
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering the HOMO–LUMO gap of indeno[1,2-b]fluorene†‡
Raquel
Casares
 a,
Álvaro
Martínez-Pinel
a,
Sandra
Rodríguez-González
b,
Irene R.
Márquez
c,
Luis
Lezama
d,
M. Teresa
González
e,
Edmund
Leary
e,
Víctor
Blanco
a,
Joel G.
Fallaque
ef,
Cristina
Díaz
g,
Fernando
Martín
efh,
Juan M.
Cuerva
*a and
Alba
Millán
*a
a,
Álvaro
Martínez-Pinel
a,
Sandra
Rodríguez-González
b,
Irene R.
Márquez
c,
Luis
Lezama
d,
M. Teresa
González
e,
Edmund
Leary
e,
Víctor
Blanco
a,
Joel G.
Fallaque
ef,
Cristina
Díaz
g,
Fernando
Martín
efh,
Juan M.
Cuerva
*a and
Alba
Millán
*a
aDepartamento de Química Orgánica, Facultad de Ciencias, Unidad de Excelencia de Química Aplicada a Biomedicina y Medioambiente (UEQ), Universidad de Granada, 18071 Granada, Spain. E-mail: jmcuerva@ugr.es; amillan@ugr.es
bDepartamento de Química Física Aplicada, Universidad Autónoma de Madrid, 28049 Madrid, Spain
cCentro de Instrumentación Científica, Universidad de Granada, 18071 Granada, Spain
dDepartamento de Química Orgánica e Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco, 48940, Leioa, Spain
eFundación IMDEA Nanociencia, 28049, Madrid, Spain
fDepartamento de Química, Módulo 13, Universidad Autónoma de Madrid, 28049, Madrid, Spain
gDepartamento de Química Física, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040, Madrid, Spain
hCondensed Matter Physics Center (IFIMAC), 28049, Madrid, Spain
First published on 28th July 2022
Abstract
A direct, efficient and versatile strategy for the modulation of optoelectronic and magnetic properties of indeno[1,2-b]fluorene has been developed. 4-Substituted-2,6-dimethylphenyl acetylene groups placed in the apical carbon of the five-membered rings lead to redshifted absorption maxima (λmax ranging from 600–700 nm) and considerable narrowing of the HOMO–LUMO energy gap (down to 1.5 eV). Experimental and theoretical data show an increase in the diradical character (y) and a decrease of the singlet-triplet energy gap. Moreover, we have investigated the single-molecule conductance of the antiaromatic indeno[1,2-b]fluorene for the first time by including thiomethyl (-SMe) anchor groups on the phenylacetylene moiety. Conductance values one order of magnitude higher than those of a reference linear 3-ring para-phenylene ethylene have been found, despite the longer length of the S-to-S molecular junction. First principles transport calculations support this high conductance value.
Introduction
Indeno[1,2-b]fluorene1–6 (IF, 1) is considered the formally antiaromatic analogue of pentacene. In contrast to the latter, IF displays high electron affinity due to the presence of the cyclopentadiene rings, thus narrowing the HOMO–LUMO energy gap.1 This fact is explained by the existence of a central proaromatic p-quinodimethane (p-QDM) unit that presents a canonical diradical contribution (Fig. 1A).6,7 The narrow HOMO–LUMO gap makes this molecule and their derivatives attractive for organic electronic devices3,8–10 and complementary to acenes.11 The challenge is to shield the highly reactive apical carbons at the five-membered ring (R group in Fig. 1A). In this sense, strategies to provide thermodynamic or kinetic stabilization are commonly used.12,13 The most widely employed R groups are 2,4,6-trimethylphenyl (mesityl, Mes) and triisopropylsilyl (TIPS) acetylene. In addition to conferring stability,14,15 these R groups have been proposed for the fine tuning of the optoelectronic properties of the systems although with a limited success. For example, Chase et al. demonstrated that the use of the electron-withdrawing TIPS acetylene produced a bathochromic shift in the UV-Vis absorption maxima in comparison to aryl groups such as Mes (Fig. 1B, 2vs.3).2,3 Modifications of the IF core would be in principle an alternative approach.3 However, the incorporation of substituents at the 2- and 8-positions have almost no impact on the HOMO–LUMO gap tuning. For example, UV-Vis absorption maxima vary by 34 nm when different arenes are used as R groups (Fig. 1B, 3) whilst the introduction of donor or acceptor groups (X) in the IF core only shifts the maxima 16 nm (Fig. 1B, 2). The reason is related to a low HOMO/LUMO orbital density in those positions. To the best of our knowledge, the state of the art is represented by the work of Nishida et al. with the inclusion of α-substituted thienyl groups at the 6- and 12-positions (Fig. 1B, 3),4 the latter compound having the most red-shifted absorption maximum (607 nm) and the lowest HOMO–LUMO gap (1.94 eV) to date. Thus, a new approach to achieve a substantial modification of energy gap values in a predictable way is most desirable.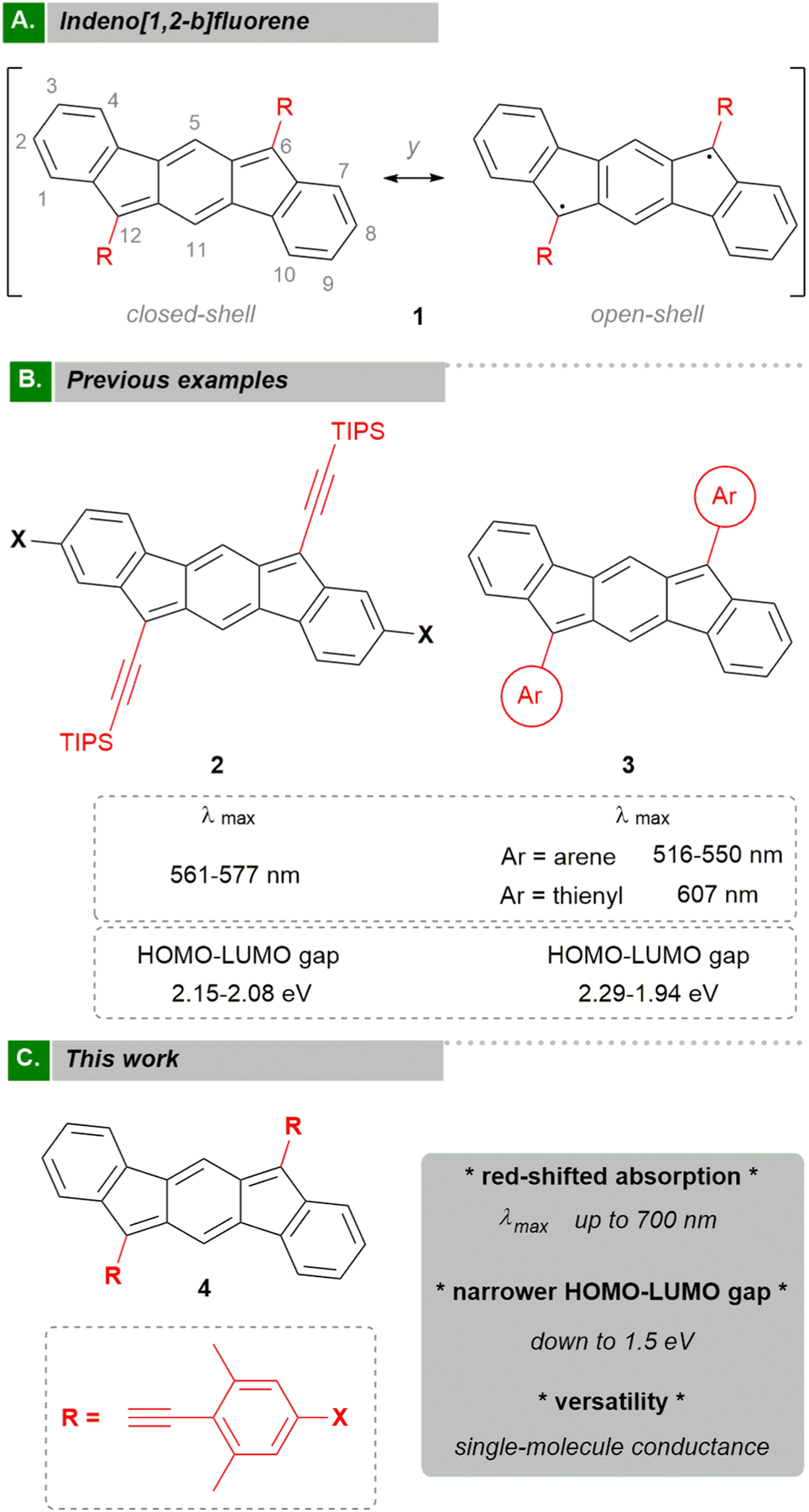 | ||
