y. Li, L.-M. Chamoreau, B. Baptiste, E. Elkaïm, N. Guillou, D. Kreher and R. Lescouëzec, Angew. Chem., Int. Ed., 2021, 60, 8803–8807 CrossRef CAS PubMed.
- M. Seredyuk, K. Znovjyak, F. J. Valverde-Muñoz, I. da Silva, M. C. Muñoz, Y. S. Moroz and J. A. Real, J. Am. Chem. Soc., 2022, 144, 14297–14309 CrossRef CAS PubMed.
- N. Suryadevara, A. Mizuno, L. Spieker, S. Salamon, S. Sleziona, A. Maas, E. Pollmann, B. Heinrich, M. Schleberger, H. Wende, S. K. Kuppusamy and M. Ruben, Chem. – Eur. J., 2022, 28, e202103853 CAS.
- D. L. Reger, J. D. Elgin, M. D. Smith, F. Grandjean, L. Rebbouh and G. J. Long, Eur. J. Inorg. Chem., 2004, 3345–3352 CrossRef CAS.
- B. Weber, W. Bauer and J. Obel, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS PubMed.
- H. J. Shepherd, T. Palamarciuc, P. Rosa, P. Guionneau, G. Molnár, J.-F. Létard and A. Bousseksou, Angew. Chem., Int. Ed., 2012, 51, 3910–3914 CrossRef CAS PubMed.
- V. Gómez, C. Sáenz de Pipaón, P. Maldonado-Illescas, J. C. Waerenborgh, E. Martin, J. Benet-Buchholz and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2015, 137, 11924–11927 CrossRef PubMed.
- E. König, G. Ritter, S. K. Kulshreshtha and N. Csatary, Inorg. Chem., 1984, 23, 1903–1910 CrossRef.
- T. Buchen, P. Gütlich, K. H. Sugiyarto and H. A. Goodwin, Chem. – Eur. J., 1996, 2, 1134–1138 CrossRef CAS.
- S. Hayami, Z.-Z. Gu, H. Yoshiki, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2001, 123, 11644–11650 CrossRef CAS PubMed.
- M. B. Bushuev, V. A. Daletsky, D. P. Pishchur, Y. V. Gatilov, I. V. Korolkov, E. B. Nikolaenkova and V. P. Krivopalov, Dalton Trans., 2014, 43, 3906–3910 RSC.
- W. Phonsri, P. Harding, L. Liu, S. G. Telfer, K. S. Murray, B. Moubaraki, T. M. Ross, G. N. L. Jameson and D. J. Harding, Chem. Sci., 2017, 8, 3949–3959 RSC.
- T. D. Roberts, F. Tuna, T. L. Malkin, C. A. Kilner and M. A. Halcrow, Chem. Sci., 2012, 3, 349–354 RSC.
- T. D. Roberts, M. A. Little, F. Tuna, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2013, 49, 6280–6282 RSC.
- C. M. Pask, S. Greatorex, R. Kulmaczewski, A. Baldansuren, E. J. L. McInnes, F. Bamiduro, M. Yamada, N. Yoshinari, T. Konno and M. A. Halcrow, Chem. – Eur. J., 2020, 26, 4833–4841 CrossRef CAS PubMed . The ‘B’, ‘C’ and ‘E’ labels for the crystal phases of the anhydrous materials used in ref. 48, and in this work, were chosen to be consistent with ref. 46.
- C. Carbonera, C. A. Kilner, J.-F. Létard and M. A. Halcrow, Dalton Trans., 2007, 1284–1292 RSC.
- H. Dote, M. Kaneko, K. Inoue and S. Nakashima, Bull. Chem. Soc. Jpn., 2018, 91, 71–81 CrossRef CAS.
- I. Capel Berdiell, R. Kulmaczewski, N. Shahid, O. Cespedes and M. A. Halcrow, Chem. Commun., 2021, 57, 6566–6569 RSC.
- I. Dance and M. Scudder, CrystEngComm, 2009, 11, 2233–2247 RSC.
- L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef CAS.
- L. J. Kershaw Cook, F. L. Thorp-Greenwood, T. P. Comyn, O. Cespedes, G. Chastanet and M. A. Halcrow, Inorg. Chem., 2015, 54, 6319–6330 CrossRef CAS PubMed.
- S. Vela, J. J. Novoa and J. Ribas-Arino, Phys. Chem. Chem. Phys., 2014, 16, 27012–27024 RSC.
