
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen light-emitting electrochemical cells based on platinum(II) complexes with a carbazole-appended carbene ligand†‡
Sara
Fuertes
 *a,
Lorenzo
Mardegan
b,
Ignacio
Martínez
a,
Silvia
Ventura
a,
Irene
Ara
a,
Daniel
Tordera
*b,
Henk J.
Bolink
b and
Violeta
Sicilia
*c
*a,
Lorenzo
Mardegan
b,
Ignacio
Martínez
a,
Silvia
Ventura
a,
Irene
Ara
a,
Daniel
Tordera
*b,
Henk J.
Bolink
b and
Violeta
Sicilia
*c
aDepartamento de Química Inorgánica, Facultad de Ciencias, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC – Universidad de Zaragoza, Pedro Cerbuna 12, 50009, Zaragoza, Spain. E-mail: s.fuertes@csic.es
bInstituto de Ciencia Molecular, Universidad de Valencia, C/J. Beltran 2, 46980, Paterna, Spain. E-mail: daniel.tordera@uv.es
cDepartamento de Química Inorgánica, Escuela de Ingeniería y Arquitectura de Zaragoza, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC – Universidad de Zaragoza, Campus Río Ebro, Edificio Torres Quevedo, 50018, Zaragoza, Spain. E-mail: sicilia@unizar.es
First published on 3rd October 2022
Abstract
Platinum(II) complexes bearing new carbazole-appended cyclometallated N-heterocyclic carbenes such as [Pt(Cbz-C^C*)Cl(NCCH3)](3), [Pt(Cbz-C^C*)Cl(PPh3)](4), [Pt(Cbz-C^C*)(PPh3)(py)]PF6 (py = pyridine, 5), [Pt(Cbz-C^C*)(P^N)]PF6 (P^N = 2-(2-(diphenylphosphino) ethyl) pyridine, 6), and [Pt(Cbz-C^C*)(P^P)]PF6 (P^P: 1,1-bis(diphenylphosphino)methane, dppm 7; 1,2-bis(diphenylphosphino)ethane, dppe, 8, 1,2-bis(diphenylphosphino) benzene, dppbz, 9) have been prepared using a stepwise procedure from 9-(4-bromo-phenyl)-9H-carbazole, imidazole, [{Pt(μ-Cl)(η3-2-Me-C3H4)}2] and the corresponding ancillary ligands. They have been fully characterized. Also, single-crystal X-ray structures, theoretical calculations and photophysical studies have been performed. These complexes have been employed as emitters in simple solution-processed light-emitting electrochemical cells (LECs). When driven under a pulsed current of 50 A m−2, devices based on 7–9 exhibited green emission, a luminance of 265 cd m−2 and a current efficiency of 5.7 cd A−1 (for the best performing emitter, 7). As far as we know, these are the first examples of green-emitting Pt(II)-based LECs.
Introduction
Light-emitting electrochemical cells (LECs) celebrated their 25th anniversary in 2020.1,2 Since the LEC technology was first discovered by Pei et al.,3 it has become an excellent candidate and a viable alternative to traditional solid state light sources.4–7 In its simplest form, it consists of a single layer of an ionic luminescent material sandwiched between two air-stable contact electrodes.8 Generally, this material consists of either a conjugated light-emitting polymer (CP)9–12 or an ionic transition-metal complex (iTMC).13 The latter presents some advantages since the ionic metal complex can perform all the necessary roles for light generation: (a) charge injection promoted by the mobile counterions, (b) transport of electrons and holes, and (c) generation of photons from triplet excited states.13–16 In this context, iTMCs have been largely investigated,13,17–19 in particular those based on Ir(III) complexes;18,20–22 however, platinum-based LECs have seldom been explored, with just a few examples found in the literature.23–26 Most of them are polynuclear complexes of Pt(II) with cyclometallated C^N or N^C^N/N^N^C pincer ligands. All the cited materials exhibit emission in the red region of the visible spectrum with no examples of green or blue emitting Pt(II)-based LECs reported to date. In this regard, Pt(II) complexes with cyclometallated N-heterocyclic carbenes (NHCs; C^C*) have been proved to emit light in different colours (blue,27–32 green-yellow,33,34 and orange-red29,30,35,36) with elevated quantum efficiencies. The cycloplatinated C^C* ligands surpass the donor capability of C^N-cyclometallated imines, increasing the energy of the empty dx2−y2 orbitals and preventing their photo- or thermal population, which would lead to severe distortions resulting in non-emissive deactivation.37 In line with this, when designing a Pt(II) emitter, it is important to minimize the structural distortions upon excitation. These promote nonemissive decays due to the electron-vibrational coupling of the excited and ground states.38 Thus, rigid or semi-rigid scaffolds with chelating coordination and strong donor atoms are desired structural features to improve emission quantum yields.With this in mind, the 9H-carbazol-9-yl (Cbz) group possesses several advantages: an intrinsic rigid molecular structure, strong electron-donating ability and hole transporting properties, which are appealing attributes for developing electroluminescent devices.39–43 Therefore, incorporating the Cbz moiety into a molecular scaffold produces a situation where the HOMO and LUMO are localized separately over the molecule, leading to donor–acceptor conjugated systems. They exhibit excellent electronic properties through the induction of a highly efficient intramolecular charge transfer process.44,45 Besides, the attachment of this bulky side-group can prevent stacking interactions of the molecules and reduce self-quenching effects, which are known limiting factors on the efficiency of LECs.46,47 Additionally, the photophysical properties of the mononuclear Pt(II) complexes can be fine-tuned by modifying the cyclometallated and the ancillary ligands.48 Incorporating electron-donating or -withdrawing substituents into the chromophoric cyclometallated fragment and into the auxiliary ligands strongly influences the energies of the frontier orbitals and consequently the MLCT/ILCT character of the lowest energy transition, thus altering the radiative energy and lifetime of the excited state.
Previous results for PhOLEDs using complexes bearing cyclometallated N-heterocyclic carbenes, such as [Pt(R-C^C*)(dppbz)]PF6 (R = H, CN), showed sky blue EL, but a poor performance. The rather deep HOMO in these complexes was considered the cause that could prevent efficient hole injection/transport processes.27 With all the above in mind and considering our interesting results with the cyclometallated C^C* complexes of Pt(II) applied in photo-and electroluminescent devices,27,29–31,33 we explored the Pt(Cbz-C^C*) system with a pendant electron-donating Cbz (Cbz-CH^C* = 1-(4-(9H-Carbazol-9-yl)phenyl)-3-methyl-1H-imidazole) group that provides suitable steric and electronic properties for its application in iTMC-LECs. As a result, herein we report the synthesis and the structural properties of these cationic compounds with non-chelating, [Pt(Cbz-C^C*)(PPh3)(py)]PF6 (5), and chelating ancillary ligands, [Pt(Cbz-C^C*)(P^N)]PF6 (6) and [Pt(Cbz-C^C*)(P^P)]PF6 (P^P = dppm 7, dppe 8, dppbz 9). Theoretical calculations and photophysical studies were also carried out on the ionic Pt(II) complexes 5–9. They showed high PL quantum yields, ranging from 47% to 83%, and were employed as emitters in simple solution-processed iTMC-LECs. The effect of different solvents was also studied showing that the solvent can have a great impact on the performance of the LECs. More importantly, the devices emit in the green region (7–9), which is, as far as we know, the first example of green-emitting Pt(II)-based LECs.
