
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general approach for all-visible-light switching of diarylethenes through triplet sensitization using semiconducting nanocrystals†
Lili
Hou‡§
 *a,
Wera
Larsson
a,
Stefan
Hecht
bc,
Joakim
Andréasson
a and
Bo
Albinsson
*a
*a,
Wera
Larsson
a,
Stefan
Hecht
bc,
Joakim
Andréasson
a and
Bo
Albinsson
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Gothenburg 412 96, Sweden. E-mail: lilihou@tju.edu.cn; balb@chalmers.se
bDepartment of Chemistry & IRIS Adlershof, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
cDWI—Leibniz Institute for Interactive Materials & Institute of Technical and Macromolecular Chemistry at RWTH Aachen University, 52074 Aachen, Germany
First published on 4th October 2022
Abstract
Coupling semiconducting nanocrystals (NCs) with organic molecules provides an efficient route to generate and transfer triplet excitons. These excitons can be used to power photochemical transformations such as photoisomerization reactions using low energy radiation. Thus, it is desirable to develop a general approach that can efficiently be used to control photoswitches using all-visible-light aiming at future applications in life- and materials sciences. Here, we demonstrate a simple ‘cocktail’ strategy that can achieve all-visible-light switchable diarylethenes (DAEs) through triplet energy transfer from the hybrid of CdS NCs and phenanthrene-3-carboxylic acid, with high photoisomerization efficiency and improved fatigue resistance. The size-tunable excitation energies of CdS NCs make it possible to precisely match the clear spectral window of the relevant DAE photoswitch. We demonstrate reversible all-visible-light photoisomerization of a series of DAE derivatives both in the liquid and solid state, even in the presence of oxygen. Our general strategy is promising for fabrication of all-visible-light activated optoelectronic devices as well as memories, and should in principle be adaptable to photopharmacology.
Introduction
Semiconducting nanocrystals (NCs), also known as quantum dots, are quantum confined inorganic crystals with high molar extinction coefficients often combined with stable and high photoluminescence (PL) quantum yields.1 They have been used in a wide range of applications such as light-emitting devices, bioimaging, etc.2–4 The absorption and emission energies of NCs critically depend on their size, which can be precisely tuned by simply varying their synthesis conditions, in particular reaction temperature and time.5,6 Moreover, the strong spin–orbit coupling of NCs mixes the singlet and triplet characters and thereby enables the harvesting of triplet excitons from the NCs to surface-anchored molecules, referred to as mediators or transmitters. The hybrid of NCs and appropriate mediators yields a long triplet lifetime through highly efficient triplet energy transfer (TET).7–9 This opens up further applications of NCs/molecule hybrids in high-performance light-emitting materials10 as well as solar energy converting devices through either singlet fission or triplet–triplet annihilation upconversion (TTA-UC).11,12Triplet excited states are critical intermediates in many photochemical transformations including photocycloaddition and photoisomerization reactions. Coupling NCs with mediators provides an efficient route to power these photochemical transformations using low energy radiation. Weiss and co-workers recently demonstrated that CdSe NCs can catalyze regio- and diastereoselective intermolecular [2+2] cycloadditions13 and that CuInS2/ZnS NCs catalyze the photoreductive deprotection of aryl sulfonyl-protected phenols.14 However, triplet sensitization from NCs to drive photoisomerization processes at longer and thus less harmful wavelengths remains a largely unexplored area. This is highly attractive for harvesting and storing solar energy with significantly longer wavelengths.15,16 Photoisomerization is the key photochemical process to operate photochromic molecular switches, often referred to as photoswitches, in which a structural change between two or more isomers is achieved by irradiation with light at different wavelengths.17 Diarylethenes (DAEs),18,19 among the most extensively used molecular switches, have been widely applied as smart materials in constructing optical memories,20 multi-responsive switchable devices21 as well as in bioimaging22 and photodynamic therapy.23
Typically, ultraviolet (UV) light is required to induce efficient colorization via ring-closing isomerization (photocyclization) of DAEs. UV light causes photooxidation and thus degradation of the molecular switches and materials thereof. In addition, the penetration depth in tissue is very poor, which is a severe downside considering the potential use of these switches in photopharmacology.24,25 Many research efforts have been made to design all-visible-light activated DAEs,26,27 such as shifting the absorption spectrum by chemically extending the π conjugation,28,29 or using upconverting nanoparticles that absorb in the visible or near-infrared region to generate UV light to drive the photoisomerization.30,31 However, directly shifting the absorption band of DAEs requires complicated synthesis, while the low efficiency of the upconverting process leads to overall low photoisomerization efficiencies. Triplet energy transfer from molecular triplet sensitizers has been proven to be another strategy,32–36 however, the typical use of molecular sensitizers imposes limitations due to the rather constrained molecular design, relatively weak light absorption and high sensitivity towards oxygen. Thus, it is desirable to develop a general and improved approach that can be applied to a large set of DAEs using visible light only, while maintaining the high efficiency of photoisomerization and excellent fatigue resistance.
Our system is based on CdS NCs combined with phenanthrene-3-carboxylic acid (3-PCA) to mediate the TET process, and it demonstrates a simple yet efficient non-covalent strategy to achieve photoisomerization of selected DAEs using light at different wavelengths in the visible region. The mechanism implies triplet-like excited states of the NCs lying only ∼20 meV below the strong excitonic absorption band, which can sensitize the ‘dark’ triplet of the surface anchored mediator.7,37 Subsequent TET from the mediator to the DAEs drives the photoisomerization along the triplet reaction pathway, as schematically illustrated in Fig. 1a. The large absorption cross sections of CdS NCs enable unusually efficient visible light absorption compared to previously reported approaches using molecular sensitizers. The visible absorption band of CdS NCs can easily be tuned by varying the size of the CdS NCs to meet the transparent windows of the relevant DAE derivatives, thereby driving the photoisomerization of diverse DAE photoswitches at desired visible wavelengths, as illustrated in Fig. 1b and c. The photoisomerization of our simple non-covalent ‘cocktail’ strategy upon visible light irradiation is as efficient as that of direct UV light irradiation, and prevents photodegradation over multiple irradiation cycles. Moreover, the system can be operated both in solution and in the solid state. The switching of our systems can be retained in an atmospheric environment, that is, it displays insensitivity to oxygen in the solid state, which is appealing in the fabrication of high-performing all-visible-light activated optoelectronics and memories.