- Y. Miyazaki, T. Nakamoto, S. Ikeuchi, K. Saito, A. Inaba, M. Sorai, T. Tojo, T. Atake, G. S. Matouzenko, S. Zein and S. A. Borshch, J. Phys. Chem. B, 2007, 111, 12508–12517 CrossRef CAS PubMed.
- M. B. Bushuev, D. P. Pishchur, V. A. Logvinenko, Y. V. Gatilov, I. V. Korolkov, I. K. Shundrina, E. B. Nikolaenkova and V. P. Krivopalov, Dalton Trans., 2016, 45, 107–120 RSC.
- A. Grosjean, N. Daro, S. Pechev, C. Etrillard, G. Chastanet and P. Guionneau, Eur. J. Inorg. Chem., 2018, 429–434 CrossRef CAS.
- Hysteresis width in SCO transitions can be scan-rate dependent. Hysteresis generally increases with faster temperature scanning, with the cooling branch of the hysteresis loop being more affected by scan rate than the warming branch. A slower scan rate was required to reveal the features of the SCO hysteresis in anhydrous 1d (0.5 K min−1; Fig. 6), compared to 1a and 1c (2 K min−1; Fig. 5). Hence, T1/2 values quoted for anhydrous 1d in Table 1 and the text will be intrinsically some degrees higher, and ΔT1/2 narrower, than if they had been measured under the same conditions as 1a and 1c. S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892.
- M. Marchivie, P. Guionneau, J.-F. Létard, D. Chasseau and J. A. K. Howard, J. Phys. Chem. Solids, 2004, 65, 17–23 CrossRef CAS.
- G. A. Craig, J. S. Costa, S. J. Teat, O. Roubeau, D. S. Yufit, J. A. K. Howard and G. Aromí, Inorg. Chem., 2013, 52, 7203–7209 CrossRef CAS PubMed.
- K. D. Murnaghan, C. Carbonera, L. Toupet, M. Griffin, M. M. Dîrtu, C. Desplanches, Y. Garcia, E. Collet, J.-F. Létard and G. G. Morgan, Chem. – Eur. J., 2014, 20, 5613–5618 CrossRef CAS PubMed.
- J. Weihermüller, S. Schlamp, B. Dittrich and B. Weber, Inorg. Chem., 2019, 58, 1278–1289 CrossRef PubMed.
- T. Boonprab, S. J. Lee, S. G. Telfer, K. S. Murray, W. Phonsri, G. Chastanet, E. Collet, E. Trzop, G. N. L. Jameson, P. Harding and D. J. Harding, Angew. Chem., Int. Ed., 2019, 58, 11811–11815 CrossRef CAS PubMed.
- R. Kulmaczewski, E. Trzop, L. J. Kershaw Cook, E. Collet, G. Chastanet and M. A. Halcrow, Chem. Commun., 2017, 53, 13268–13271 RSC.
- Y. S. Ye, X. Q. Chen, Y. D. Cai, B. Fei, P. Dechambenoit, M. Rouzières, C. Mathonière, R. Clérac and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 18888–18891 CrossRef CAS PubMed.
- N. Paradis, G. Chastanet, T. Palamarciuc, P. Rosa, V. Varret, K. Boukheddaden and J.-F. Létard, J. Phys. Chem. C, 2015, 119, 20039–20050 CrossRef CAS.
- V. A. Money, C. Carbonera, J. Elhaïk, M. A. Halcrow, J. A. K. Howard and J.-F. Létard, Chem. – Eur. J., 2007, 13, 5503–5514 CrossRef CAS PubMed.
- J.-F. Létard, S. Asthana, H. J. Shepherd, P. Guionneau, A. E. Goeta, N. Suemura, R. Ishikawa and S. Kaizaki, Chem. – Eur. J., 2012, 18, 5924–5934 CrossRef PubMed.
- E. Milin, V. Patinec, S. Triki, E. Bendeif, S. Pillet, M. Marchivie, G. Chastanet and K. Boukheddaden, Inorg. Chem., 2016, 55, 11652–11661 CrossRef CAS PubMed.
- Y.-C. Chen, Y. Meng, Y.-J. Dong, X.-W. Song, G.-Z. Huang, C.-L. Zhang, Z.-P. Ni, J. Navařík, O. Malina, R. Zbořil and M.-L. Tong, Chem. Sci., 2020, 11, 3281–3289 RSC.
- V. Jornet-Mollá, C. Giménez-Saiz, L. Cañadillas-Delgado, D. S. Yufit, J. A. K. Howard and F. M. Romero, Chem. Sci., 2021, 12, 1038–1053 RSC.