Experimental section
Materials, instrumentation, and synthesis of 9-[4-(1H-Imidazol-1-yl)phenyl]-9H-carbazole (1), 1-(4-(9H-carbazol-9-yl)phenyl)-3-methyl-1H-imidazolium iodide (2), [Pt(Cbz-C^C*)Cl(NCCH3)] (3) and NMR spectroscopic data for 4–9 are included in the ESI.‡Synthesis of [Pt(Cbz-C^C*)(Cl)(PPh3)] (4)
Triphenylphosphine (66.3 mg, 0.253 mmol) was added to a suspension of 3 (150 mg, 0.253 mmol) in dichloromethane (20 mL) at rt. After 90 min. of the reaction, the solution was filtered through Celite and evaporated to dryness. The residue was treated with methanol (5 mL) and filtered to give 4 as a yellow solid. Yield: 176.0 mg, 85.3%. Anal. calcd for C40H31ClN3PPt: C, 58.93; H, 3.83; N, 5.15. Found: C, 58.58; H, 3.66; N, 5.01. IR (ATR, cm−1): ν = 691 (s, PPh3). MS (MALDI+): m/z 779.2 [Pt(C^C*)(PPh3)]+.Synthesis of [Pt(Cbz-C^C*)(PPh3)(py)]PF6 (5)
Pyridine (19.6 mg, 20 μL, 0.247 mmol) and KPF6 (33.9 mg, 0.184 mmol) were added to a suspension of 4 (150 mg, 0.184 mmol) in a mixture of dichloromethane/acetone (10/20 mL) at rt. After 2 hours of the reaction, the solution was evaporated to dryness. Then, 15 mL of dichloromethane was added and the solution was filtered through Celite and evaporated to dryness again. The residue was treated with diethyl ether (5 mL) and filtered to give 5 as a yellow solid. Yield: 117.8 mg, 63.8%. Anal. calcd. for C45H36F6N4P2Pt·CH2Cl2: C, 50.75; H, 3.52; N, 5.15. Found: C, 50.57; H, 3.48; N, 5.23. IR (ATR, cm−1): ν = 833 (s, PF6), 697 (s, PPh3), 556 (s, PF6). MS (MALDI+): m/z 779.2 [Pt(C^C*)(PPh3)]+. ΛM (5 × 10−4 acetone): 100.7 Ω−1 cm2 mol−1.Synthesis of [Pt(Cbz-C^C*)(P^N)]PF6 (6)
2-(2-(Diphenylphosphino)ethyl)pyridine (40.1 mg, 0.138 mmol) and KPF6 (25.3 mg, 0.138 mmol) were added to a suspension of 3 (81.7 mg, 0.138 mmol) in a mixture of dichloromethane/acetone (10/5 mL) at rt., under an argon atmosphere. After 2 hours of the reaction, the solution was evaporated to dryness. Then, 10 mL of dichloromethane was added and the solution was filtered through Celite and evaporated to dryness again. The residue was treated with diethyl ether (5 mL) and filtered. The solid was purified by dissolving in dichloromethane/methanol (2/2 mL) and then adding 20 mL of diethyl ether to precipitate 6 as a yellow solid. Yield: 91.6 mg, 69.8%. Anal. calcd. for C41H34F6N4P2Pt: C, 51.63; H, 3.59; N, 5.87. Found: C, 51.49; H, 3.44; N, 5.64. IR (ATR, cm−1): ν = 834 (s, PF6), 556 (s, PF6). MS (MALDI+): m/z 808.3 [Pt(C^C*)(P^N)]+. ΛM (5 × 10−4 acetone): 67.5 Ω−1 cm2 mol−1.Synthesis of [Pt(Cbz-C^C*)(dppm)]PF6 (7)
dppm (49 mg, 0.13 mmol) and KPF6 (23 mg, 0.13 mmol) were added to a suspension of 3 (75 mg, 0.13 mmol) in acetone (25 mL). After 3 h of the reaction at room temperature, the mixture was evaporated to dryness. The residue was treated with dichloromethane (3 × 15 mL) and filtered through Celite. Then, the solution was evaporated to a volume of ca. 2 mL and the addition of diethyl ether (15 mL) rendered 7 as a pale-yellow solid. Yield: 90 mg, 68.2%. Anal. calcd. for C47H38N3P3F6Pt: C, 53.93; H, 3.66; N, 4.01. Found: C, 53.73; H, 3.70; N, 3.72. IR (ATR, cm−1): ν = 834 (s, PF6), 556 (s, PF6). MS (MALDI+): m/z 901.4 [Pt(C^C*)(dppm)]+. ΛM (5 × 10−4 M acetone) = 64.54 Ω−1 cm2 mol−1.Synthesis [Pt(Cbz-C^C*)(dppe)]PF6 (8)
It was prepared following the method described for 7. dppe (83 mg, 0.21 mmol), KPF6 (38 mg, 0.21 mmol) and 3 (124 mg, 0.21 mmol). Compound 8 (159 mg, 72%). Anal. calcd for C48H40N3P3F6Pt: C, 54.35; H, 3.80; N, 3.96. Found: C, 54.14; H, 3.84; N, 4.01. IR (ATR, cm−1): ν = 835 (s, PF6), 556 (s, PF6). MS (MALDI+): m/z 915.1 [Pt(C^C*)(dppe)]+. ΛM (5 × 10−4 M acetone) = 64.37 Ω−1 cm2 mol−1.Synthesis of [Pt(Cbz-C^C*)(dppbz)]PF6 (9)
It was prepared following the method described for 7. dppbz (83 mg, 0.18 mmol), KPF6 (34 mg, 0.18 mmol) and 3 (110 mg, 0.19 mmol). Compound 9 (178 mg, 86.3%). Anal. calcd for C52H40N3P3F6Pt: C, 56.32; H, 3.64; N, 3.79. Found: C, 56.07; H, 3.69; N, 3.79. IR (ATR, cm−1): ν = 833 (s, PF6), 547 (s, PF6). MS (MALDI+): m/z 963.6 [Pt(C^C*)(dppbz)]+. ΛM (5 × 10−4 M acetone) = 64.99 Ω−1 cm2 mol−1.Results and discussion
Synthesis and characterization of platinum(II) complexes
The new Pt(II) complex with a cyclometallated N-heterocyclic carbene (NHC) containing an appended carbazole, 3, was synthetized following a procedure similar to a previously reported one29,33,49 (see the ESI,‡ part 1.6 and Fig. S1–S3 for the NMR spectra) and it was used as the starting material to synthesize new complexes containing the Pt(Cbz-C^C*) fragment. As shown in Scheme 1, compound 4 (path a) was prepared by adding PPh3 to a suspension of 3 in dichloromethane at room temperature. After the reaction work-up, a yellow solid was obtained in a very good yield (85%). Compound 5 was obtained as a yellow solid by reacting 4 with pyridine and KPF6 in a mixture of acetone and dichloromethane at rt (Scheme 1, path b). On the other hand, compounds 6–9 were obtained directly from compound 3, KPF6 and the corresponding chelating phosphine (see Scheme 1, paths c and d).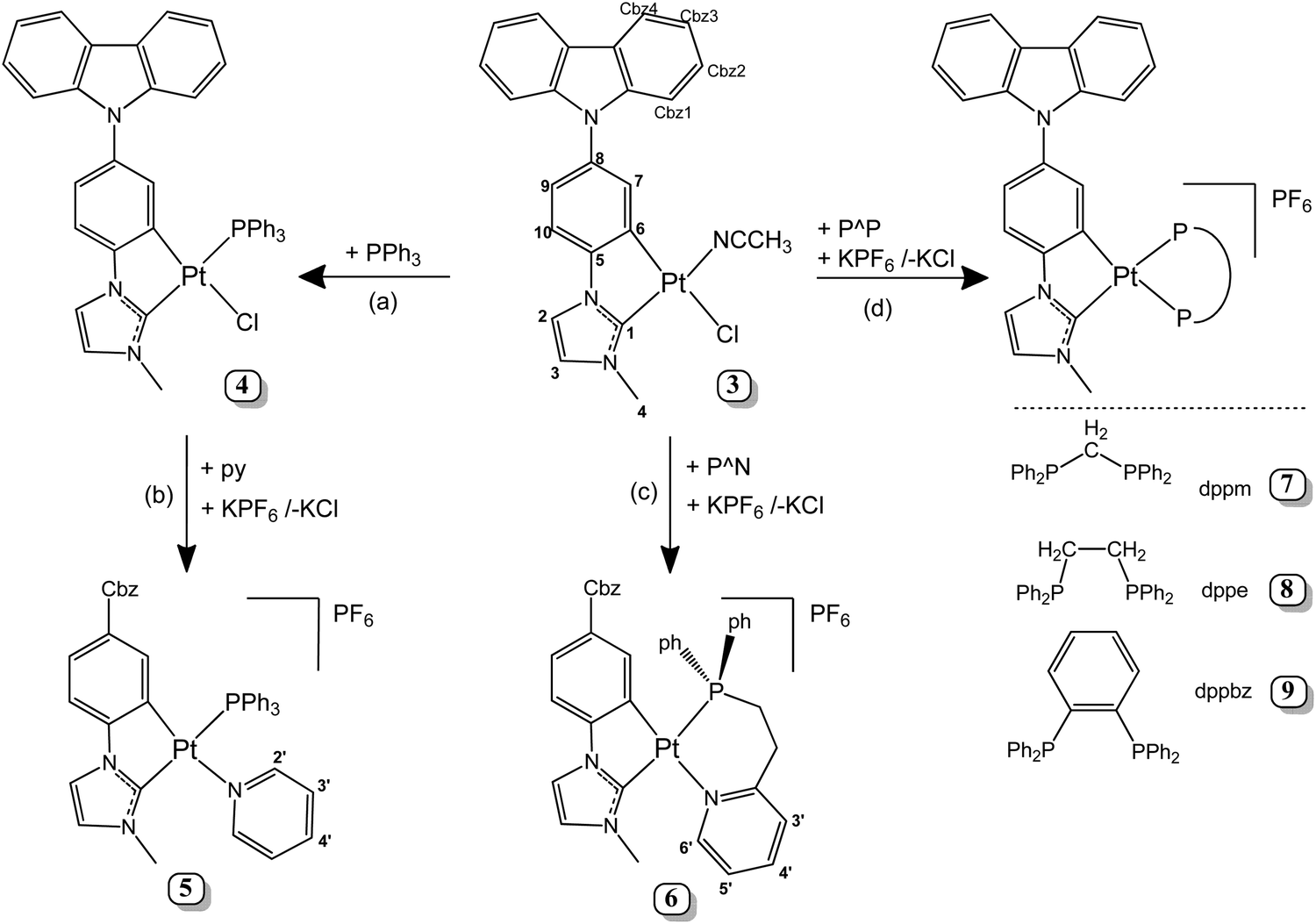 | ||