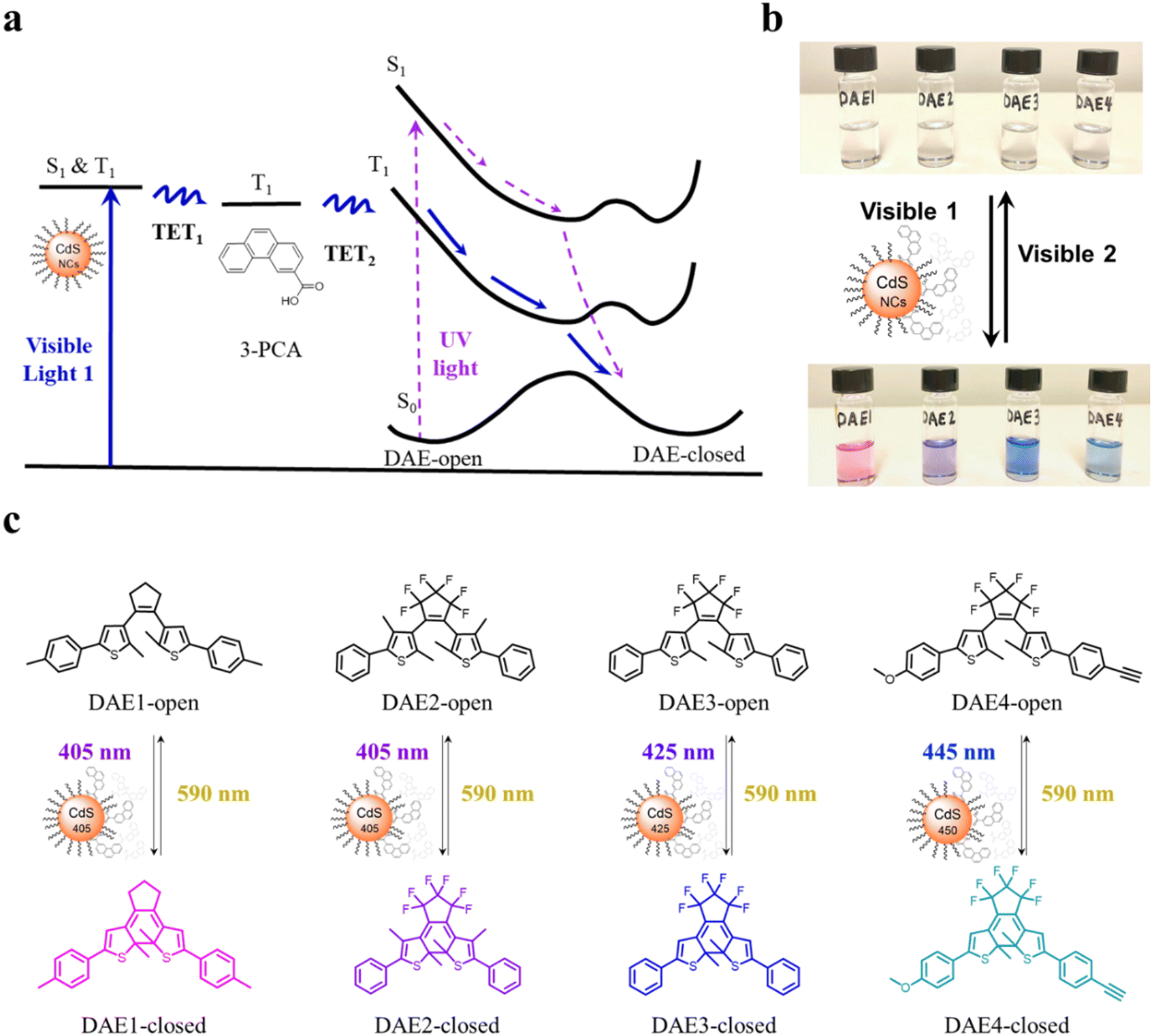 | ||
| Fig. 1 Schematic mechanism, color change and chemical structures of all-visible-light activated photoisomerization of DAEs through triplet energy transfer from CdS NCs and 3-PCA. (a) Mechanism of the photoisomerization from DAE-open to DAE-closed under visible light irradiation via two triplet energy transfer steps (TET1 and TET2), blue lines. The purple dashed lines indicate the direct UV-induced isomerization through the first singlet excited state. (b) Color changes of DAE derivatives under visible light irradiation in both isomerization directions in the presence of CdS NCs and 3-PCA. (c) Chemical structures of the DAEs. In view of the different absorption (color) of DAEs, CdS NCs with varied absorption bands are used to achieve irradiation at specific visible wavelengths. | ||
Results and discussion
Materials and mechanism
CdS NCs were synthesized according to previous reports,38,39 see the details in Methods and Experiments in the ESI.† The growth of CdS NCs during the reaction was monitored by measuring UV-visible absorption spectra of aliquots at different time intervals. Once the first absorption peak of CdS NCs reached the desired wavelength for the visible light irradiation study, the reaction was stopped by quickly cooling the solution to room temperature. Three batches of CdS NCs were prepared with the first absorption peak at 405 nm, 425 nm, and 450 nm, labeled CdS 405, CdS 425, and CdS 450, respectively. Transmission electron microscopy (TEM) images, together with UV-visible absorption and PL spectra of the synthesized CdS NCs are shown in Fig. S1 (ESI†). 3-PCA was synthesized to mediate the triplet energy of CdS NCs.9 The carboxylic acid functional group enables the anchoring of 3-PCA to the surface of CdS NCs, which leads to efficient harvesting of long-lived 3-PCA localized triplets. Four DAE derivatives (Fig. 1c) purchased or synthesized according to previous reports40–42 were used to investigate the photoswitching behavior upon visible light irradiation.All-visible-light switchable DAEs are achieved through TETs from CdS NCs via 3-PCA, as schematically indicated in Fig. 1a. Visible light irradiation populates the excited state of the CdS NCs, from where the excitation energy is transferred to 3-PCA via the first step of triplet energy transfer (TET1). The triplet energy of 3-PCA is then transferred to DAE-open via the second step of triplet energy transfer (TET2), thereby sensitizing the photocyclization reaction along the triplet state pathway. Compared to directly driving the photocyclization of DAE through the singlet excited state upon UV light irradiation (the dashed purple path in Fig. 1a), the energy transfer from the triplet state allows for the use of light at longer wavelengths with all the advantages mentioned above.