| Fig. 1 (A) Resonance structures of indeno[1,2-b]fluorene; y: diradical character index (0 (closed-shell) ≤ y ≤ 1 (open-shell)). (B) Previous examples of indeno[1,2-b]fluorenes; X = donor/acceptor groups; Ar = arene or thienyl; HOMO–LUMO energy gap values estimated from optical properties. (C) Our approach to modulate optoelectronic properties of indeno[1,2-b]fluorene; X = donor/acceptor groups. | ||
This HOMO–LUMO gap engineering would be useful to modulate the optical properties and also the electron transport through the molecule even at the single-molecule level. In this latter context, the energy gap narrowing offers the possibility of bringing the HOMO/LUMO levels closer to the Fermi level of the metal, in order to achieve high conductance potentially via a resonant transport mechanism. In a simple view of electron transport this is an optimal situation and it has been proposed as the origin of the greater conductance of antiaromatic systems over aromatic ones.16–18 Nevertheless, the electron transport through antiaromatic hydrocarbons at the single-molecule level is still understudied, and experimental results do not always follow this expectation. For example, the 12π-electron antiaromatic biphenylene core was thoroughly investigated by Gantenbein et al.19 They observed slight differences between conductance (G) values for biphenylene and its non-antiaromatic counterpart, fluorene (log(G/G0) =–4.9 vs.–4.8, where G0 = 2e2/h). These results agreed with those reported previously by Schneebeli et al.20 and show that for the smallest systems any differences due to antiaromaticity do not appear in the measured conductance.16 Clearer results were observed by Schmidt et al. for the 16π-electron dibenzo[a,e]pentalene (DBP). They measured the conductance through the pentalene central core and compared it with an aromatic anthracene analogue. Here, a small increase was found between the anthracene and antiaromatic DBP (log(G/G0) = −4.4 vs. −4.3, respectively).21 Fujii et al. found that an antiaromatic norcorrole nickel complex was about one order of magnitude more conductive than an aromatic nickel-porphyrin analogue.18 The norcorrole compound is, however, slightly smaller than the porphyrin, so at least some of the conductance gain could be accounted for from the length change. Apart from these examples, there are no other conductance measurements on antiaromatic polycyclic hydrocarbons,22,23 mainly due to the demanding synthesis and inherent instability.
Here, we present a set of novel indeno[1,2-b]fluorenes (20π-electrons), the next in the Hückel's 4n π-electron counting series, in which the HOMO–LUMO gap is tuned from ∼1.8 eV down to 1.5 eV resulting in an absorption maxima displacement of up to ∼100 nm with respect to the indeno[1,2-b]fluorene described by Nishida et al.4 Our approach is based on a stabilizing group consisting of 2,4,6-substituted phenyl acetylenes, which permits the fine-tuning of the optoelectronic and magnetic properties of indeno[1,2-b]fluorene (Fig. 1C). It is worth noting that the common core can be electronically coupled with different donor and acceptor groups in the 4-position of the phenyl acetylene moiety, which can be subsequently used for different purposes. In particular, the inclusion of anchoring groups for gold electrodes (SMe) allows us to measure the single-molecule conductance of indeno[1,2-b]fluorene for the first time, observing an unusually high conductivity in comparison with that of the dibenzopentalene analogue owing to the HOMO–LUMO gap narrowing. All these results have been rationalized using quantum chemical calculations.