| Scheme 1 Synthesis of compounds 4–9. The numerical scheme for NMR purposes. | ||
Regarding structural characterization, multinuclear NMR spectra (Fig. S4–S9, ESI‡ and Table 1) provided the most significant information, which revealed that only one isomer was obtained for compounds 4–6, the trans-(C1, P). Their 31P{1H} resonances appear flanked by 195Pt satellites with Pt–P coupling constants around 2900 Hz, according to the P atom being located trans to the N-Heterocyclic carbene (C1).50 Compared to the diphosphine complexes, the two inequivalent P atoms appear as an AB system for the dppm derivative (7) or as two doublets for 8 and 9. 31P and 195Pt chemical shifts and the 1JPt,P values are similar to those found in the related compounds [Pt(R-C^C*)(PPh3)(py)]PF6 (R = –CN, –CO2Et)31 and [Pt(R-C^C*)(P^P)]PF6 (R = –H,27 –CN,27 and –CO2Et29). Also, it deserves to be noted that the H4 resonance appears more shielded (δ ∼ 2.9–3.2 ppm) in 5–9 than that in compound 4 (δ = 4.3 ppm) due to the anisotropic shielding effect caused by the proximity in space of the aromatic ring current of the pyridine or phenyl rings.
The structures of 5–7 and 9 were confirmed by X-ray diffraction (Fig. 1 and Fig. S10 and Tables S1 and S2, ESI‡). In 5 and 6, the asymmetric unit contains two molecules (Pt01 and Pt02) with very similar structural details; thus, only one set of data (Pt01) is included in Table S2 (ESI‡) for discussion. As expected, they are mononuclear complexes with the platinum(II) center in a distorted square-planar environment due to the small bite angle of the C^C* ligand, which is under 80° in all cases. PPh3 and pyridine complete the coordination sphere of the metal center in 5, whereas in the rest these positions are filled in by the P and X (N (6) and P (7, 9)) atoms from the chelating phosphine ligands. Comparing the bite angle of these chelate ligands (P–Pt–X), it can be observed that it is lower for compound 7 (71.37(10)°), which forms a four-membered ring, than those observed for 6 and 9, with six- and five-membered rings.
 | ||
| Fig. 1 Molecular structures of the cationic complexes 5 and 9. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms, solvent molecules and PF6− have been omitted for clarity. | ||
Bond distances and angles (Table S2, ESI‡) are similar to those observed in other complexes with the same kinds of ligands.27,29,31,50 The Cbz moiety (N3 and C13–C24) appears twisted with respect to the platinum coordination plane forming dihedral angles of 59.21(3)° for 5, 61.47(2)° for 6, 54.39(17)° for 7 and 75.76(4)° for 9. In 5, the interplanar angle between the platinum coordination plane and that of the pyridine is 79.8(2)°, while it is somewhat lower (53.7(1)°) for the complex with the chelate ligand, 6. As commented before, this orientation of the pyridine ring has a great influence on the H4 signal in the 1H NMR spectra. Also, in their supramolecular structure, there are no close intermolecular Pt⋯Pt or π–π interactions between the adjacent molecules, probably due to the great steric demand of the Cbz and phosphine groups.
Photophysical properties and theoretical calculations
The photophysical properties of the ionic compounds 5–9 have been investigated. Their absorption spectra in CH2Cl2 solution are depicted in Fig. 2 and their data are listed in Table S3 (ESI‡). They exhibit very similar absorption profiles with relatively intense bands around 290–350 nm that are normally attributed to intraligand transitions of the NHC ligand. Additionally, they show rather weak absorptions at λ > 370 nm that appear slightly redshifted for the diphosphine derivatives (see the inset of Fig. 2). These bands do not experience any significant wavelength shift with solvent polarity (Fig. S11, ESI‡ for 9).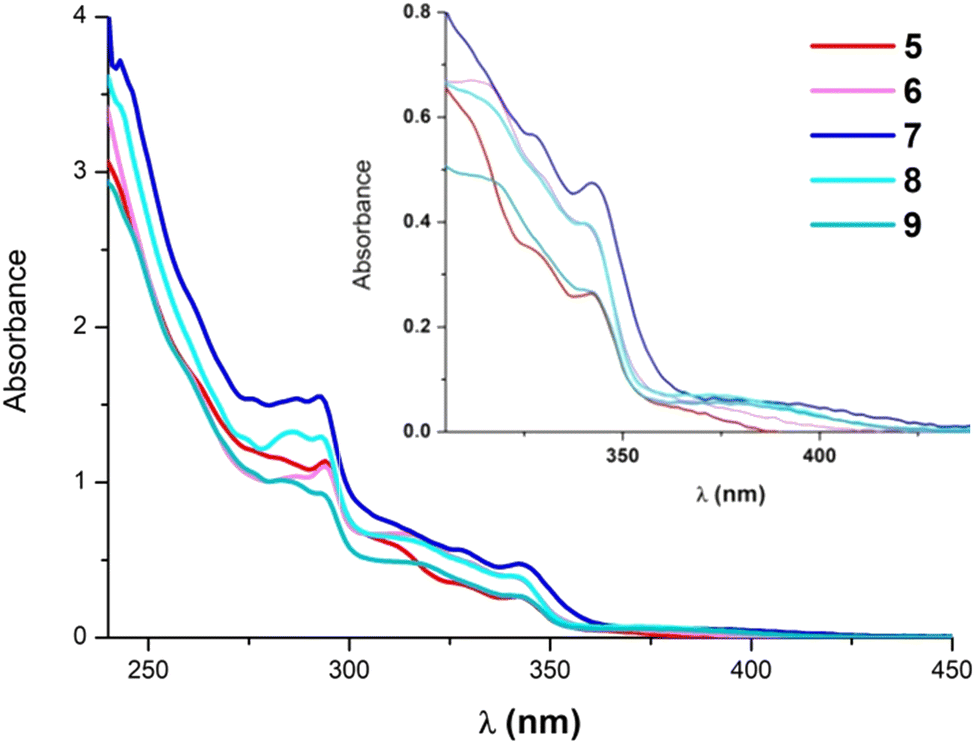 | ||
| Fig. 2 UV-Vis spectra of 5–9 in CH2Cl2 at 5 × 10−5 M. Inset: close-up for the 350–450 nm range. | ||
A concentration dependence study of 5 and 7 in CH2Cl2 showed that no significant ground state aggregation takes place in the concentration range from 10−6 to 10−3 M (see Fig. S12 and S13, ESI‡). Therefore, the low energy absorptions at λ > 370 nm are originated from electronic transitions within the mononuclear species. Besides, when comparing compounds with the same ancillary ligands but different C^C* groups [Pt(R-C^C*)(PPh3)(py)]PF6, (R = –CN, –CO2Et)31 and [(R-C^C*)Pt(P^P)]PF6, (R = –H, –CN, –CO2Et),27,29 the absorption bands appear shifted to lower energies in these electron donating carbazolyl derivatives (5 and 7–9). Thus, these observations are indicating a great participation of the C^C* fragment in the electronic transitions.