- The two steps might also reflect a transformation to a new phase of the material, containing two unique iron environments that undergo SCO independently.92–96 However, for the reasons described in the text, the data are more consistent with the aged sample being a kinetically frozen intermediate form of the phase transformation of 1d.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed Engl., 1994, 33, 2024–2054 CrossRef.
- M. A. Halcrow, Polyhedron, 2007, 26, 3523–3576 CrossRef CAS.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS.
- G. A. Craig, O. Roubeau and G. Aromí, Coord. Chem. Rev., 2014, 269, 13–31 CrossRef CAS.
- D. J. Harding, P. Harding and W. Phonsri, Coord. Chem. Rev., 2016, 313, 38–61 CrossRef CAS.
- K. Senthil Kumar, Y. Bayeh, T. Gebretsadik, E. Elemo, M. Gebrezgiabher, M. Thomas and M. Ruben, Dalton Trans., 2019, 48, 15321–15337 RSC.
- M. Hostettler, K. W. Törnroos, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Angew. Chem., Int. Ed., 2004, 43, 4589–4594 CrossRef CAS PubMed.
- M. Yamada, H. Hagiwara, H. Torigoe, N. Matsumoto, M. Kojima, F. Dahan, J.-P. Tuchagues, N. Re and S. Iijima, Chem. – Eur. J., 2006, 12, 4536–4549 CrossRef CAS PubMed.
- T. Sato, K. Nishi, S. Iijima, M. Kojima and N. Matsumoto, Inorg. Chem., 2009, 48, 7211–7229 CrossRef CAS PubMed.
- M. A. Al-Azzani, F. Al-Mjeni, R. Mitsuhashi, M. Mikuriya, I. A. Al-Omari, C. C. Robertson, E. Bill and M. S. Shongwe, Chem. – Eur. J., 2020, 26, 4766–4779 CrossRef CAS PubMed.
- R. Díaz-Torres, W. Phonsri, K. S. Murray, L. Liu, M. Ahmed, S. M. Neville, P. Harding and D. J. Harding, Inorg. Chem., 2020, 59, 13784–13791 CrossRef PubMed.
- N. Galland, C. Laurence and J.-Y. Le Questel, J. Org. Chem., 2022, 87, 7264–7273 CrossRef CAS PubMed.
- The volume of a ClO4− ion (47 Å3) is 24% larger than for a BF4− ion (38 Å3). D. M. P. Mingos and A. L. Rohl, J. Chem. Soc., Dalton Trans., 1991, 3419–3425 RSC.
- Y. Zhou, W. Chen and D. Wang, Dalton Trans., 2008, 1444–1453 RSC.
- G. M. Sheldrick, Acta Crystallogr. Sect. C, 2015, 71, 3–8 CrossRef PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141–1146 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. J. O’Connor, Prog. Inorg. Chem., 1982, 29, 203–283 Search PubMed.
- M. S. Shongwe, B. A. Al-Rashdi, H. Adams, M. J. Morris, M. Mikuriya and G. R. Hearne, Inorg. Chem., 2007, 46, 9558–9568 CrossRef CAS PubMed.
- J. Tang, J. S. Costa, S. Smulders, G. Molnár, A. Bousseksou, S. J. Teat, Y. Li, G. A. van Albada, P. Gamez and J. Reedijk, Inorg. Chem., 2009, 48, 2128–2135 CrossRef CAS PubMed.
- Y.-Y. Zhu, H.-Q. Li, Z.-Y. Ding, X.-J. Lü, L. Zhao, Y.-S. Meng, T. Liu and S. Gao, Inorg. Chem. Front., 2016, 3, 1624–1636 RSC.
- S. Chorazy, T. Charytanowicz, D. Pinkowicz, J. Wang, K. Nakabayashi, S. Klimke, F. Renz, S. Ohkoshi and B. Sieklucka, Angew. Chem., Int. Ed., 2020, 59, 15741–15749 CrossRef CAS PubMed.
- O. I. Kucheriv, S. I. Shylin, V. Y. Sirenko, V. Ksenofontov, W. Tremel, I.-A. Dascălu, S. Shova and I. A. Gural'skiy, Chem. – Eur. J., 2022, 28, e202200924 CrossRef CAS PubMed.
Footnotes |
| † Data supporting this study are available at DOI: https://doi.org/10.5518/1233 |
| ‡ Electronic supplementary information (ESI) available: Previously published magnetic susceptibility data for the 1a·2H2O and 1b·2H2O, illustrating their different spin state behaviours; crystallographic data, refinement details, figures and tables; and other solid state characterisation data for the new materials in this study. CCDC 2195223–2195228. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc03654a |
| This journal is © The Royal Society of Chemistry 2022 |