Results and discussion
Firstly, we synthesized the known diketone 524 that provides the common IF core. Then different alkynes 6a–h were prepared following a general strategy based on the Sonogashira cross-coupling reaction25 using trimethylsilylacetylene and the corresponding iodoarenes, and finally deprotection of the alkyne-protecting group (see ESI,‡ for details). With both building blocks in our hands, we prepared a variety of indeno[1,2-b]fluorenes (4a–h) bearing both electron withdrawing and electron donating groups at the 4-position of the arene (X group in Scheme 1). For that, we carried out nucleophilic addition of the corresponding lithiated alkynes 6a–h to diketone 5 to give diols 7a–h. Subsequent dearomatization using SnCl2 gave the final products 4a–h as deep blue air-stable solids.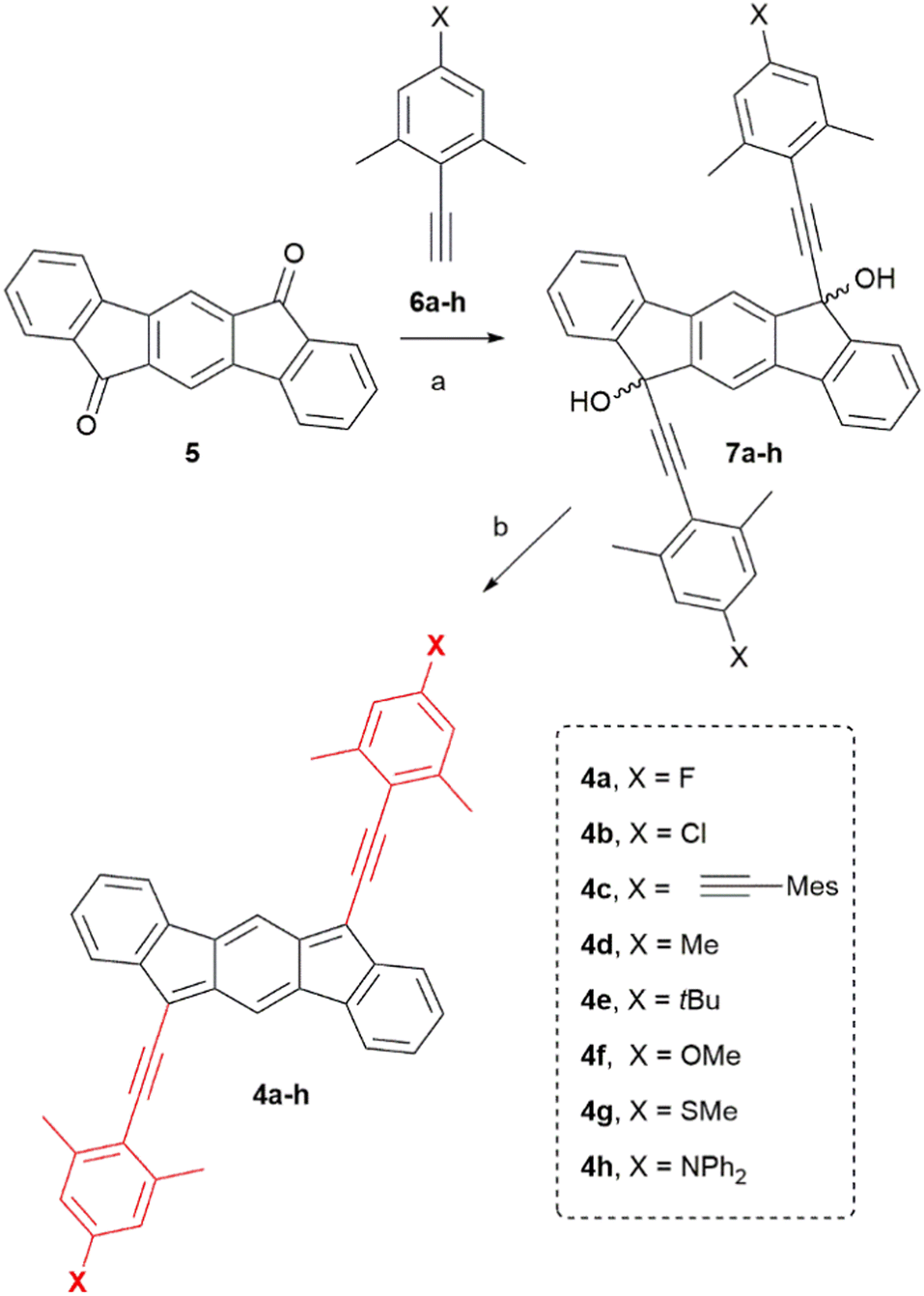 | ||
| Scheme 1 Synthesis of compounds 4a–h. (a) n-BuLi, THF, 0 °C to rt, 3 h; (b) SnCl2, toluene, 40 °C, 15 min. | ||
In general, compounds 4a–h are highly insoluble in a wide range of organic solvents at typical NMR concentrations, which precludes characterization by 13C-NMR in most of the cases. Nevertheless, the exact mass and isotopic distribution pattern of the peak corresponding to the [M]+ species in the high-resolution mass spectra were in good agreement with the theoretical data in all cases. It is worth mentioning that blocking the ortho- and para-position of the arene is key in order to ensure air stability. On the one hand, dearomatization reactions on products bearing either 4-tBu-phenylacetylene or 4-SMe-phenylacetylene fragments at the 6- and 12-positions gave messy reaction crudes. On the other hand, IF bearing 2,6-dimethylphenylacetylene decomposed in minutes. Increased stability by blocking ortho- and para-positions in the arene has been inspected from a theoretical point of view, through the spin density distribution (see Section S9.6 in the ESI‡). In addition, 4d and 4e were checked by UV-Vis after several days not observing any significant change in their UV-Vis absorption profiles (see ESI,‡ Section S6.1).
Then we investigate the optoelectronic properties. Fig. 2 shows the UV-Vis absorption spectra of each compound recorded in CH2Cl2 solution.26 In general, the phenylacetylene group in the 6,12-IF position induces a larger bathochromic shift in absorption maxima compared to 2 and 3 (Fig. 1B and Table 1). We can see low-energy absorption maxima spanning almost 100 nm from λmax = 608 nm for the electron-withdrawing fluorine substituent (4a) to λmax = 694 nm for the electron-donating diphenylamine group (4h). Remarkably, these data are comparable to those reported for much more extended indeno[1,2-b]fluorene cores.27–29
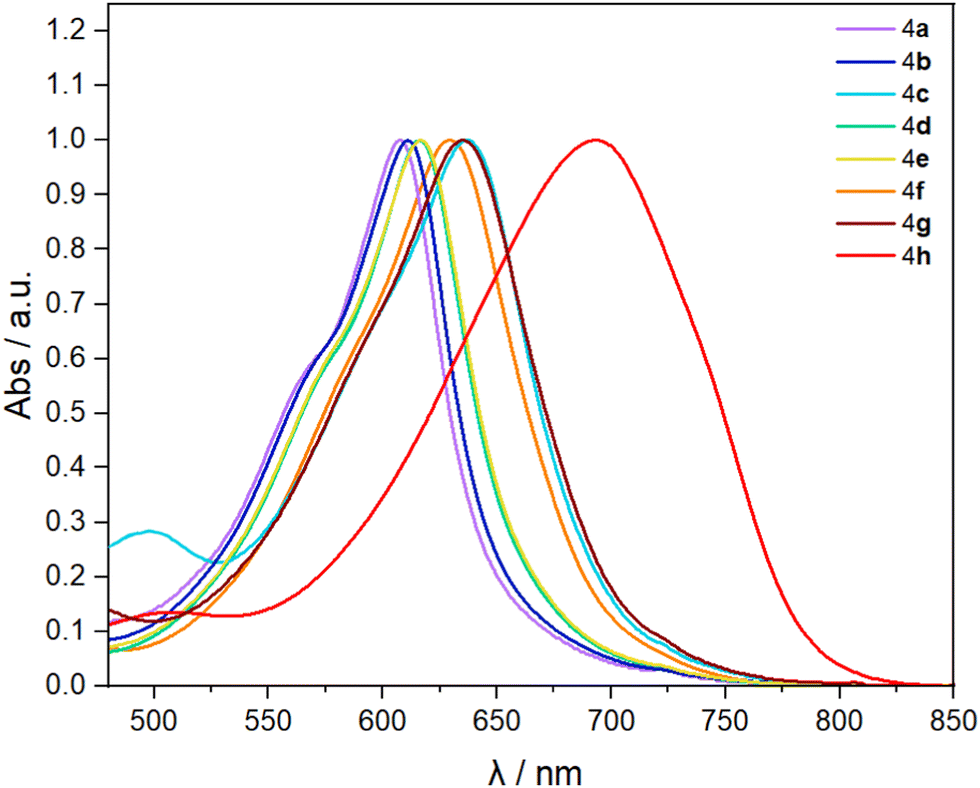 | ||
| Fig. 2 Normalized electronic absorption spectra for compounds 4a–h. All spectra were recorded in CH2Cl2 at 25 μM. | ||
| Comp. | Optical dataa | Electrochemical datab | Computational datac | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ max | E gap | E ox/Epa | E red/Epc | E HOMO | E LUMO | E gap | E HOMO | E LUMO | E gap | |
| a Spectra were obtained in CH2Cl2; wavelengths are in nm. The optical HOMO–LUMO gap was determined as the intersection of the x axis and a tangent line passing through the inflection point of the lowest-energy absorption; energies are in eV. b CVs were recorded using 1.5 mM of compound in 0.1 M of Bu4NPF6 in CH2Cl2 at a scan rate of 50 mV s−1. A glassy carbon disc was used as the working electrode, a Pt wire as the counter electrode, and a Ag wire as the pseudo-reference electrode. Eox/red is the half-wave potential (E1/2) for reversible/quasi-reversible processes or the potential of the peak anodic (Epa) or cathodic current (Epc) for irreversible processes. Potential values are reported in V vs. Fc+/Fc. HOMO and LUMO energies were estimated from first oxidation/reduction peak assuming an ionization energy of 4.8 eV for ferrocene. c Calculations performed at the the B3LYP/6-311+G** level of theory; energies in eV. d Data taken from ref. 3. | ||||||||||