All-visible-light activated photoisomerization
To demonstrate the all-visible-light control, we first examined the photocyclization of a DAE derivative referred to as DAE1 (2-bis(2-methyl-5-(p-tolyl)thiophen-3-yl)cyclopentene), in the presence of CdS 405 and 3-PCA upon visible light irradiation. Fig. 2a shows the individual UV-visible absorption spectra of DAE1-open, DAE1-closed, CdS 405 and 3-PCA in toluene. DAE1-open displays no detectable absorption at wavelengths longer than around 350 nm (black spectrum in Fig. 2a). Irradiation of DAE1-open at 302 nm (30 s, see details in Methods and Experiments/Light sources in the ESI†) leads to the formation of DAE1-closed via the ring-closing isomerization, with the characteristic appearance of an absorption band in the visible region (red spectrum in Fig. 2a). Concomitant change of the solution color from transparent to pink is observed. No spectral changes were observed for DAE1-open alone upon 405 nm visible light irradiation (Fig. S2, ESI†). This is expected, as DAE1-open displays no detectable absorbance at 405 nm and it is also in agreement with the estimated energy of the singlet excited state (4.2 eV)35,43versus the photon energy at 405 nm (3.06 eV). CdS 405 was used as its first absorption band corresponds to a wavelength where DAE1-open and DAE1-closed have no or very low absorbance. Fig. 2b shows UV-visible absorption spectra of the mixed solution of DAE1-open with CdS 405 and 3-PCA in deaerated toluene. The spectral features of DAE1 remain unchanged after mixing, indicating that there is no ground state interaction. Due to the high molar absorption coefficient of CdS 405, ε405 = 4.0 × 105 M−1 cm−1, only 0.5 μM is required to initiate the photoisomerization of 50 μM DAE1 under visible light exposure. Although the concentration of the mediator is 100 μM, it is completely transparent for wavelengths longer than 370 nm, see the purple spectrum in Fig. 2a. Upon visible light irradiation (405 nm, 120 s), the formation of DAE1-closed follows as manifested by the appearance of an absorption band in the visible region (red spectrum in Fig. 2b). The observed changes in the UV-visible absorption spectrum are identical to those observed for DAE1 alone upon direct UV light irradiation, i.e., the visible bands display the same peak position and absorbance at the concentration of 50 μM of DAE-open. Thus, the photoconversion of DAE1 in the presence of CdS 405 and 3-PCA upon exposure to 405 nm light yields a photostationary state (PSS) the same as that resulting from irradiation of DAE1 alone with UV light, that is, 94% DAE1-closed as reported.43 The photocyclization quantum yield of DAE1 in the presence of CdS 405 and 3-PCA under 405 nm irradiation was determined to be 35%, which is comparable to the quantum yield of DAE1 under direct 313 nm UV light irradiation (43%).43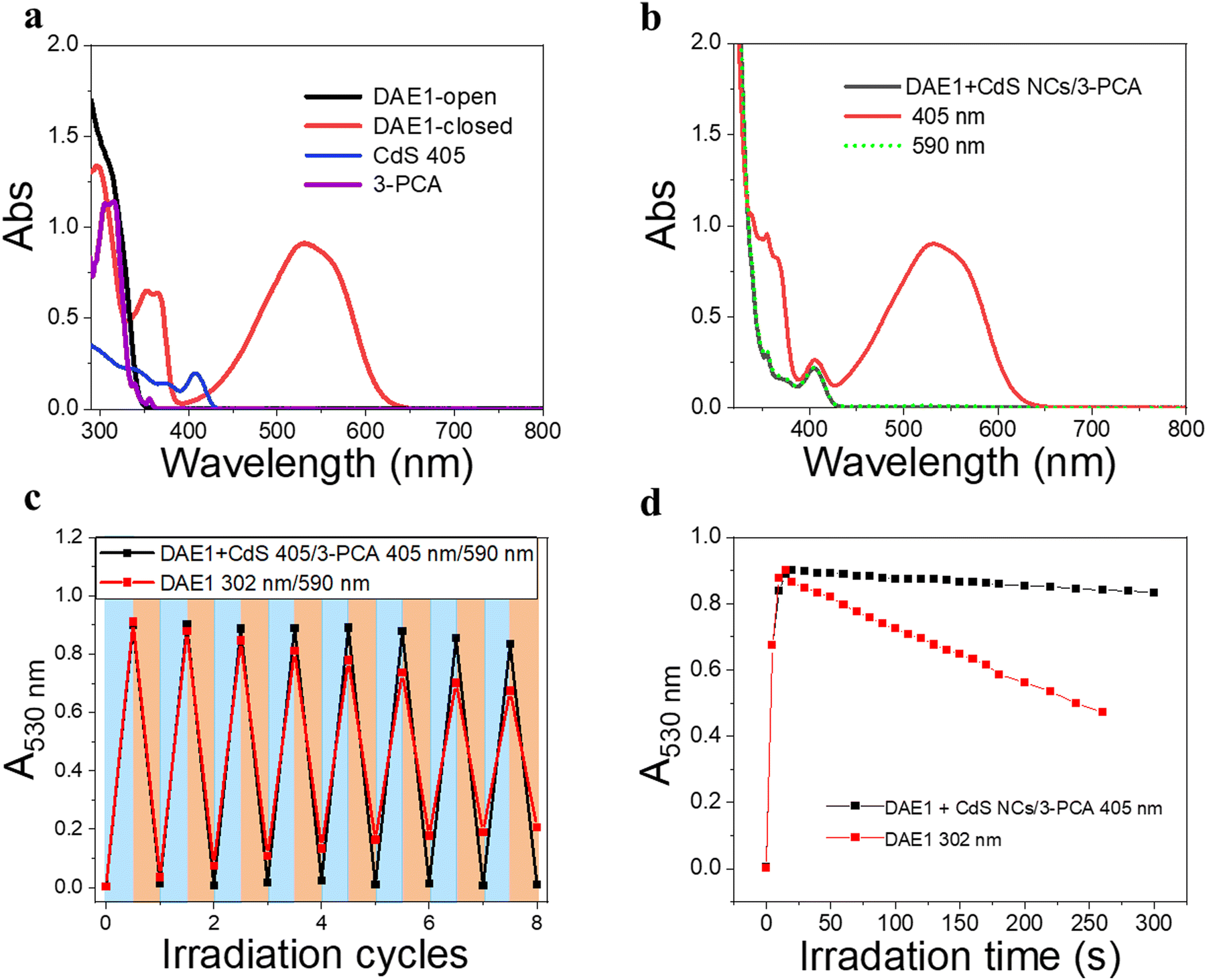 | ||
| Fig. 2 UV-visible absorption spectra of visible-light activated photoisomerization of DAE1. (a) UV-visible absorption spectra of the individual components: DAE1-open (50 μM, black), DAE1-closed converted from the open form (50 μM) under 302 nm UV light irradiation (red), CdS 405 (0.5 μM, blue) and 3-PCA (100 μM, purple) in toluene. (b) UV-visible absorption spectra of a mixed solution of DAE1-open (50 μM), CdS 405 (0.5 μM), and 3-PCA (100 μM) in deaerated toluene, before (black) and after light irradiation at 405 nm (120 s, red), and then after 590 nm light irradiation (90 s, green-dot). (c) Reversibility of the photoswitching of the mixed solution as followed by the absorbance at 530 nm over eight irradiation cycles of 405 nm and 590 nm light (black), and DAE1 only over the irradiation cycles of 302 nm and 590 nm (red). Note that the maxima and minima truly correspond to the respective PSSs of forward and backward switching. (d) Evolution of the 530 nm absorbance of DAE1-open in the mixture under 405 nm (black) and DAE1 only under 302 nm (red) irradiation over time. | ||
