
Real-time views of morphological evolution in solution-processed organic photovoltaics
 *a
*a
Abstract
The nanoscale morphology of the photoactive layer in solution-processed organic photovoltaics plays a critical role in device performance. Such an intricate morphology is sensitive to the processing conditions during film formation. From such a perspective, in situ characterizations stand out as they are able to provide insights for screening the film's transition from the solution state to the solid state. In this review, we summarize the state-of-the-art in situ characterization methods that are specifically designed to observe the drying kinetics of photoactive layers and discuss how they deepen our understanding of morphological evolution under different processing conditions. In particular, key factors such as the evolution of fine structure and interplay between solutes and solvents upon film formation could be revealed. We therefore use them as indicators to guide the future optimization of film morphology. We believe that a thorough understanding of morphological evolution, such as kinetics of crystallization and phase separation, through the above methodologies, can further push the efficiency of devices.
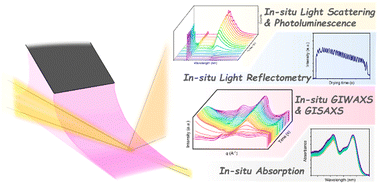
- This article is part of the themed collection: Journal of Materials Chemistry C Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