To gain further knowledge on this matter, theoretical calculations (density functional theory (DFT) and time dependent density functional theory (TD-DFT)) were carried out for 5, 7 and 9 in CH2Cl2 (Tables S4 and S5, ESI‡). Considering that the computational results of the diphosphine derivatives are very similar, those corresponding to 9 are available in the ESI.‡ As shown in Fig. 3, the lowest energy calculated absorption (S1) fits well with the experimental one and its main contribution is the HOMO → LUMO transition (69% 5 and 92% 7, see Fig. 3). The assignment of these electronic transitions is based on the analysis of the frontier orbitals (Table S5, ESI‡).
 | ||
| Fig. 3 UV-Vis absorption spectra, S1 calculated transitions in CH2Cl2 (bars) and molecular orbital plots (isovalue 0.03) of compounds 5 (left) and 7 (right). | ||
For both complexes, the HOMO is centered on the NHC ligand (99%), essentially on the Cbz fragment whereas the LUMO is distributed over the ancillary ligands (50% 5 and 28% 7), the NHC (32% 5 and 43% 7) and the Pt center (18% 5 and 29% 7), with no participation of the Cbz fragment. Thus, the lowest energy absorption for both is attributed to a mixed transition LL′CT [π(NHC) → π*(L′)]/ILCT [(NHC)] and LMCT [π(NHC) → d(Pt)], with the latter having a greater contribution in 7.
The photoluminescence properties were examined in poly(methyl methacrylate) (PMMA) films at a 5 wt% doping concentration of the Pt compound (Table 2). As shown in Fig. 4, compounds 5 and 6 show blue emissions whereas the diphosphine derivatives (7–9) display green ones. Their emission lifetime decays fit to one rather long component (∼20 μs) and their quantum yield (QY) measurements reach very high values under an Ar atmosphere, in particular those corresponding to the P^P derivatives (QY > 0.75). The excitation spectra match the absorption ones, even though there is a remarkable increase in the intensity of the lowest energy band.
| Comp. | λ em(nm) | τ(μs)a | Φ Ar/ΦAir | k r | k nr |
|---|---|---|---|---|---|
| a In air; kr = Φ/τexp and knr = (1 − Φ)/τexp. | |||||
| 5 | 461, 481max | 22.6 | 0.47/0.39 | 1.7 × 104 | 2.7 × 104 |
| 6 | 467, 484max | 18.1 | 0.49/0.35 | 1.9 × 104 | 3.6 × 104 |
| 7 | 492sh, 522max | 22.1 | 0.76/0.55 | 2.5 × 104 | 2.0 × 104 |
| 8 | 487sh, 507max | 26.6 | 0.77/0.50 | 1.8 × 104 | 1.8 × 104 |
| 9 | 478sh, 509max | 23.1 | 0.83/0.63 | 2.7 × 104 | 1.6 × 104 |
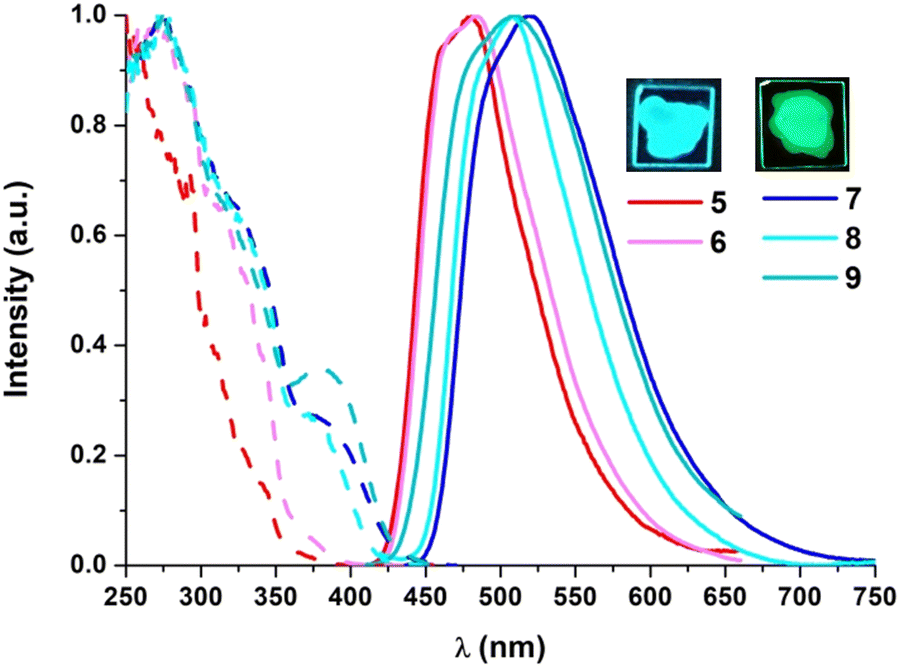 | ||
| Fig. 4 Normalized excitation (—) and emission (—) spectra of 5–9 in 5 wt% PMMA films (right; λexc = 340 nm). Pictures of 5 and 7 were taken under 365 nm light. | ||
To assist in the photoluminescence discussion, the lowest energy triplet states for the optimized structures at the ground state have been calculated by TD-DFT in CH2Cl2 (see Tables S4 and S5, the ESI‡ for the three lowest ones: T1, T2 and T3). In all cases, T1 energies are considerably lower than those for T2 (0.16 eV for 5, 0.28 eV for 7 and 0.17 eV for 9); thus, there are no close low-lying triplets. The calculated wavelengths of these T1 states follow the trend observed for the experimental phosphorescence data (calculated 429 5, 449 7, and 431 nm 9vs. experimental 481 5, 522 7, and 509 nm 9). Also, the geometry optimizations of the T1 states were carried out and their spin-density distributions calculated (Fig. S15, ESI‡). They are mainly located on the C^C* ligand (1.803 5, 1.712 7, and 1.753 9) and to a minor extent on the Pt center (0.150 5, 0.166 7, and 0.155 9) and the ancillary ligands (0.047 5, 0.122 7, and 0.092 9). This great contribution of C^C* to the emissive state T1 agrees with the obtained Kr values (104 order, Table 2).51
Therefore, the emission for all complexes would be attributed mainly to metal-perturbed 3IL [NHC] transitions with some contribution of LL′CT [π(NHC) → π*(L′)]. Similar to what occurs in the UV-vis absorptions, the emissions of 5 (λmax = 481 nm) and 7–9 (λmax = 522–507 nm) appear shifted to lower energies in relation to those bearing different substituents in the C^C* ligand [Pt(R-C^C*)(py)(PPh3)]PF6 (R = –CN, –CO2Et; λmax ∼ 470 nm)31 and [(R-C^C*)Pt(P^P)]PF6 (R = –H, –CN, –CO2Et; λmax ∼ 490–476 nm).27,29 Thus, based on these spectroscopic observations and on the DFT calculations, the presence of an electron donating group (R = Cbz) in the C^C* skeleton increases the energy of the HOMO and results in a red-shift in the emission bands.