Upon subsequent irradiation of the mixed solution with long-wavelength visible light (590 nm) for 90 s, the band in the visible region disappears and the spectrum recovers to the initial state, see green-dot spectrum in Fig. 2b. These observations confirm that when DAE1 is mixed with CdS 405 and 3-PCA, it can undergo reversible photoisomerization using visible light irradiation, that is, both the ring-opening and the ring-closing reactions are triggered without employing UV light. DAE1 shows fatigue over the switching cycles upon direct UV light irradiation, because of the formation of annulated ring byproducts.44 Over eight irradiation cycles of alternating 302 nm light (30 s) and 590 nm (90 s), nearly 50% degradation was observed (red in Fig. 2c). The absorbance evolution also indicates that ∼50% DAE1 was degraded over 5 min of constant UV light irradiation (red in Fig. 2d). In comparison, over eight irradiation cycles of alternating 405 nm (120 s) and 590 nm (90 s) light exposure, the fatigue resistance of DAE1 is improved (black in Fig. 2c), which is also evidenced under long time irradiation experiments (black in Fig. 2d). The photoswitching behavior mentioned above indicates that triplet sensitization from CdS NCs and 3-PCA not only provides a strategy to achieve reversible all-visible-light activated DAEs, but can substantially improve the resistance to fatigue as the byproduct is generated mainly on the singlet surface.43
Mechanistic investigation of the triplet state sensitization
As the excited state lifetime of CdS NCs is too short (with an amplitude-weighted average lifetime of ∼16 ns, Section 4 in the ESI†) to directly sensitize the triplet state of DAEs, a mediator that can anchor to the surface of CdS NCs with a long-lived triplet state (on the microsecond or millisecond timescale) is required. The triplet energy of the mediator should be located in between those of CdS NCs and DAE-open. The triplet-like state of CdS 405 is about 3.0 eV, and the triplet state energies of DAE1-open and DAE1-closed are reported to be 2.5 eV and 0.7 eV, respectively.43 3-PCA with a triplet state energy (2.6 eV) close to DAE1-open was chosen as the mediator.9 The efficiency of TET1 from CdS 405 to 3-PCA was determined to be close to unity (91%, see Section 4 in the ESI†). In the absence of 3-PCA as the mediator, 405 nm light irradiation did not result in any detectable ring-closing isomerization in a solution of DAE1 and CdS 405 (Fig. S4a, ESI†). When using phenanthrene as the mediator for CdS 405, no isomerization of DAE1 under visible light irradiation was observed (Fig. S4b, ESI†), suggesting that the TET1 step only occurs when the carboxylic acid group enables anchoring of the mediator to the surface of CdS NCs, because of the short-range Dexter-type TET.7 When a mediator with a triplet state much lower than that of DAE1-open, such as 1-pyrenecarboxylic acid (PyCOOH, T1 = 2.0 eV)45 was used, no ring-closing isomerization was observed upon 405 nm irradiation (Fig. S4c, ESI†) even if efficient TET1 from CdS NCs to PyCOOH occurred (Fig. S5, ESI†).The TET process was further investigated by time-resolved transient absorption (TA) spectroscopy. Our recent work shows that the triplet state of 3-PCA has a broad positive absorption band between 450 nm and 650 nm.9 415 nm pulsed laser excitation (2.0 mJ, 10 ns) of CdS 405 mixed with an excess of 3-PCA induced the formation of triplet 3-PCA via the TET1 step, evidenced by the initial rise in the TA signal at 470 nm (black dots in Fig. 3a). The subsequent monoexponential decay of triplet 3-PCA corresponds to a lifetime of 22 μs, consistent with our previous observation.9 Adding DAE1 to the CdS 405/3-PCA solution results in a decrease in the triplet lifetime of 3-PCA, due to the TET2 step from 3-PCA to DAE1-open. Fig. 3b shows an analysis by the Stern–Volmer equation, τ0/τ = 1 + τ0kq[Q], where τ0 and τ are the triplet lifetimes of 3-PCA without and with DAE1, respectively, kq is the bimolecular quenching rate constant, and [Q] is the concentration of the quencher, i.e. DAE1-open. The linear fit of the Stern–Volmer plot in Fig. 3b gives kq = 1.4 × 109 M−1 s−1, which is expected for an efficient diffusion-controlled process. Thus, the TA study demonstrates that the triplet states of 3-PCA formed in the TET1 step are effectively transferred to DAE1 in the diffusion-controlled TET2 step.