| 2a | 568 | 2.12 | 1.20 | −0.69, −1.20 | −5.88 | −4.00 | 1.88 | −5.51 | −3.47 | 2.04 |
| 3a | 516 | 2.29 | 1.10, 1.59 | −1.12, −1.73 | −5.78 | −3.56 | 2.22 | −5.34 | −2.92 | 2.42 |
| 4a | 608 | 1.84 | 0.61 | −1.11, −1.47 | −5.41 | −3.69 | 1.72 | −5.17 | −3.47 | 1.71 |
| 4b | 611 | 1.80 | 0.61 | −1.07, −1.36 | −5.41 | −3.73 | 1.68 | −5.23 | −3.53 | 1.70 |
| 4c | 637 | 1.72 | — | −0.80, −1.00 | n.d. | −4.00 | n.d. | −4.99 | −3.42 | 1.57 |
| 4d | 616 | 1.78 | 0.59 | −1.33 | −5.39 | −3.47 | 1.92 | −4.98 | −3.29 | 1.69 |
| 4e | 616 | 1.79 | 0.62 | −1.24 | −5.42 | −3.56 | 1.86 | −4.98 | −3.28 | 1.69 |
| 4f | 630 | 1.73 | 0.66 | −1.13 | −5.46 | −3.67 | 1.79 | −4.83 | −3.18 | 1.65 |
| 4g | 636 | 1.70 | 0.41 | −1.14 | −5.21 | −3.66 | 1.55 | −4.90 | −3.28 | 1.62 |
| 4h | 694 | 1.55 | 0.23, 0.64 | −1.11, −1.55 | −5.03 | −3.69 | 1.34 | −4.71 | −3.17 | 1.54 |
Vertical excitation energies were calculated by means of the TD-DFT approach, predicting absorption spectra (Fig. S27 and Table S4, ESI‡), with energies within 0.3 eV of the measured values. The lowest-lying energy electronic transition corresponds to a HOMO → LUMO excitation, which redshifts with the strength of the electron-donating substituent in the 4-position (X group) of the phenyl group, according to the predicted narrowing of the HOMO–LUMO gap (see Table 1 and Table S2 in the ESI‡); and in full agreement with the experimental trend observed for the absorption maxima and the optical gap. A full description of the employed computational methods is presented in the ESI.‡
In general, our optical energy gap values are considerably lower (1.55–1.84 eV) than those reported before for indeno[1,2-b]fluorenes (1.9–2.2 eV).1–6 We highlight the case of 4h, for which we estimated an optical gap of 1.55 eV, the lowest reported to date for non-extended indeno[1,2-b]fluorene. As for other IF derivatives, compounds 4a–h are all non-emissive.30
Cyclic voltamograms (CVs) for compounds 4a and 4h are shown in Fig. 3 (see ESI,‡ Section S7 for CVs of the other compounds). Data are depicted in Table 1, which shows the half-potentials (Eox/red) of the reversible waves or the potential of peak current (Epa/pc) for irreversible ones. We observed that every compound (4a-h) can accept at least one electron. In addition, 4a–4c and 4h show a second reduction event. In general, the potential values of the first cathodic peaks at approximately −0.8 to −1.33 V are in agreement with other related compounds.2–4 In principle, more electron-withdrawing groups should stabilize anionic species, giving less negative values. However, this trend is not observed in our compounds 4a–h as can be seen in the representative examples shown in Fig. 3. Both compound 4a and 4h display a first reversible wave at Ered= –1.11 V, making the presence of either acceptor or donor group inconsequential (4a (F) and 4h (NPh2) respectively). This is consistent with the distribution of the LUMO orbital over the system, which is mainly located on the central core (see ESI,‡ Section S9.3). Remarkably, compounds 4a, 4b and 4d–4h gave an oxidation peak at a low-voltage potential (from +0.23 to +0.66 V) that translates as a raising of the HOMO energy. These observations can be explained by an increase in overall electron-density on the systems provided by the phenylacetylene moiety.
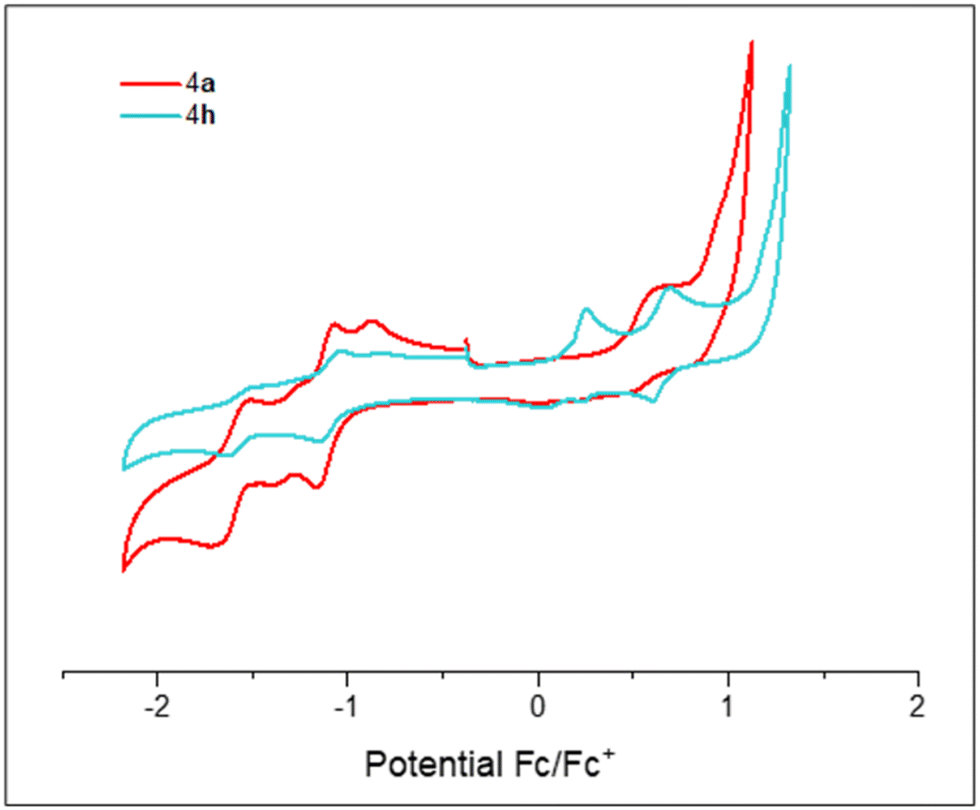 | ||
| Fig. 3 Cyclic voltammograms (CVs) for 4a and 4h. | ||
Consequently, the estimated electrochemical HOMO–LUMO gaps are reduced considerably in comparison to other indeno[1,2-b]fluorenes.1–6 Interestingly, the electrochemically determined energy gaps are lower than optical and computational values which have been also observed for other examples of indeno[1,2-b]fluorenes.2–4 In any case, the trend is quite consistent for all three data sets.