Light-emitting electrochemical cells
Light-emitting electrochemical cells using complexes 5–9 as active materials were fabricated. Complexes were mixed with the ionic liquid (IL) 1-butyl-3-methyl-imidazolium-hexafluorophosphate (BMIM+PF6−) in a molar ratio of 4 to 1 (iTMC:IL). In the case of complexes 7, 8 and 9, they were dissolved in two different solvents, acetonitrile (ACN) and dichloromethane (DCM). Due to their low solubility in these solvents, complexes 5 and 6 were tested in dichloroethane (DCE) solutions. The active layers (iTMC:IL) were characterized by means of photoluminescence (PL) and quantum yield (PLQY). Devices were then fabricated from the solutions adopting the archetypical stack configuration: ITO/PEDOT:PSS (80 nm)/iTMC + IL (90–120 nm)/Al (100 nm) (where ITO is indium tin oxide and PEDOT:PSS is poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate)). The details of the processing can be found in the Experimental Section of the ESI.‡ Devices were tested by measuring their performance over time and their electroluminescence (EL) spectra were recorded.We will start the discussion focusing on the emitters 7, 8 and 9. The principal figures of merit are collected in Table 3. First, the PL and PLQY of the active layers prepared from ACN and DCM solutions of 7, 8 and 9 were measured (λex = 340 nm), showing no significant solvent effect on either λem or PLQY (Fig. S16, ESI‡ and Table 3). These PLQY values are far lower than the ones obtained in PMMA, most probably due to the higher concentration of iTMC in the active layer. In fact, a self-quenching process is often observed, taking place in highly concentrated iTMC thin films.52 These results are in agreement with the lower PLQYs of the solid samples of compounds 7–9 (Table S6, ESI‡) compared to those obtained in PMMA. The QY values are generally a good indicator of the luminance performance of iTMCs when employed in a light-emitting device. In view of these results, a similar peak luminance is expected regardless of the solvent used.
| Emitter in ACN | PL λem [nm] | PLQY [%] | EL [nm] | Peak Lum. [cd m−2] | t 50 [min] | CE [cd A−1] | PE [lm W−1] | EQE [%] |
|---|---|---|---|---|---|---|---|---|
| 7 | 531 | 11 | 523 | 61 | 13 | 1.2 | 0.61 | 0.34 |
| 8 | 516 | 8 | 528 | 60 | 35 | 1.2 | 0.61 | 0.33 |
| 9 | 521 | 14 | 519 | 57 | 43 | 1.1 | 0.58 | 0.32 |
| Emitter in DCM | PL λem [nm] | PLQY [%] | EL [nm] | Peak Lum. [cd m−2] | t 50 [min] | CE [cd A−1] | PE [lm W−1] | EQE [%] |
|---|---|---|---|---|---|---|---|---|
| 7 | 525 | 12 | 542 | 265 | 15 | 5.7 | 2.24 | 1.46 |
| 8 | 517 | 6 | 535 | 187 | 13 | 3.7 | 1.26 | 0.97 |
| 9 | 517 | 11 | 528 | 180 | 17 | 3.6 | 1.13 | 1.00 |
The LEC devices were operated under a pulsed current driving with an average current density of 50 A m−2 (1000 Hz block-wave, 50% duty cycle with a peak current of 100 A m−2) and, in contrast to what was expected from the PLQY results, they behaved quite differently depending on the solvent used to process the active layers (Fig. 5).
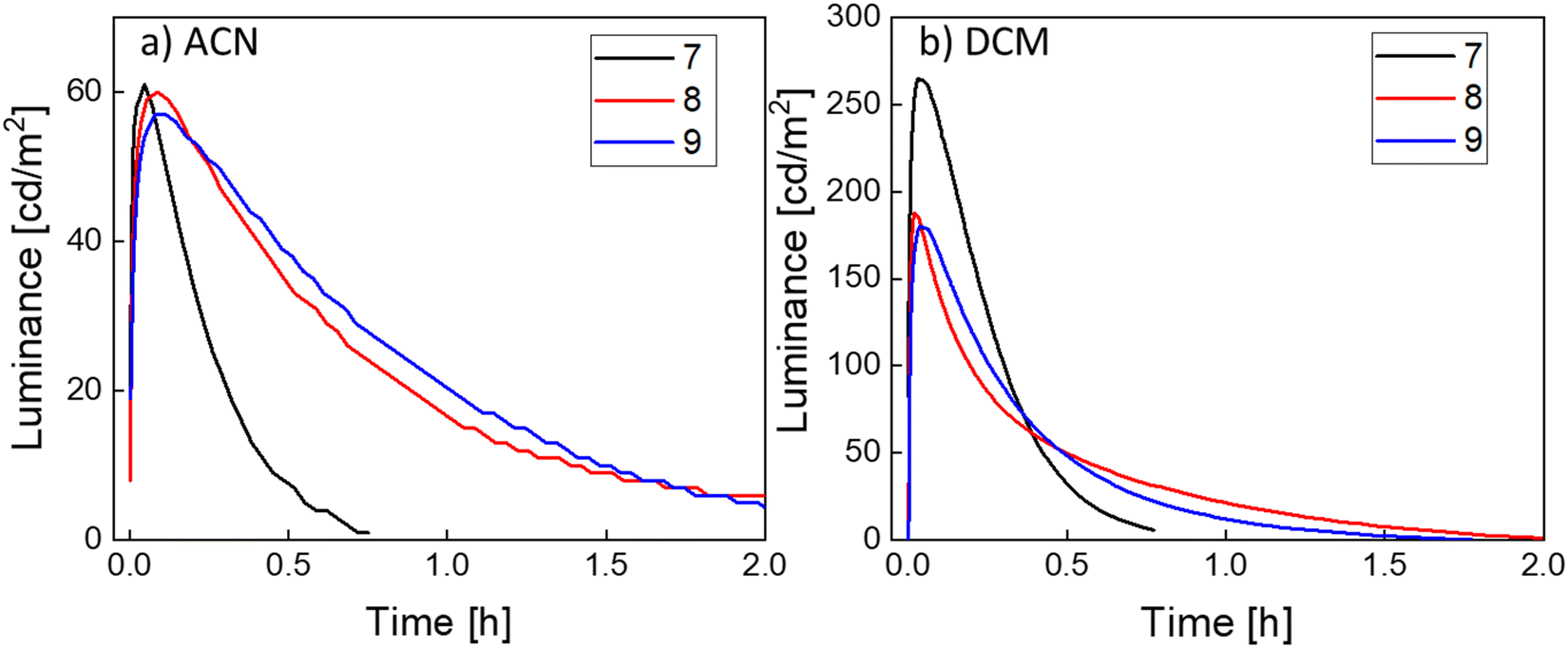 | ||
Fig. 5 Time-dependence of the luminance [cd m−2] of LECs ITO/PEDOT:PSS/iTMC:IL(4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/Al, (iTMC: 7 (black), 8 (red) and 9 (blue)) with the active layer prepared from solutions in ACN (a) and DCM (b). All the devices shown were driven with a pulsed current with an average current density of 50 A m−2. :1)/Al, (iTMC: 7 (black), 8 (red) and 9 (blue)) with the active layer prepared from solutions in ACN (a) and DCM (b). All the devices shown were driven with a pulsed current with an average current density of 50 A m−2. | ||
Devices made from ACN solutions (Fig. 5a) showed in general lower efficiency with maximum luminance values of about 60 cd m−2, current efficiency (CE) values of around 1.2 cd A−1 and power efficiency (PE) values of around 0.6 lm W−1. When devices were fabricated from DCM instead, a three- to four-fold increase of the luminance and efficiency values was observed, achieving luminance values of 265 cd m−2, 187 cd m−2 and 180 cd m−2 with peak CEs of 5.7 cd A−1, 3.7 cd A−1 and 3.6 cd A−1 and peak PEs of 2.2 lm W−1, 1.3 lm W−1 and 1.1 lm W−1 for compound 7, 8 and 9, respectively. These effects were also reflected in the external quantum efficiency (EQE), as expected since it depends directly on the CE (see Table 3). When compared with the theoretical maximum EQEs of these devices extracted from eqn S1 and shown in Table S7 (ESI‡), we notice that the EQE derived from the EL data is substantially lower than the calculated EQE. The main reason behind this could be a quench of the emitter during the device operation. A possible cause for this phenomenon can be an unbalanced current injection because of the impartial screening by the mobile ions accumulated at the electrodes.53,54 Another reason, as discussed above, is the effect of the concentration that eventually leads to a self-quenching of the emission.