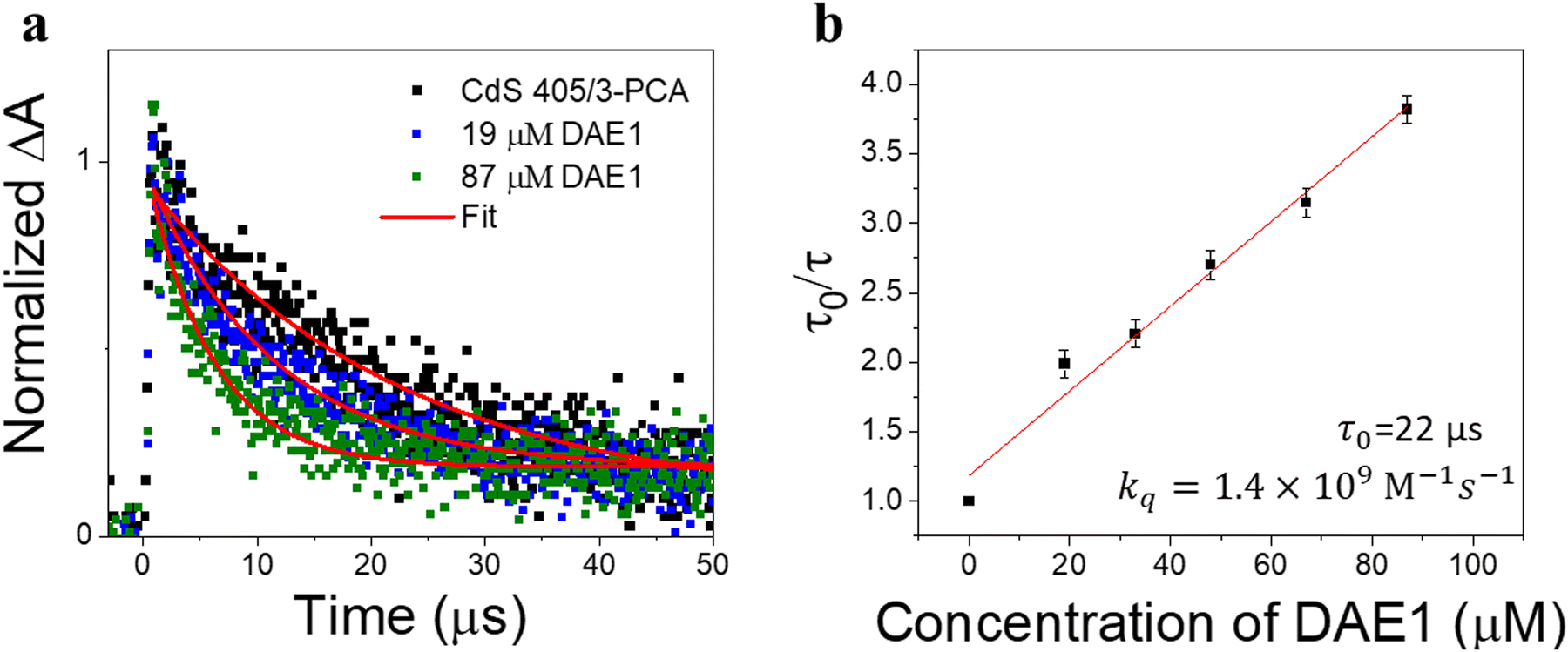 | ||
| Fig. 3 Quenching the triplet state of the CdS 405/3-PCA hybrid by adding DAE1. (a) Time-resolved transient absorption kinetics at 470 nm upon 415 nm pulsed excitation of CdS 405/3-PCA, and CdS 405/3-PCA mixed with DAE1-open in deaerated toluene. (b) Stern–Volmer plot and linear fit for triplet lifetime quenching of CdS 405/3-PCA by DAE1. | ||
General application to DAE derivatives
Our simple non-covalent cocktail strategy allows the hybrid of CdS NCs and 3-PCA to be used as a general triplet sensitizer for a variety of DAE derivatives under all-visible-light control. To demonstrate the general applicability, we further tested visible-light-activated photoswitching of a commercially available DAE derivative, 1,2-bis(2,4-dimethyl-5-phenyl-3-thienyl)-3,3,4,4,5,5-hexafluoro-1-cyclopentene (DAE2). The sample was conveniently prepared by adding DAE2 into the solution of CdS 405 and 3-PCA in accordance with the procedure for DAE1. Fig. S6 (ESI†) shows the individual UV-visible absorption spectra of DAE2-open, DAE2-closed, CdS 405 and 3-PCA in toluene. Fig. 4a shows the UV-visible absorption spectra of the mixed solution and its response to visible light exposure. Upon 405 nm irradiation, DAE2 undergoes photoisomerization from the open form to the closed form, evidenced by the formation of the absorption band in the visible region and purple colorization of the sample, see the red spectrum in Fig. 4a and the photo in Fig. 1b. The photoconversion of DAE2 in the presence of CdS 405 and 3-PCA upon exposure to 405 nm light yields a PSS that contains 58% DAE2-closed, which is lower than that of direct UV irradiation at 313 nm (79%).46 We assign this observation to the relatively large spectral overlap between DAE2-closed and the excitation light at 405 nm, resulting in a direct ring-opening reaction on the singlet excited manifold. The photocyclization quantum yield of DAE2 in the presence of CdS 405 and 3-PCA under 405 nm irradiation was determined to be 39%, which is comparable to the quantum yield of DAE2 under direct 313 nm UV light irradiation (46%).46 Upon 590 nm irradiation, the visible band of DAE2-closed disappears and the spectrum completely recovers to the initial state. The all-visible-light activated photoswitching of DAE2 is fully reversible without obvious fatigue as shown in Fig. 4b. During submission of this version of the manuscript, but after the original preprint publication online in December 2021,47 we noticed that Kaifeng Wu and co-workers reported a ring-closing isomerization of the same DAE molecule using lead halide perovskites NCs upon 450 nm irradiation with a quantum yield of 20.1%.48 The lower quantum yield is possibly because of even larger spectral overlap between the closed form and the excitation light at 450 nm. | ||
| Fig. 4 UV-visible absorption spectra and triplet quenching of CdS 405/3-PCA mixed with DAE2. (a) UV-visible absorption spectra of the solution of DAE2-open (50 μM), CdS 405 (0.5 μM), and 3-PCA (100 μM) in deaerated toluene, before and after light irradiation at 405 nm (black and red) and then after 590 nm light irradiation (green-dot). (b) Reversibility of the photoswitching as followed by the absorbance at 575 nm over eight irradiation cycles of 405 nm and 590 nm light. (c) Time-resolved transient absorption kinetics at 450 nm upon 410 nm pulsed excitation of CdS 405/3-PCA and mixed with DAE2 in deaerated toluene. (d) Stern–Volmer plot for triplet lifetime quenching of CdS 405/3-PCA by DAE2. | ||
From density functional theory (DFT) calculations, the T1 state of DAE2-open is estimated to be 2.8 eV (Table S1, ESI†), about 0.2 eV higher than that of 3-PCA. A TET reaction will occur even under slightly endergonic conditions as described by the Sandros equation49 albeit with a slower rate. Triplet quenching experiments of the CdS 405/3-PCA hybrid by DAE2 and Stern–Volmer plot yield kq = 2.7 × 107 M−1 s−1 (See Fig. 4c and d), which is almost two orders of magnitude smaller than that observed for DAE1, in accordance with a less effective TET2 step due to endergonic TET.