As explained in the introduction (Fig. 1A), the resonance between the closed-shell and the open-shell forms implies that indeno[1,2-b]fluorene could be a potential diradical. Diradicals have been postulated as building blocks for magnetically active materials.31–33 For that, a transition between the singlet and the triplet state (low- to high-spin) should occur and its energy should be low in order to facilitate the switching processes. As the HOMO–LUMO gap is supposed to trend with singlet–triplet energy gaps34 (ΔEST, the energy difference between the singlet ground electronic state and the lowest-lying triplet excited state), we wondered whether our phenylacetylene groups could serve to tune this key parameter. We select 4d as a model compound due to its structural resemblance to other reported indeno[1,2-b]fluorenes (e.g.2a and 3a).2,3 We analysed compound 4d by electron paramagnetic resonance (EPR) spectroscopy. Interestingly, 4d is not silent, indicating the presence of paramagnetic species. We observed a very weak signal for 4d at room temperature which could correspond to a small population of the triplet species (see ESI,‡ Section S8). This phenomenon has not been observed in other indeno[1,2-b]fluorenes before.35 To gain insight into this result, first, the open-shell singlet character for 2a (X = H), 3a (Ar = Mes) and 4d were evaluated with DFT using the diradical character index (y). A negligible y value was calculated for 3a (0.090); meanwhile a similar value of 0.30 and 0.36 has been estimated for 2a and 4d, respectively. DFT calculations of ΔEST for 2a, 3a and 4d36 estimate a decrease in the singlet-triplet energy gaps of over 7 kcal mol−1 compared to other reported indeno[1,2-b]fluorene structures (10.7 kcal mol−1 for 4dvs. 18.24 kcal mol−1 for 3a) (see ESI,‡ Sections S9.4 and S9.5).
This greater diradical character should reflect in the C–C bond distances of the central p-QDM unit. To evaluate this, we attempted the crystallization of compound 4d. However, this compound was challenging to crystallize, and no suitable crystals were obtained in a variety of solvent mixtures and/or applying different crystallization techniques. Fortunately, we did obtain single crystals of 4e (bearing a tBu instead of Me) and this compound was characterized by X-ray diffraction (Fig. 4).37 The compound 4e has helped to complete the study since it shows the same electronic and diradical properties as 4d, (see ESI,‡ for comparison, Sections S9.1 and S9.5). The structure of 4e was confirmed and bond lengths were analysed.
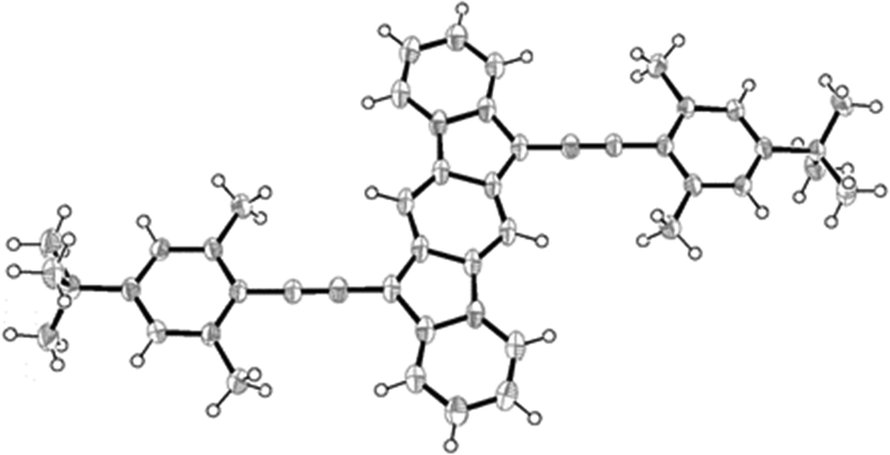 | ||
| Fig. 4 ORTEP drawing of the X-ray crystal structure of compound 4e, showing the thermal ellipsoids at 50% probability. | ||
The molecule features the distinct p-QDM unit within the core, with alternating long (1.418 Å and 1.451 Å) and short (1.397 Å and 1.361 Å) C–C bond lengths. However, 4e contains longer double bonds and shorter single bonds in comparison to previously reported ones (see ESI,‡ Table S1).2–4 promoting a slight gain in aromatic character for the central p-QDM unit. Quantum chemical DFT calculations reproduce the observed lengthening/shortening of double/single bonds, (see Table S3 in ESI‡) testing the structural similarity with 4d. In addition, calculated nucleus independent chemical shift (NICS) indices for the open-shell singlet state are consistent with an increase in the aromaticity of the central benzenoid ring of 4e (and therefore 4d) in comparison to 3a (see ESI,‡ Fig. S40), supporting also the higher diradical character and smaller singlet–triplet energy gap presented above. All these results suggest that 4e has a modest open-shell (diradical) singlet system in the ground state. Although the triplet state is only partially accessible at room temperature and these structures are better described as closed-shell molecules, these results open a promising approach to tune ΔEST in a straightforward manner, just by changing the stabilizing group.
Finally, to show the versatility of this stabilizing group, we focus on compound 4g. The introduction of the thiomethyl group (-SMe) makes this molecule suitable for single-molecule conductance measurements by means of the scanning tunneling microscopy break-junction technique (STM-BJ).38 These experiments are based on a push–pull process between the STM gold tip and the substrate. During the pulling, G decreases with the distance between electrodes (z), displaying regions of constant conductance (plateaus) at G < G0 if molecular junctions are formed between the metallic electrodes. SMe anchor groups have been widely used in conductance studies39,40 and avoid known drawbacks related to the use of thiol groups and gold electrodes.41 In addition, the introduction of the alkyne spacer offers a double advantage: (i) to avoid a permanent torsion of the phenyl group which would hamper electron transport,42,43 and (ii) to promote a narrowing of the HOMO–LUMO gap as has been formerly proved. Fig. 5 displays 2D G vs. z and 1D G histograms obtained for 4g. We found that 18% of the pulling G vs. z traces showed plateaus (total number of traces ∼11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800), which produced a well-defined cloud in their 2D histogram (Fig. 5b) in contrast with the smooth exponential decay of the traces without plateaus (Fig. 5a). This cloud translates into a quasi-symmetric Gaussian type curve in the 1D G histogram (Fig. 5c) with a peak centered at log (G/G0) = −3.6, with a half width at half maximum (HWHM) of 0.36. Fig. 5c (inset) shows that the plateau-length distribution peaks at 1.1 nm (HWHM = 0.5 nm), while it reaches its maximum values at the expected Au-to-Au molecular bridge length, once the typical gold retraction after breakage is subtracted (2.1 nm minus 0.4 nm), indicated by a vertical black line in the figure. These plateau-length distribution features are typical for SMe-terminated compounds, with molecules fully extended between the gold electrodes for the longest conductance plateaus.