Lower driving currents have been shown to improve the LEC device efficiency.55 We biased devices fabricated from DCM and using emitter 7 with a pulsed current driving with average current densities of 25 and 12.5 A m−2 (Fig. S17a and b, ESI‡). The results show that, even using these lower biases, the efficiencies remain very similar, with peak efficiencies of 4.7 and 5.2 cd A−1 for current densities of 25 and 12.5 A m−2, respectively, which indicates that phenomena such as self-heating of the device is not the limiting factor on the efficiency in these devices, at least in the current density range studied.56 A similar trend was observed when biasing devices fabricated from ACN (using emitter 7) with average current densities of 25 and 12.5 A m−2 (Fig. S17c and d, ESI‡). Generally, the half-luminance lifetime (t50) value is defined as the time needed to reach 50% of the peak luminance and it is used to determine the stability of a device over time, albeit it is also dependent on the driving conditions. In this case, devices made from ACN have t50 values of 13, 35 and 43 minutes, for emitters 7, 8 and 9, respectively, while those from DCM have t50 values of 15, 13 and 17 minutes, respectively. The increase in luminance together with the reduced t50 for DCM devices, despite the same PLQY values, could be explained by two main factors. The first point to take into consideration is the difference in the boiling point of the two solvents, ∼81 °C for ACN and ∼40 °C for DCM and the temperature at which the films were annealed before the cathode vacuum deposition, 70 °C for 15 min. These annealing conditions could allow all the DCM to evaporate and escape from the samples but in the case of the ACN residual solvent could remain in the active films. The effect of retained solvents has been reported in the past as detrimental for the devices as it can lead to degradation pathways or quenching.57 In view of this, and once the stability of the emitters was checked by NMR in MeCN-d3 at 70 °C for at least 1 hour (Fig. S18, ESI‡ for 7), we increased the annealing conditions of devices prepared from ACN in terms of time and temperature. To do this, we prepared a device using emitter 7 with a longer annealing time (4 h) at the same temperature (70 °C) and a device using emitter 9 with a higher temperature annealing process (90 °C) and a longer time (1 h). The performance of the device processed at 70 °C for 4 h slightly improved with a maximum luminance of 75 cd m−2 (vs. 61 cd m−2), a CE of 1.5 cd A−1 (vs. 1.2 cd A−1) and a t50 of 11 min (vs. 13 min) (Fig. S19a, ESI‡). On the other hand, the device processed at higher annealing temperature not only did not improve but it decreased, showing a maximum luminance of 48 cd m−2 (vs. 57 cd m−2), a CE of 0.96 cd A−1 (vs. 1.1 cd A−1) and a t50 of 7 min (vs. 43 min) (Fig. S19b, ESI‡). These results, in particular the much lower t50, could be an indication that the harsher annealing conditions are inducing a degradation of the emitter. The second aspect to be pointed out is the voltage during operation (Fig. S20, ESI‡). When compared, the voltage of the devices prepared from ACN is significantly lower than the DCM, with values between 3 V and 3.5 V for ACN and values between 4 V and 5 V for DCM. The origin of this effect can also be related to the residual solvent present on the active layer, where it has been seen that the presence of a solvent decreases the resistance of the film, resulting in a lower turn-on voltage.57 Lower voltages notoriously allow for longer lifetimes, and therefore higher t50, but at the expense of lower luminance levels. All in all, these results point that the origin of the difference in the performance of devices processed from ACN and from DCM might reside in their different morphology. Indeed, in previous works, it has been seen that morphology can affect the performance of LEC devices.58–60 However, atomic force microscopy (AFM) images show no significant differences between active films using emitter 7 processed from different solvents (Fig. S21, ESI‡). Both layers are very flat and homogeneous with no apparent phase separation or aggregation and with just small differences in the root mean square (RMS) roughness of the films (0.5 nm and 0.3 nm for ACN and DCM, respectively).
Finally, the EL spectra were characterized in both ACN and DCM (Fig. 6a, b and Table 3). In ACN, the EL peaks of 7, 8 and 9 are centered at 523 nm, 528 nm and 519 nm while in DCM at 542 nm, 535 nm and 528 nm, respectively. The slight difference on the EL peaks between ACN- and DCM-processed devices is ascribed to optical effects arising from the differences in the thicknesses of the active layers (90 and 120 nm, respectively). The EL is very similar to the PL of the active layer films. In some cases, 8 from ACN and 7, 8 and 9 from DCM, a slight red-shift of the EL can be observed. The red-shift of the EL of iTMCs can be common and was also previously observed in LECs based on Ir(III) and Cu(I) complexes.61,62 The corresponding emission colours, expressed as CIE (Commission Internationale de l’Éclairage) coordinates, are shown in Fig. S22 (ESI‡) and compiled in Table S8 (ESI‡). The EL was also measured over time (for devices made from emitter 7), down to their t50, in order to observe the EL evolution during the degradation (Fig. S23, ESI‡). The λmax of the EL remains constant for devices processed from ACN and only slightly changes for devices processed from DCM (from 542 to 549 nm), an indication of the high stability of the EL over the lifetime of the device. Notably, as seen by the EL spectra and the CIE coordinates, the three devices emit in the green region. To our knowledge, these are the first reported green-emitting Pt(II)-based LECs.
