
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Timing performance of lead halide perovskite nanoscintillators embedded in a polystyrene matrix†
Kateřina
Děcká
 ab,
Fiammetta
Pagano
*cd,
Isabel
Frank
ce,
Nicolaus
Kratochwil
c,
Eva
Mihóková
ab,
Etiennette
Auffray
c and
Václav
Čuba
a
ab,
Fiammetta
Pagano
*cd,
Isabel
Frank
ce,
Nicolaus
Kratochwil
c,
Eva
Mihóková
ab,
Etiennette
Auffray
c and
Václav
Čuba
a
aFaculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague, Břehová 7, 115 19 Prague, Czech Republic
bInstitute of Physics of the Czech Academy of Sciences, Cukrovarnická 10, 162 53 Prague, Czech Republic
cCERN, Esplanade des Particules 1, 1211 Meyrin, Switzerland. E-mail: fiammetta.pagano@cern.ch
dUniversità degli Study Milano Bicocca, Piazza dell’Ateneo Nuovo, 1-20126 Milano, Italy
eLMU Munich, Geschwister-Scholl-Platz 1, 80539 Munich, Germany
First published on 22nd August 2022
Abstract
Nanomaterials like CsPbBr3, benefiting from quantum confinement effects to feature ultra-fast decay time and tunable emission, are paving the way for the next generation of fast timing detectors. However, an ongoing challenge is to exploit their favorable properties in a full detector, given their size and instability. Embedding halide perovskite nanocrystals in solid matrices like organic polymers can provide the required stability and, in the case of high nanoparticle filling factors with little aggregation, results in a flexible scintillator, featuring sub-ns decay times. In this work, we present the production, characterization, and – for the first time – time resolution measurements of CsPbBr3 nanocrystals embedded in polystyrene, using two different surface ligands (OA + OAm and DDAB) and three different filling factors of up to 10%. The samples were characterized by spectroscopic methods, namely photo- and radio-luminescence as well as transmittance, while scintillation decay kinetics was measured in a time correlated single photon counting setup upon X-ray excitation. The characterization results suggest that, for both ligands, a 10% filling factor with little to no aggregation can be obtained. In addition, the time resolution of these materials was measured using a novel setup coupled to analog silicon photomultipliers and low energy pulsed X-ray excitation. When comparing with the state of the art inorganic (LYSO:Ce) crystal, more than twofold time resolution improvement was obtained, despite the lower light transport and small energy deposition. These first promising results represent the starting point for the optimization of CsPbBr3 nanocrystals embedded in polymer matrices and their application in fast timing detectors for TOF-CT, TOF-PET and high energy physics.
1 Introduction
Lead halide perovskite nanocrystalline phosphors with the formula CsPbX3 (X = Cl, Br, I) have received significant attention from the scientific community in the last few years, mostly due to their high light output, fast decay times, and narrow tunable emission bands with wide application potential in optoelectronics.1–4 Recently, the attention shifted toward their implementation as scintillation detectors as well,5–10 specifically for applications in fast timing detectors. Indeed, the capability of ultra-fast detection of X-rays, 511 keV gamma-rays, and high energy particles has triggered interest in high energy physics11,12 and medical imaging applications, such as time-of-flight positron emission tomography (TOF-PET)13,14 and time-of-flight computed tomography (TOF-CT).15 Among many concepts to boost the production of ultra-fast photons – such as Cherenkov emission,16 hot intraband luminescence,17 or cross-luminescence18 – quantum confinement effects in nanocrystals result in a unique combination of fast emission with relatively high light output.10,19,20 As a rough estimate, the timing capability of a material scales with the square root of the ratio of effective decay time and light output,21 justifying the increasing interest in nanocrystalline materials as ultra-fast radiation detectors.Despite their excellent fast emission, several drawbacks have to be addressed for halide perovskite nanocrystals to be effectively used as radiation detectors. Among them, limited stability against air moisture and oxygen22 and nanometer size, with consequent low energy deposition upon particle interaction, are the main drawbacks. Both issues can be partially overcome by the immobilization of nanocrystals in solid matrices such as glass or organic polymers or in other composites such as silica/alumina monoliths.23 The first two techniques are already well established: glass matrices guarantee better radiation hardness,24,25 while polymer matrices are much more cost-effective. Furthermore, the applicability of CsPbBr3 embedded in polystyrene has already been demonstrated, with encouraging results about radiation hardness as well.6 The main drawback of polymer matrices for ionizing radiation detectors is the low stopping power, but the incorporation of dense nanocrystals, such as CsPbBr3, will automatically enhance it. Moreover, concerning TOF-PET applications, encouraging results on heterostructured scintillators made of alternating layers of a dense inorganic material (e.g. BGO whose density is 7 g cm−3) and fast organic polymers (e.g. BC422 and EJ232 whose decay time is about 1.5 ns) have been published in the last few years.26–28 Lead halide perovskites embedded in polystyrene constitute an alternative candidate as a fast performing material for heterostructures with potentially better time resolution than BC422 or EJ232 as a result of faster scintillation decay kinetics and higher light yield (about 24 ph keV−129 instead of 8.4 ph keV−130).