Due to the size-tunable absorption of CdS NCs, it is possible to selectively activate the photoisomerization also of other DAE derivatives in the desired visible wavelength window where both the ring-open and closed form are more or less transparent. As a demonstration, herein we used two other DAE molecules, referred to as DAE3 and DAE4, respectively. The wavelength window for these two derivatives allows for convenient excitation at 425 nm and 445 nm, respectively, as seen in Fig. 5a and c. Direct irradiation at 425 nm and 445 nm of the samples containing DAE3-open alone and DAE4-open alone, respectively, did not result in any ring-closing photoisomerization (see Fig. S7, ESI†). CdS 425 and CdS 450 were synthesized to meet the absorption at the above-mentioned wavelengths (blue spectra in Fig. 5a and c), and were used for the purpose of all-visible-light switching of DAE3 and DAE4. Fig. 5b shows UV-visible absorption spectra of DAE3 mixed with CdS 425 and 3-PCA in deaerated toluene. Upon 425 nm visible light irradiation, the formation of DAE3-closed was evidenced by the appearance of the typical visible absorption band and the color of the solution changed to blue, see the red spectrum in Fig. 5b and the photo in Fig. 1b. Fig. 5d shows the corresponding spectra of DAE4 mixed with CdS 450 and 3-PCA. Indeed, irradiation at 445 nm clearly triggers the photocyclization reaction for DAE4, as shown again by the appearance of the absorption band in the visible region and the solution color changed to cyan, see the red spectrum in Fig. 5d and the photo in Fig. 1b. After subsequent irradiation at 590 nm of the mixed solutions, the visible bands of DAE3 and DAE4 disappeared, and the spectra recovered to their initial states (green-dot spectra in Fig. 5b and d). The above-mentioned observations illustrate that the ring-closing photoisomerization of a variety of DAE derivatives can be activated at desired visible wavelengths by simply varying the sizes of CdS NCs, and the all-visible-light photoswitching processes are fully reversible.
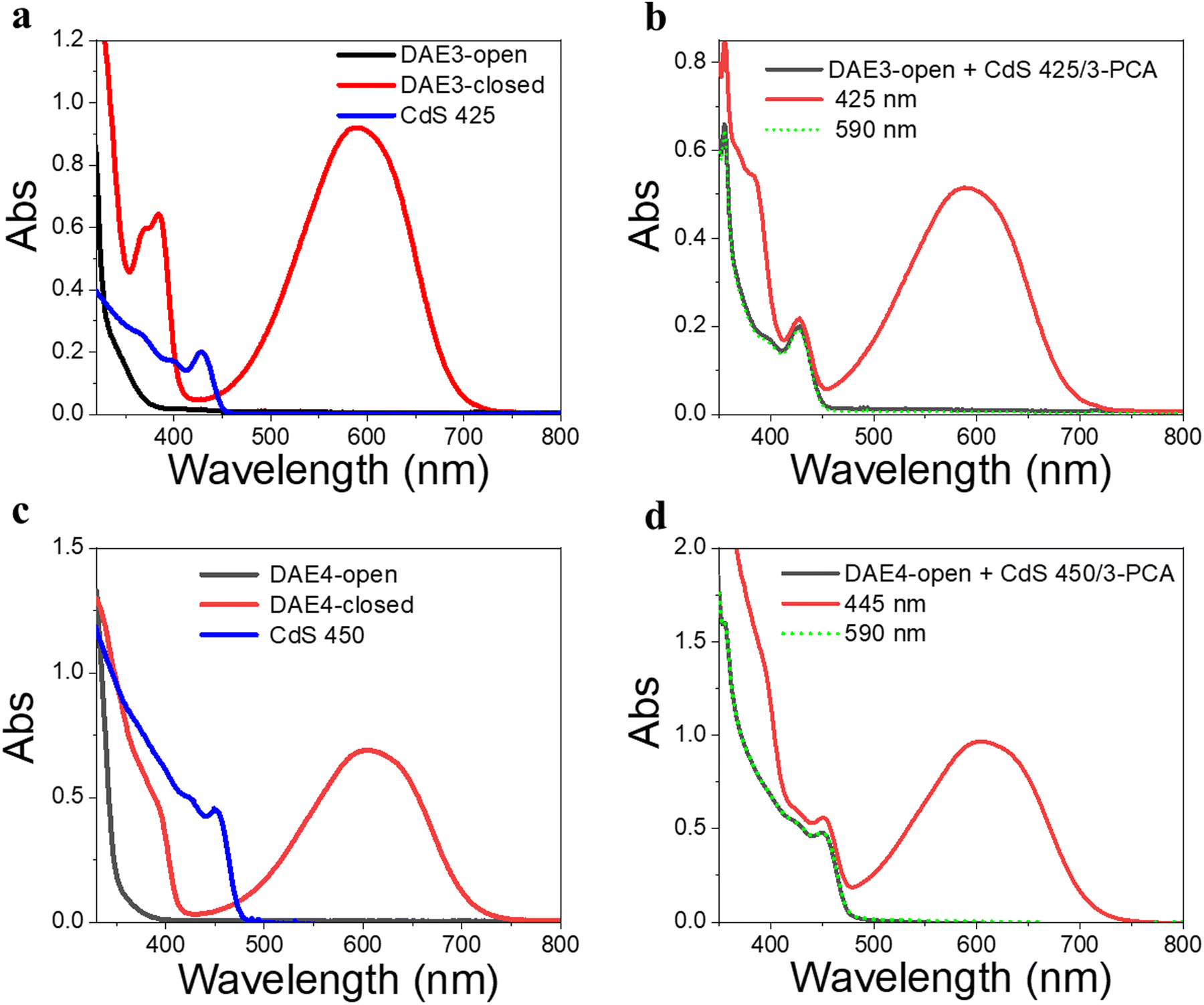 | ||
| Fig. 5 UV-visible absorption spectra of two DAE derivatives under tunable visible irradiation. (a) Individual UV-visible absorption spectra of DAE3-open (50 μM, black), DAE3-closed (formed under UV light irradiation, red), and CdS 425 (0.3 μM, blue) in toluene. (b) UV-visible absorption spectra of the mixed solution of DAE3-open, CdS 425, and 3-PCA in deaerated toluene, before and after irradiation at 425 nm (pulsed laser, 10 min) and then after 590 nm light irradiation (60 s). (c) Individual UV-visible absorption spectra of DAE4-open (50 μM, black), DAE4-closed (formed under UV light irradiation, red), and CdS 450 (0.5 μM, blue) in toluene. (d) UV-visible absorption spectra of the mixed solution of DAE4-open, CdS 450, and 3-PCA in deaerated toluene, before and after light irradiation at 445 nm (continuous laser, 9 min) and then after 590 nm light irradiation (60 s). | ||
All-visible-light switching in the solid state
Our excitation strategy also performs well in the solid state. A solid sample was prepared by soaking a piece of filter paper in a solution containing CdS 405, 3-PCA, and commercially available DAE2, with the same concentrations that were used for the UV-visible absorption study. The solvent slowly evaporated at 60 °C in the dark. Different patterns can be generated and erased reversibly on this solid sample as demonstrated in Fig. 6. Initially, the dry sample paper is white, and 405 nm light irradiation stimulates a white-to-purple color change of the entire paper. Subsequent irradiation with visible light at 590 nm fully reverses the color change. Moreover, the patterns of ‘L’ and ‘H’ shapes can be written and erased consecutively using masks on the same paper by simply altering between the two visible wavelengths. Although the TET reactions require diffusion to occur, which is limited in the solid state, we hypothesize that the slow evaporation of the solvent can cause the mediator 3-PCA to aggregate around the surface of CdS NCs. This would lead to a local concentration enhancement of the molecules, and the