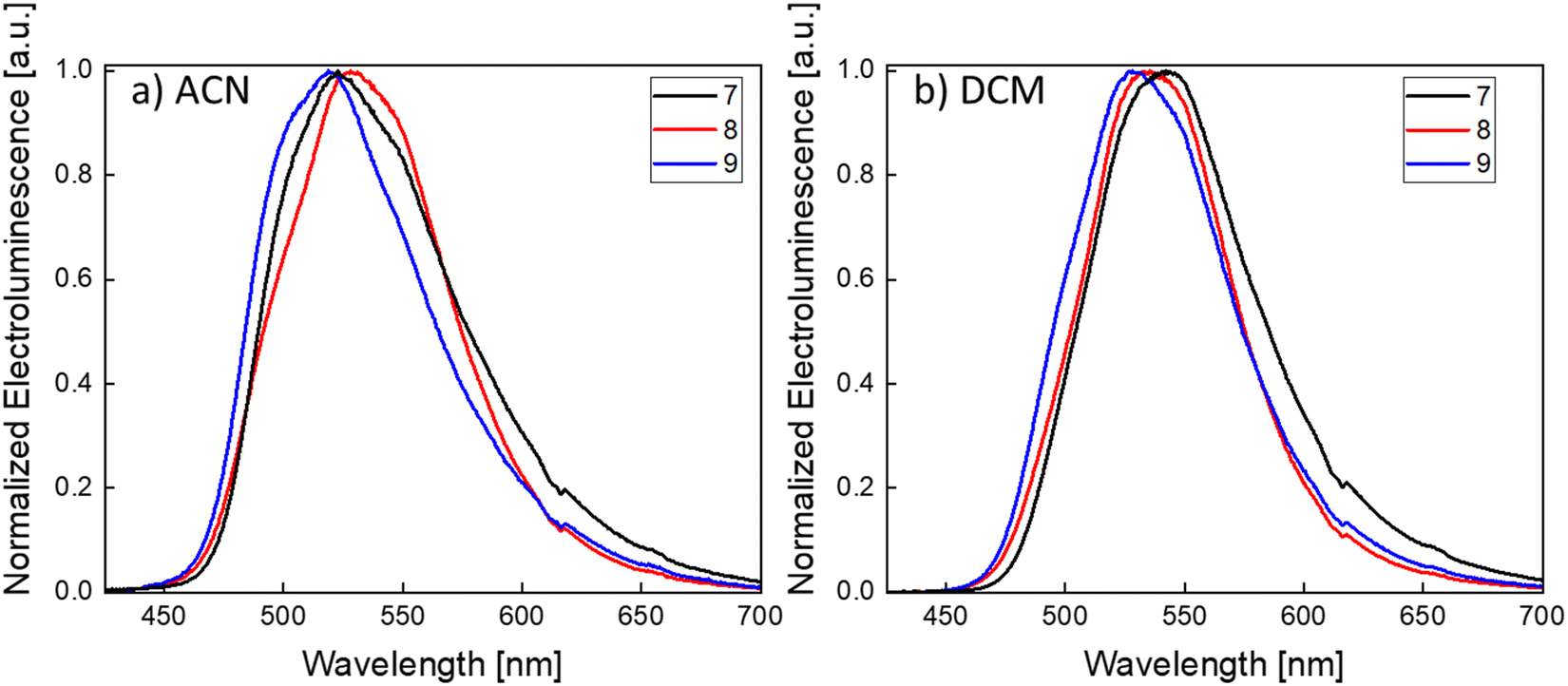 | ||
| Fig. 6 Normalized electroluminescence spectra of LEC devices ITO/PEDOT:PSS/iTMC:IL(4:1)/Al, (iTMC: 7 (black), 8 (red) and 9 (blue)) with the active layer prepared from solutions in ACN (a) and DCM (b). | ||
In spite of their simplicity, iTMC-LECs fabricated from DCM solutions of the carbazole-appended compounds, 7–9, showed an improved performance than PhOLEDs based on similar complexes bearing R-C^C* (R = H, CN), since the optimal OLED, based on compound [Pt(H-C^C*)(dppbz)]PF6 achieved a maximum luminance of 200 cd m−2 with a peak CE of 0.4, at 10 V.27
Similarly, devices made from compounds 5 and 6 were also fabricated. The active layers (iTMC:IL) showed very low PLQYs with values of 1% for both emitters. Devices made with 5 and 6 turned-on, showing the classic voltage evolution over time of a LEC (Fig. S24, ESI‡); however their performance was extremely poor. For emitter 6, the maximum observed luminance was 4 cd m−2 (Fig. S24b, ESI‡) while for emitter 5 the luminance was lower than the sensitivity of our LEC lifetime characterization setup (less than ca. 1 cd m−2). For emitter 6, it was also possible to measure the EL spectra, which showed a blue emission centered at 491 nm (CIE 0.2343, 0.4776). The very low QY of emitters 5 and 6 somewhat predicts the very low EL observed on these devices.
Conclusions
Platinum(II) complexes bearing new carbazole-appended cyclometallated N-heterocyclic carbenes such as [Pt(Cbz-C^C*)Cl(NCCH3)](3), [Pt(Cbz-C^C*)Cl(PPh3)](4), [Pt(Cbz-C^C*)(PPh3)(py)]PF6 (5), [Pt(Cbz-C^C*)(P^N)]PF6 (6), and [Pt(Cbz-C^C*)(P^P)]PF6 (P^P: dppm 7; dppe, 8; dppbz, 9) have been prepared by a stepwise procedure from 9-(4-bromo-phenyl)-9H-carbazole, imidazole, [{Pt(μ-Cl)(η3-2-Me-C3H4)}2] and the corresponding ancillary ligands and then exhaustively characterized. The presence of an electron donating group (R = Cbz) in the R-C^C* skeleton of these complexes increases the energy of the HOMO compared with those of similar complexes bearing R-C^C* (R = H, CN, and CO2Et), shifting their emissions from blue to green. In spite of the simplicity, iTMC-LECs fabricated from the DCM solutions of the carbazole-appended compounds, 7–9, operate at a 4–5 V voltage, and achieve luminance values of 265, 187 and 180 cd m−2 with peak CEs of 5.7, 3.7 and 3.6 cd A−1, respectively. Thus, we have tactically brought together the strong electron-donating abilities and good hole transporting properties from Pt(C^C*) and Cbz groups to prepare the first green-emitting iTMC-LECs based on mononuclear Pt(II) complexes.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the “Ministerio de Ciencia Innovación y Universidades”/FEDER (Project PGC2018-094749-B-I00) and the Gobierno de Aragón (Grupo E17_20R: Quimica Inorgánica y de los Compuestos Organometálicos). The authors thank the Centro de Supercomputación de Galicia (CESGA) for generous allocation of computational resources. The authors acknowledge support from Comunitat Valenciana (IDIFEDER/2020/063, PROMETEU/2020/077 and CIGE/2021/027), the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program Grant agreement No. 834431 and the Spanish Ministry of Science, Innovation and Universities (MICIU) (RTI2018-095362-A-I00 and EQC2018-004888-P).References
- K. Schlingman, Y. Chen, R. S. Carmichael and T. B. Carmichael, Adv. Mater., 2021, 33, 2006863 CrossRef CAS PubMed.
- Q. B. Pei and R. D. Costa, Adv. Funct. Mater., 2020, 30 Search PubMed.
- Q. B. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086–1088 CrossRef CAS PubMed.
- E. Fresta and R. D. Costa, Adv. Funct. Mater., 2020, 30, 1908176 CrossRef CAS.
- K. Youssef, Y. Li, S. O'Keeffe, L. K. Li and Q. Pei, Adv. Funct. Mater., 2020, 30, 1909102 CrossRef CAS.
- S. B. Meier, D. Tordera, A. Pertegas, C. Roldan-Carmona, E. Orti and H. J. Bolink, Mater. Today, 2014, 17, 217–223 CrossRef CAS.
- S. Tang, A. Sandström, P. Lundberg, T. Lanz, C. Larsen, S. van Reenen, M. Kemerink and L. Edman, Nat. Commun., 2017, 8, 1190 CrossRef PubMed.
- Q. J. Sun, Y. F. Li and Q. B. Pei, J. Disp. Technol., 2007, 3, 211–224 CAS.
- T. Lanz, E. M. Lindh and L. Edman, J. Mater. Chem. C, 2017, 5, 4706–4715 RSC.
- E. M. Lindh, P. Lundberg, T. Lanz and L. Edman, Sci. Rep., 2019, 9, 10433 CrossRef PubMed.
- J. Ràfols-Ribé, X. Zhang, C. Larsen, P. Lundberg, E. M. Lindh, C. T. Mai, J. Mindemark, E. Gracia-Espino and L. Edman, Adv. Mater., 2022, 34, 2107849 CrossRef.
- L. Edman, Electrochim. Acta, 2005, 50, 3878–3885 CrossRef CAS.
- R. D. Costa, E. Orti, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- A. F. Henwood and E. Zysman-Colman, Top. Curr. Chem., 2016, 374, 36 CrossRef PubMed.
- C. H. Lyons, E. D. Abbas, J. K. Lee and M. F. Rubner, J. Am. Chem. Soc., 1998, 120, 12100–12107 CrossRef CAS.
- G. U. Mahoro, J. Fernandez-Cestau, J.-L. Renaud, P. B. Coto, R. D. Costa and S. Gaillard, Adv. Opt. Mater., 2020, 8, 2000260 CrossRef CAS.
- K. Matsuki, J. Pu and T. Takenobu, Adv. Funct. Mater., 2020, 30, 1908641 CrossRef CAS.
- R. B. Bai, X. W. Meng, X. X. Wang and L. He, Adv. Funct. Mater., 2020, 30, 1907169 CrossRef CAS.
- S. Keller, A. Pertegas, G. Longo, L. Martinez, J. Cerda, J. M. Junquera-Hernandez, A. Prescimone, E. C. Constable, C. E. Housecroft, E. Orti and H. J. Bolink, J. Mater. Chem. C, 2016, 4, 3857–3871 RSC.