800), which produced a well-defined cloud in their 2D histogram (Fig. 5b) in contrast with the smooth exponential decay of the traces without plateaus (Fig. 5a). This cloud translates into a quasi-symmetric Gaussian type curve in the 1D G histogram (Fig. 5c) with a peak centered at log (G/G0) = −3.6, with a half width at half maximum (HWHM) of 0.36. Fig. 5c (inset) shows that the plateau-length distribution peaks at 1.1 nm (HWHM = 0.5 nm), while it reaches its maximum values at the expected Au-to-Au molecular bridge length, once the typical gold retraction after breakage is subtracted (2.1 nm minus 0.4 nm), indicated by a vertical black line in the figure. These plateau-length distribution features are typical for SMe-terminated compounds, with molecules fully extended between the gold electrodes for the longest conductance plateaus.
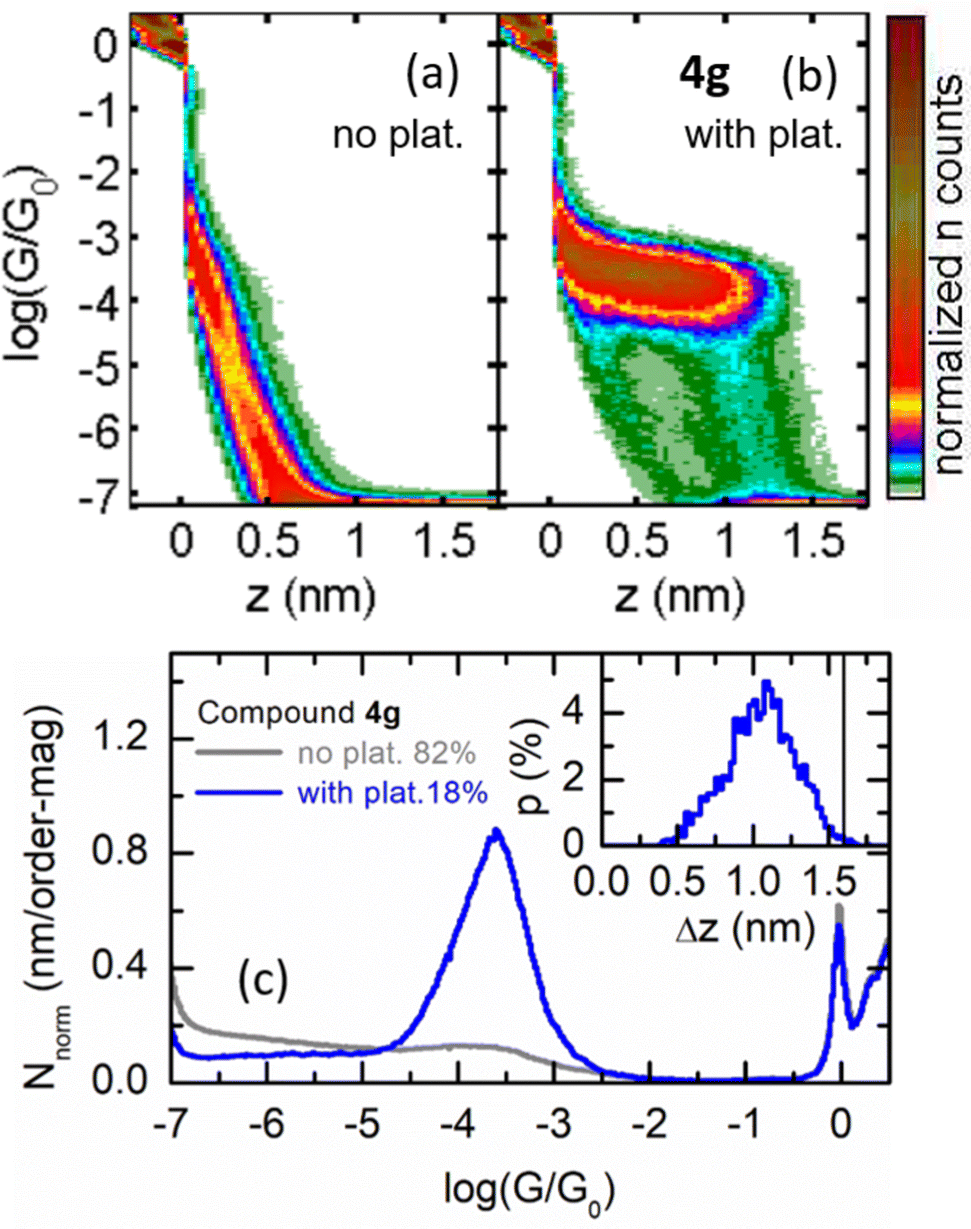 | ||
| Fig. 5 (a) and (b) 2D histograms for compound 4g built from traces without (a) and with (b) plateaus. (c) Corresponding 1D histograms. The inset in (c) shows the plateau-length distribution for the traces with plateaus, where the black vertical line corresponds to the expected plateau length for a S-to-S molecular bridge with the molecule fully stretched between the apexes of the electrodes. For details of sample preparation and methodology, see ESI,‡ Section S10. | ||
It is remarkable that the conductance value observed for 4g is one order of magnitude higher than that of the slightly shorter 3-ring reference p-phenylene ethylene (OPE3-SMe).44 In addition, the conductance of 4g is more than half order of magnitude greater than that observed for a similar dibenzopentalene (DBP) compound, also with SMe anchor groups (log(G/G0) = −4.3) which has two fewer bonds along the shortest S-to-S pathway.21 This behaviour is noteworthy because the relationship between the degree of aromaticity/antiaromaticity and molecular conductance in neutral molecules is under debate and there is no clear consensus in the literature. For example, Chen et al. concluded that for a family of amine-terminated wires containing either thiophene, oxazole or cyclopentadiene, the conductance was lowest for the thiophene derivative, which is formally the most aromatic of the three.16 On the other hand, Yang et al. studied a similar family of 2,5-disubstituted 5-membered heterocycles, this time with pyridyl anchor groups. Here, however, they did not find any correlation between conductance and aromaticity.23a
It is insightful to compare the properties of compound 4g with the DBP-SMe previously studied.21 The electrochemical HOMO–LUMO gaps of the two are 1.55 eV and 1.99 eV respectively. Our NICS calculations show that the central 5–6–5 rings are quite strongly antiaromatic which is reflected in the low electrochemical HOMO–LUMO gap of 1.55 eV. The gap is about 20% narrower than for the dibenzopentalene previously studied which suggests 4g has greater antiaromatic character. Our results thus demonstrate that despite the slight length increase of 4g over DBP-SMe, the reduced HOMO–LUMO gap as a consequence of the greater antiaromatic character is more than enough to outweigh this difference.