shorter intermolecular distances that follow would still allow for TET reactions to occur even in the presence of oxygen, despite that the photoswitching efficiency might be reduced due to aggregation in the solid state. We also observed that all-visible-light activated photoisomerization was possible in an air-equilibrated solution (Fig. S8, ESI†), although the photocyclization efficiency was a factor of 3–5 times lower than that in the deaerated case. Thus, our strategy of combining CdS NCs, 3-PCA and DAEs in the solid state can be used to generate different patterns reversibly and reproducibly, which holds great potential in all-visible-light responsive materials for optical memory and data storage. | ||
| Fig. 6 All-visible-light switching in the solid state. Images of a filter paper prepared from CdS 405, 3-PCA and DAE2 solution, and its subsequent color (white and purple) and pattern change (‘L’ shape and ‘H’ shape), upon visible light irradiation by simply varying the wavelength between 405 nm and 590 nm in a normal atmospheric environment. | ||
Conclusion
In summary, we have demonstrated that the hybrid of CdS NCs and 3-PCA can induce all-visible-light activated photoisomerization of diarylethene photoswitches through triplet energy transfer. This represents a novel strategy to achieve all-visible-light controlled photoswitchable systems at selective wavelengths both in solution and in the solid state, even in the presence of oxygen. Our systems are prepared by a ‘cocktail’ approach, that is, simply by mixing the molecular building blocks and the NCs via non-covalent assembly. The quantum yield of photocyclization through triplet energy transfer from CdS NCs/3-PCA is almost as high as that of direct UV light irradiation, and the hybrid system displays better fatigue resistance over multiple irradiation cycles. NCs are par excellence candidates for this approach due to their appealing photophysical properties (high molar absorptivity, small singlet–triplet energy gap, and near unity efficiency of triplet energy transfer). It should be stressed that our strategy is generally applicable to a large variety of DAE derivatives with different absorption spectra and energy levels, due to the ability to precisely adjust the absorption spectra of the NCs by size variations. Further development of DAE derivatives with carboxylic acid functional groups can simplify the current three-component system to a bi-component system by excluding the mediator, and also allow to develop covalent coupling of NCs and the switching unit via ligand exchange. Our simple yet effective non-covalent assembly approach paves the way to convenient fabrication of all-visible-light controlled photoswitchable optoelectronic devices and memories. The use of visible purple light can reduce the cellular apoptosis caused by direct UV light irradiation to the photoswitches, which allows non-invasive addressing of bioactive switches in the context of superficial photopharmacology. Further applying two-photon absorption benefiting from the large absorption cross sections of the NCs provides the capability of current design to achieve deeper penetration into tissues. Further coating with nontoxic organic molecules or polymers,50 such as poly(ethylene glycol), which can eliminate the toxicity of the NCs and aromatic hydrocarbon materials, will promote current proof-of-concept design for future bioimaging applications.Author contributions
L. H. conceived the ideas and designed the project. S. H. and J. A. provided the diarylethene samples. L. H. synthesized CdS NCs. L. H. and W. L. performed the spectroscopy measurements and the data analysis. B. A. performed theoretical calculations. All authors discussed the results and contributed to interpretation of data. L. H. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Xiaoyan Zhang and Dr Shameel Thurakkal for synthesis of 3-PCA, Jutta Schwarz for synthesis of DAE1, and Dr Shiming Li for synthesis of DAE3 and DAE4. B.A. acknowledges support from the Swedish Energy Agency (contracts: 46526-1 and 36436-2).Notes and references
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS
.