- Q. Zeng, F. Li, Z. Chen, K. Yang, Y. Liu, T. Guo, G.-G. Shan and Z. Su, ACS Appl. Mater. Interfaces, 2020, 12, 4649–4658 CrossRef CAS PubMed.
- A. F. Henwood and E. Zysman-Colman, A Comprehensive Review of Luminescent Iridium Complexes Used in Light-Emitting Electrochemical Cells (LEECs), 2017 Search PubMed.
- C. E. Housecroft and E. C. Constable, Coord. Chem. Rev., 2017, 350, 155–177 CrossRef CAS.
- W. Lu, M. C. W. Chan, N. Y. Zhu, C. M. Che, C. N. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS PubMed.
- K. T. Weber, K. Karikis, M. D. Weber, P. B. Coto, A. Charisiadis, D. Charitaki, G. Charalambidis, P. Angaridis, A. G. Coutsolelos and R. D. Costa, Dalton Trans., 2016, 45, 13284–13288 RSC.
- M. Z. Shafikov, S. Tang, C. Larsen, M. Bodensteiner, V. N. Kozhevnikov and L. Edman, J. Mater. Chem. C, 2019, 7, 10672–10682 RSC.
- L. M. Cinninger, L. D. Bastatas, Y. L. Shen, B. J. Holliday and J. D. Slinker, Dalton Trans., 2019, 48, 9684–9691 RSC.
- V. Sicilia, L. Arnal, A. J. Chueca, S. Fuertes, A. Babaei, A. M. Igual Munoz, M. Sessolo and H. J. Bolink, Inorg. Chem., 2020, 59, 1145–1152 CrossRef CAS PubMed.
- S. Fuertes, A. J. Chueca, A. Martín and V. Sicilia, J. Organomet. Chem., 2019, 889, 53–61 CrossRef CAS.
- V. Sicilia, S. Fuertes, A. J. Chueca, L. Arnal, A. Martín, M. Perálvarez, C. Botta and U. Giovanella, J. Mater. Chem. C, 2019, 7, 4509–4516 RSC.
- S. Fuertes, A. J. Chueca, M. Perálvarez, P. Borja, M. Torrell, J. Carreras and V. Sicilia, ACS Appl. Mater. Interfaces, 2016, 8, 16160–16169 CrossRef CAS PubMed.
- S. Fuertes, A. J. Chueca, L. Arnal, A. Martín, U. Giovanella, C. Botta and V. Sicilia, Inorg. Chem., 2017, 56, 4829–4839 CrossRef CAS PubMed.
- S. Stipurin and T. Strassner, Inorg. Chem., 2021, 60, 11200–11205 CrossRef CAS PubMed.
- S. Fuertes, H. García, M. Perálvarez, W. Hertog, J. Carreras and V. Sicilia, Chem. – Eur. J., 2015, 21, 1620–1631 CrossRef CAS.
- H. Leopold, M. Tenne, A. Tronnier, S. Metz, I. Munster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2016, 55, 15779–15782 CrossRef CAS PubMed.
- P. Pinter, J. Soellner and T. Strassner, Eur. J. Inorg. Chem., 2021, 3104–3107 CrossRef CAS.
- P. Pinter, J. Soellner and T. Strassner, Organometallics, 2021, 40, 557–563 CrossRef CAS.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312–319 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Y. Xie, Y. Zhang, S. Liu, J. F. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- W.-J. Xu, S.-J. Liu, T.-C. Ma, Q. Zhao, A. Pertegás, D. Tordera, H. J. Bolink, S.-H. Ye, X.-M. Liu, S. Sun and W. Huang, J. Mater. Chem., 2011, 21, 13999–14007 RSC.
- A. Chan, M. Ng, Y.-C. Wong, M.-Y. Chan, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 10750–10761 CrossRef CAS PubMed.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 6281–6291 CrossRef CAS.
- L. K. Li, W. K. Kwok, M. C. Tang, W. L. Cheung, S. L. Lai, M. Ng, M. Y. Chan and V. W. W. Yam, Chem. Sci., 2021, 12, 14833–14844 RSC.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- F. S. Kim, X. G. Guo, M. D. Watson and S. A. Jenekhe, Adv. Mater., 2010, 22, 478 CrossRef CAS PubMed.
- D. Tordera, A. Pertegás, N. M. Shavaleev, R. Scopelliti, E. Ortí, H. J. Bolink, E. Baranoff, M. Grätzel and M. K. Nazeeruddin, J. Mater. Chem., 2012, 22, 19264–19268 RSC.
- H.-C. Su, F.-C. Fang, T.-Y. Hwu, H.-H. Hsieh, H.-F. Chen, G.-H. Lee, S.-M. Peng, K.-T. Wong and C.-C. Wu, Adv. Funct. Mater., 2007, 17, 1019–1027 CrossRef CAS.
- J. a G. Williams, Photochemistry and Photophysics of Coordination Compounds: Platinum, Springer, Berlin, 2007 Search PubMed.
- S. Jaime, L. Arnal, V. Sicilia and S. Fuertes, Organometallics, 2020, 39, 3695–3704 CrossRef CAS.
- S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885–9895 CrossRef CAS PubMed.
- T. Ogawa, W. M. C. Sameera, D. Saito, M. Yoshida, A. Kobayashi and M. Kato, Inorg. Chem., 2018, 57, 14086–14096 CrossRef CAS.
- J. D. Slinker, J. Rivnay, J. S. Moskowitz, J. B. Parker, S. Bernhard, H. D. Abruna and G. G. Malliaras, J. Mater. Chem., 2007, 17, 2976–2988 RSC.
- H. J. Bolink, E. Coronado, R. D. Costa, N. Lardies and E. Orti, Inorg. Chem., 2008, 47, 9149–9151 CrossRef CAS PubMed.
- J. D. Slinker, J. A. DeFranco, M. J. Jaquith, W. R. Silveira, Y.-W. Zhong, J. M. Moran-Mirabal, H. G. Craighead, H. D. Abruña, J. A. Marohn and G. G. Malliaras, Nat. Mater., 2007, 6, 894–899 CrossRef CAS PubMed.
- D. Tordera, J. Frey, D. Vonlanthen, E. Constable, A. Pertegás, E. Ortí, H. J. Bolink, E. Baranoff and M. K. Nazeeruddin, Adv. Energy Mater., 2013, 3, 1338–1343 CrossRef CAS.
- J. Ràfols-Ribé, N. D. Robinson, C. Larsen, S. Tang, M. Top, A. Sandström and L. Edman, Adv. Funct. Mater., 2020, 30, 1908649 CrossRef.
- W. Zhao, C. Y. Liu, Q. Wang, J. M. White and A. J. Bard, Chem. Mater., 2005, 17, 6403–6406 CrossRef CAS.
- Y. Cao, Q. Pei, M. R. Andersson, G. Yu and A. J. Heeger, J. Electrochem. Soc., 1997, 144, L317–L320 CrossRef CAS.
- L. Edman, B. Liu, M. Vehse, J. Swensen, G. C. Bazan and A. J. Heeger, J. Appl. Phys., 2005, 98, 044502 CrossRef.
- M. L. D. Weber, E. Fresta, M. Elie, M. E. Miehlich, J.-L. Renaud, K. Meyer, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2018, 28, 1707423 CrossRef.
- H. J. Bolink, L. Cappelli, S. Cheylan, E. Coronado, R. D. Costa, N. Lardies, M. K. Nazeeruddin and E. Orti, J. Mater. Chem., 2007, 17, 5032–5041 RSC.
- M. Meyer, L. Mardegan, D. Tordera, A. Prescimone, M. Sessolo, H. J. Bolink, E. C. Constable and C. E. Housecroft, Dalton Trans., 2021, 50, 17920–17934 RSC.
Footnotes |
| † Dedicated to Paul R. Raithby, celebrating a career in inorganic and organometallic chemistry, on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2151291–2151294. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc02539f |
| This journal is © The Royal Society of Chemistry 2022 |