Fig. S41 (ESI‡) compares conductance vs voltage (G–V) curves for both compounds, recorded by sweeping the voltage between −1 V and +1 V at different positions along the conductance plateaus.45 The increase in conductance above ±0.6 V observed for 4g is in contrast with flatter G–V curves of dibenzopentalene,21 and points towards the closest molecular level of 4g being closer in energy to the gold Fermi level than that of dibenzopentalene, in agreement with the greater observed low-bias conductance.
Overall, this comparison suggests that the reason for the higher conductance of 4g over DBP-SMe is its smaller HOMO–LUMO gap, which is a direct consequence of its enhanced antiaromatic character. Perhaps this is not quite the reasoning suggested by Breslow as to why conductance should correlate with the degree of aromatic/antiaromatic character, but it suggests that the general intuition is nonetheless correct.
In order to rationalize this experimental result, first principles calculations were carried out to compute the zero-bias DFT+Σ transmission function within the Landauer formalism. The end-to-end molecule arrangement within the junction was considered. All the details are described in Section S9.7 of the ESI.‡ From the single-molecule junction optimized geometry, an S-to-S vertical distance of 2.12 nm is obtained, in full agreement with the value estimated experimentally. In addition, a conductance value of log (G/G0) = −3.27 is calculated as the logarithm of the transmission probability at the Fermi level, in reasonable agreement with the experimental value.
The structure of compound 4g in the junction has been evaluated through the NICS index, and compared with that of molecule 4d (4e). As is shown in Fig. S40 and Table S6 in the ESI,‡ molecule 4g shows the same aromatic/antiaromatic character profile than compound 4d (4e) in the singlet state: aromatic external six-member rings, anti-aromatic five-member rings and less/more aromatic central p-QMD unit than the six/five rings. Quantitatively, external, and central six-member rings are slightly less aromatic than in the 4d (4e) case, indicating the closed-shell nature of this system into the junction.
Conclusions
In conclusion, a new strategy for narrowing the HOMO–LUMO energy gap of indeno[1,2-b]fluorene has been developed. The approach is based on the inclusion of different 4-substituted-2,6-dimethylphenyl acetylenes at the five-membered ring apical carbon atoms. This substitution maintains the stability while shifting absorption maxima to the 600–700 nm region corresponding to a narrower HOMO–LUMO gap. Thus, in comparison to other reported indeno[1,2-b]fluorene, a considerable overall decrease (down to ∼1.8–1.5 eV) is achieved. Interestingly, this approach also allowed us to tune the diradical character and the singlet-triplet energy gap and could be an appealing strategy to apply to other potential diradical structures. This is something we will be pursuing in future studies. In addition, we report single-molecule conductance measurements for the antiaromatic indeno[1,2-b]fluorene for the first time thanks to the introduction of anchoring groups (-SMe) in the phenylacetylene fragment. Our results show an unusually high conductance value for such a long molecule. This remarkable example adds to the very few studies of single-molecule conductance on antiaromatic molecular bridges and reinforces the idea that the smaller the energy gap, the larger the conductance value due to a better alignment of the HOMO/LUMO molecular orbitals with respect to the Fermi level of the metal. Our experimental results fit remarkably well with the theoretical calculations.Author contributions
Conceptualization: J. M. C., A. M.; data curation: S. R. G., J. G. F., C. D., F. M.; formal analysis: R. C., A. M P., S. R. G., I. R. M., L. L., M. T. G., E. L., V. B., J. M. C., A. M.; funding acquisition: F. M. J. M. C., A. M.; investigation: R. C., A. M. P., I. R. M.; methodology: R. C., A. M. P.; project administration: J. M. C., A. M.; resources: J. M. C., A. M., F. M.; software: S. R. G., M. T. G., E. L., J. G. F., C. D., F. M.; supervision: J. M. C., A. M.; validation: R. C., A. M. P.; visualization: R. C., S. R. G., M. T. G., A. M.; writing – original draft: J. M. C., A. M.; writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from MCIN/AEI/10.13039/501100011033 and “ERDF A way of making Europe” (PGC2018-101873-A-I00), FEDER/Junta de Andalucía-Consejería de Economía y Conocimiento (A-FQM-221-UGR18), and the Ministerio de Ciencia e Innovación (PID2019-106732GB-I00, PID2019-105458RB-I00), ‘Severo Ochoa’ Programme for Centers of Excellence in R&D SEV-2016-0686, and ‘María de Maeztu’ Programme for Units of Excellence in R&D CEX2018- 000805-M). R. C. thanks the Ministerio de Ciencia e Innovación for a FPI contract (PRE2018-083406). A. M.-P. thanks the Ministerio de Universidades for a FPU contract (FPU19/03751). I. R. M. thanks MCIN/AEI/10.13039/501100011033 for a Personal Técnico de Apoyo contract (PTA2017-13681-I). E. L. thanks the Comunidad de Madrid Atracción de Talento grant 2019-T1/IND-16384. J. G. F. thanks the FPI program of the MCIN co-financed by the European Social Found. We acknowledge the allocation of computer time by the Red Española de Supercomputación and the Centro de Computación Científica at Universidad Autónoma de Madrid (CCC-UAM).References
- D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127 CrossRef CAS PubMed.
- D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 11103 CrossRef CAS PubMed.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349 CrossRef CAS PubMed.
- J.-i Nishida, S. Tsukaguchi and Y. Yamashita, Chem. – Eur. J., 2012, 18, 8964 CrossRef CAS PubMed.
- B. D. Rose, N. J. Sumner, A. S. Filatov, S. J. Peters, L. N. Zakharov, M. A. Petrukhina and M. M. Haley, J. Am. Chem. Soc., 2014, 136, 9181 CrossRef CAS PubMed.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977 CrossRef CAS PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed.
- I. Martínez, X. Zarate, E. Schott, C. Morales-Verdejo, F. Castillo, J. M. Manríquez and I. Chávez, Chem. Phys. Lett., 2015, 636, 31 CrossRef.
- A. M. Zeidell, L. Jennings, C. K. Frederickson, Q. Ai, J. J. Dressler, L. N. Zakharov, C. Risko, M. M. Haley and O. D. Jurchescu, Chem. Mater., 2019, 31, 6962 CrossRef CAS.
- A. Can, A. Facchetti and H. Usta, J. Mater. Chem. C, 2022, 10, 8496 RSC.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed.
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978 CrossRef CAS PubMed.
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem. Sci., 2020, 11, 1192 RSC.
- The less hindered trimethylsilylacetylene R group gives di-, tri- and oligomerization of [1,2-b]IF. See: X. Fu and D. Zhao, Org. Lett., 2015, 17, 5694 CrossRef CAS PubMed.