- Q. Sun, Y. A. Wang, L. S. Li, D. Y. Wang, T. Zhu, J. Xu, C. H. Yang and Y. F. Li, Nat. Photonics, 2007, 1, 717–722 CrossRef CAS
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS
- X. L. Dai, Z. X. Zhang, Y. Z. Jin, Y. Niu, H. J. Cao, X. Y. Liang, L. W. Chen, J. P. Wang and X. G. Peng, Nature, 2014, 515, 96–99 CrossRef CAS
- R. Rossetti, S. Nakahara and L. E. Brus, J. Chem. Phys., 1983, 79, 1086–1088 CrossRef CAS
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 437, 121–124 CrossRef CAS PubMed
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed
- N. J. Thompson, M. W. B. Wilson, D. N. Congreve, P. R. Brown, J. M. Scherer, T. S. Bischof, M. F. Wu, N. Geva, M. Welborn, T. Van Voorhis, V. Bulovic, M. G. Bawendi and M. A. Baldo, Nat. Mater., 2014, 13, 1039–1043 CrossRef CAS
- L. Hou, A. Olesund, S. Thurakkal, X. Y. Zhang and B. Albinsson, Adv. Funct. Mater., 2021, 31, 2106198 CrossRef CAS
- C. J. Qin, T. Matsushima, W. J. Potscavage, A. S. D. Sandanayaka, M. R. Leyden, F. Bencheikh, K. Goushi, F. Mathevet, B. Heinrich, G. Yumoto, Y. Kanemitsu and C. Adachi, Nat. Photonics, 2020, 14, 70–75 CrossRef CAS
- M. Tabachnyk, B. Ehrler, S. Gelinas, M. L. Bohm, B. J. Walker, K. P. Musselman, N. C. Greenham, R. H. Friend and A. Rao, Nat. Mater., 2014, 13, 1033–1038 CrossRef CAS
- M. F. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulovic, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31–34 CrossRef CAS
- Y. S. Jiang, C. Wang, C. R. Rogers, M. S. Kodaimati and E. A. Weiss, Nat. Chem., 2019, 11, 1034–1040 CrossRef CAS
- K. A. Perez, C. R. Rogers and E. A. Weiss, Angew. Chem., Int. Ed., 2020, 59, 14091–14095 CrossRef CAS PubMed
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS
- Z. Y. Zhang, Y. X. He, Z. H. Wang, J. L. Xu, M. C. Xie, P. Tao, D. Y. Ji, K. Moth-Poulsen and T. Li, J. Am. Chem. Soc., 2020, 142, 12256–12264 CrossRef CAS PubMed
-
B. L. Feringa and W. R. Browne, Molecular switches, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed
- H. Tian and S. J. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC
- M. Irie, T. Fulcaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS
- T. Leydecker, M. Herder, E. Pavlica, G. Bratina, S. Hecht, E. Orgiu and P. Samori, Nat. Nanotechnol., 2016, 11, 769–775 CrossRef CAS
- L. Hou, X. Y. Zhang, G. F. Cotella, G. Carnicella, M. Herder, B. M. Schmidt, M. Patzel, S. Hecht, F. Cacialli and P. Samori, Nat. Nanotechnol., 2019, 14, 347–353 CrossRef CAS PubMed
- Y. Zou, T. Yi, S. Z. Xiao, F. Y. Li, C. Y. Li, X. Gao, J. C. Wu, M. X. Yu and C. H. Huang, J. Am. Chem. Soc., 2008, 130, 15750–15751 CrossRef CAS
- L. Hou, X. Zhang, T. C. Pijper, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 910–913 CrossRef CAS PubMed
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS
- D. Bleger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef CAS
- Z. Zhang, W. Wang, M. O'Hagan, J. Dai, J. Zhang and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202205758 CAS
- G. M. Tsivgoulis and J. M. Lehn, Adv. Mater., 1997, 9, 627–630 CrossRef CAS
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef CAS
- J. C. Boyer, C. J. Carling, B. D. Gates and N. R. Branda, J. Am. Chem. Soc., 2010, 132, 15766–15772 CrossRef CAS PubMed
- T. Wu and N. R. Branda, Chem. Commun., 2016, 52, 8636–8644 RSC
- R. T. F. Jukes, V. Adamo, F. Hartl, P. Belser and L. De Cola, Inorg. Chem., 2004, 43, 2779–2792 CrossRef CAS PubMed
- V. W. W. Yam, C. C. Ko and N. Y. Zhu, J. Am. Chem. Soc., 2004, 126, 12734–12735 CrossRef CAS
- S. Fredrich, R. Gostl, M. Herder, L. Grubert and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef CAS PubMed
- Z. W. Zhang, J. J. Zhang, B. Wu, X. Li, Y. Chen, J. H. Huang, L. L. Zhu and H. Tian, Adv. Opt. Mater., 2018, 6, 1700847 CrossRef
- Z. W. Zhang, W. H. Wang, P. P. Jin, J. D. Xue, L. Sun, J. H. Huang, J. J. Zhang and H. Tian, Nat. Commun., 2019, 10, 4232 CrossRef PubMed
- A. L. Efros, M. Rosen, M. Kuno, M. Nirmal, D. J. Norris and M. Bawendi, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 4843–4856 CrossRef CAS
- W. W. Yu and X. G. Peng, Angew. Chem., Int. Ed., 2002, 41, 2368–2371 CrossRef CAS
- Z. Li, Y. J. Ji, R. G. Xie, S. Y. Grisham and X. G. Peng, J. Am. Chem. Soc., 2011, 133, 17248–17256 CrossRef CAS PubMed
- E. Orgiu, N. Crivillers, M. Herder, L. Grubert, M. Patzel, J. Frisch, E. Pavlica, D. T. Duong, G. Bratina, A. Salleo, N. Koch, S. Hecht and P. Samori, Nat. Chem., 2012, 4, 675–679 CrossRef CAS PubMed
- P. A. Liddell, G. Kodis, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2002, 124, 7668–7669 CrossRef CAS PubMed
- S. Hermes, G. Dassa, G. Toso, A. Bianco, C. Bertarelli and G. Zerbi, Tetrahedron Lett., 2009, 50, 1614–1617 CrossRef CAS
- M. Herder, B. M. Schmidt, L. Grubert, M. Patzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed
- M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747–748 RSC
- C. Mongin, P. Moroz, M. Zamkov and F. N. Castellano, Nat. Chem., 2018, 10, 225–230 CrossRef CAS
- M. Irie, K. Sakemura, M. Okinaka and K. Uchida, J. Org. Chem., 1995, 60, 8305–8309 CrossRef CAS
- L. Hou, W. Larsson, S. Hecht, J. Andreasson and B. Albinsson, PREPRINT (Version 1) available at Research Square, 2021, December DOI:10.21203/rs.3.rs-1148200/v1.
- M. Liu, P. Xia, G. Zhao, C. Nie, K. Gao, S. He, L. Wang and K. Wu, Angew. Chem., Int. Ed., 2022, 61, e202208241 CAS
- K. Sandros, Acta Chem. Scand., 1964, 18, 2355–2374 CrossRef
- E. B. Voura, J. K. Jaiswal, H. Mattoussi and S. M. Simon, Nat. Med., 2004, 10, 993–998 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc03582k |
| ‡ Present address: School of Precision Instruments and Optoelectronics Engineering, Tianjin University, Tianjin 300072, China. |
| § Present address: Key Laboratory of Optoelectronics Information Technology, Ministry of Education, Tianjin 300072, China. |
| This journal is © The Royal Society of Chemistry 2022 |