- The bare [1,2-b]-IF structure has been generated on-surface. See: Z. Majzik, N. Pavliček, M. Vilas-Varela, D. Pérez, N. Moll, E. Guitián, G. Meyer, D. Peña and L. Gross, Nat. Commun., 2018, 9, 1198 CrossRef PubMed.
- W. Chen, H. Li, J. R. Widawsky, C. Appayee, L. Venkataraman and R. Breslow, J. Am. Chem. Soc., 2014, 136, 918 CrossRef CAS PubMed.
- R. Breslow and F. W. Foss, J. Phys.: Condens. Matter, 2008, 20, 374104 CrossRef.
- S. Fujii, S. Marqués-González, J.-Y. Shin, H. Shinokubo, T. Masuda, T. Nishino, N. P. Arasu, H. Vázquez and M. Kiguchi, Nat. Commun., 2017, 8, 15984 CrossRef CAS PubMed.
- M. Gantenbein, X. Li, S. Sangtarash, J. Bai, G. Olsen, A. Alqorashi, W. Hong, C. J. Lambert and M. R. Bryce, Nanoscale, 2019, 11, 20659 RSC.
- S. Schneebeli, M. Kamenetska, F. Foss, H. Vazquez, R. Skouta, M. Hybertsen, L. Venkataraman and R. Breslow, Org. Lett., 2010, 12, 4114 CrossRef CAS PubMed.
- M. Schmidt, D. Wassy, M. Hermann, M. T. González, N. Agraït, L. A. Zotti, B. Esser and E. Leary, Chem. Commun., 2021, 57, 745 RSC.
- For other experimental studies based on antiaromatic molecules see ref. 18 and: X. Yin, Y. Zang, L. Zhu, J. Z. Low, Z.-F. Liu, J. Cui, J. B. Neaton, L. Venkataraman and L. M. Campos, Sci. Adv., 2017, 3, eaao2615 CrossRef PubMed.
- For experimental studies concerning aromaticity-conductance relationship: (a) Y. Yang, M. Gantenbein, A. Alqorashi, J. Wei, S. Sangtarash, D. Hu, H. Sadeghi, R. Zhang, J. Pi, L. Chen, X. Huang, R. Li, J. Liu, J. Shi, W. Hong, C. J. Lambert and M. R. Bryce, J. Phys. Chem. C, 2018, 122, 14965 CrossRef CAS; (b) M. Gantenbein, L. Wang, A. A. Al-jobory, A. K. Ismael, C. J. Lambert, W. Hong and M. R. Bryce, Sci. Rep., 2017, 7, 1794 CrossRef PubMed; (c) A. Mahendran, P. Gopinath and R. Breslow, Tetrahedron Lett., 2015, 56, 4833 CrossRef CAS.
- S. Merlet, M. Birau and Z. Y. Wang, Org. Lett., 2002, 13, 2157 CrossRef PubMed.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- Individual UV-Vis spectra are provided in the ESI,‡ Section S6.
- C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827 CrossRef CAS PubMed.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753 CrossRef CAS PubMed.
- C. K. Frederickson, J. E. Barker, J. J. Dressler, Z. Zhou, E. R. Hanks, J. P. Bard, L. N. Zakharov, M. A. Petrukhina and M. M. Haley, Synlett, 2018, 2562 CAS.
- B. D. Rose, L. E. Shoer, M. R. Wasielewski and M. M. Haley, Chem. Phys. Lett., 2014, 616-617, 137 CrossRef CAS.
- A. Rajca, Chem. Rev., 1994, 94, 871 CrossRef CAS.
- X. Hu, W. Wang, D. Wang and Y. Zheng, J. Mater. Chem. C, 2018, 6, 11232 RSC.
- P. Murto and H. Bronstein, J. Mater. Chem. C, 2022, 10, 7368 RSC.
- (a) M. Nakano, Top. Curr. Chem., 2017, 375, 47 CrossRef PubMed; (b) A. S. Hacker, M. Pavano, J. E. Wood, H. Hashimoto, K. M. D’Ambrosio, C. K. Frederickson, J. L. Zafra, C. J. Gómez-García, V. Postils, A. R. McDonald, D. Casanova, D. K. Frantz and J. Casado, Chem. Commun., 2019, 55, 14186 RSC.
- Although we cannot rule out the presence of a radical impurity, the detection of a signal in the Δms = ±2 region suggests that the species should be a triplet. Thus, monoradical impurity can be almost discarded. We submitted the sample to ICP-MS analysis before the EPR measurement to detect inorganic impurities. Different metals were detected but in ppm concentrations .
- Usually, these calculations are carried out replacing the R groups for simpler groups such as methyl in order to reduce calculation time. In this case, we have calculated ΔEST for the whole structures.
- Deposition number 2177496‡ contains the supplementary crystallographic data for this paper. ORTEP-3 software was used to create the ellipsoid picture: L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- B. Xu and J. N. Tao, Science, 2003, 301, 1221 CrossRef CAS PubMed.
- Y. S. Park, A. C. Whalley, M. Kamenetska, M. L. Steigerwald, M. S. Hybertsen, C. Nuckolls and L. Venkataraman, J. Am. Chem. Soc., 2007, 129, 15768 CrossRef CAS PubMed.
- M. Frei, S. V. Aradhya, M. S. Hybertsen and L. Venkataraman, J. Am. Chem. Soc., 2012, 134, 4003 CrossRef CAS PubMed.
- E. Leary, L. A. Zotti, D. Miguel, I. R. Márquez, L. Palomino-Ruiz, J. M. Cuerva, G. Rubio-Bollinger, M. T. González and N. Agraït, J. Phys. Chem. C, 2018, 122, 3211 CrossRef CAS.
- L. Venkataraman, J. E. Klare, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nature, 2006, 442, 904 CrossRef CAS PubMed.
- A. Mishchenko, D. Vonlanthen, V. Meded, M. Bürkle, C. Li, I. V. Pobelov, A. Bagrets, J. K. Viljas, F. Pauly, F. Evers, M. Mayor and T. Wandlowski, Nano Lett., 2010, 10, 156 CrossRef CAS PubMed.
- D. Miguel, L. Álvarez de Cienfuegos, A. Martín-Lasanta, S. P. Morcillo, L. A. Zotti, E. Leary, M. Bürkle, Y. Asai, R. Jurado, D. J. Cárdenas, G. Rubio-Bollinger, N. Agraït, J. M. Cuerva and M. T. González, J. Am. Chem. Soc., 2015, 137, 13818 CrossRef CAS PubMed.
- G–V curves of the dibenzopentalene were reported in ref. 21 and replotted in the ESI‡ to compare with compound 4g.
Footnotes |
| † Dedicated to Professor Joan Bosch on occasion of his retirement. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and characterisation of new compounds, NMR spectra, high-resolution mass spectra, single-crystal X-Ray diffraction details, UV-Vis spectra, voltammograms, electron spin resonance spectrum, computational details, STM-BJ experiments and analysis and cartesian coordinates. CCDC 2177496. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc02475f |
| This journal is © The Royal Society of Chemistry 2022 |