This study is the first step toward the characterization and development of lead halide perovskite-based scintillators suitable for radiation detectors. For the first time, we provide time resolution measurements of nanocrystal-based samples upon pulsed X-ray irradiation. While the scintillation decay kinetics of these samples was already accessible using laser or X-ray irradiation, the time resolution measurements investigate a different physical quantity; they encompasses not only the simple scintillation decay kinetics, but also the scintillation light yield.21,31 The coincidence time resolution (CTR) is already an established quantity usually measured using higher energy sources, like 22Na emitting 511 keV γ-rays. Due to low detection efficiency, the standard characterization procedure is not possible for these samples, therefore we used a novel experimental setup32,33 to measure the time resolution of scintillators upon low energy (about 10 keV) X-ray irradiation.
In this study, we explore CsPbBr3 nanocrystals capped with different surface ligands embedded in a polystyrene matrix with various weight filling factors (up to 10%). The chosen ligands were the standard combination of oleic acid and oleylamine (OA + OAm) and DDAB (didodecyldimethylammonium bromide), which was reported to exhibit better surface passivation capability compared to OA + OAm, resulting in higher light yields of CsPbBr3 nanocrystals.34,35 We characterize them focusing on their potential for applications in fast timing detectors. Therefore, we provide time resolution measurements upon X-ray excitation and discuss integration aspects for applicability as radiation detectors.
2 Experimental section
2.1 CsPbBr3@PS composite fabrication
CsPbBr3 nanocrystals were synthesized using the standard hot injection procedure.1 DDAB capped nanocrystals were prepared by a ligand exchange method. The size of nanocrystals used for nanocomposite fabrication was determined to be 10 ± 1 nm. For more information, please refer to the ESI.†For the polystyrene (PS) nanocomposite samples, about 210 mg of PS pellets were dissolved in toluene in a Petri dish of 5 cm in diameter. Then, the calculated amount of the CsPbBr3 solution was pipetted to achieve the desired final concentration in PS, namely 1%, 5%, and 10%. For example, for 1% filling factor, 40 μL of 52.5 mg mL−1 CsPbBr3 solution was pipetted. The viscous solution was thoroughly mixed until homogenized completely. Finally, the toluene was left to evaporate in air at room temperature. After one week, the already solid samples were taken out of the Petri dishes and the residual toluene was left to evaporate in air at room temperature for another week.
Before characterization, all samples were cleaned with a cellulose swab soaked in hexane, finally resulting in 5 cm diameter discs with a thickness of 100 μm, as measured by a caliper with 20 μm resolution.
Ultimately, two sets of samples were obtained, each with three different filling factors – 1%, 5%, and 10% – giving a total of six samples. In the first set, CsPbBr3 nanocrystals were capped with oleic acid and oleylamine (OA + OAm), while in the second one, the nanoparticles were capped with DDAB. All samples are summarized and shown in Table 1. Note that the transparency is good even for 10% filling.

|
2.2 Characterization
Before embedding CsPbBr3 nanocrystals in the polystyrene matrix, X-ray powder diffraction, PL, and RL spectra were obtained. After the embedding, RL and transmission spectra, as well as scintillation decay kinetics and time resolution under X-ray excitation were recorded for all samples.X-ray powder diffraction (XRPD) was measured using a Rigaku Miniflex 600 diffractometer equipped with a Cu X-ray tube (Kα1,2 average wavelength 0.15418 nm, voltage 40 kV, current 15 mA). Data were collected with a speed of 2° min−1 and compared with the ICDD PDF-2 database, version 2013. The Halder–Wagner method with the Scherrer constant value of 0.94 was used to determine the linear crystallite size.
Transmission spectra, PL excitation, and emission spectra were obtained using a FluoroMax spectrofluorometer (Horiba Jobin Yvon). RL spectra were obtained using a 5000M spectrofluorometer (Horiba Jobin Yvon) with a monochromator, a TBX-04 (IBH Scotland) photodetector, and a Seifert X-ray tube (40 kV, 15 mA) as an excitation source.
The decay time was measured in time correlated single photon counting (TCSPC) mode.36 The samples were excited using a Hamamatsu pulsed X-ray tube, and the emission light was collected using a Hybrid Photomultiplier Tube (HPMT). The overall instrumental response function (IRF), which takes into account the HPMT together with the laser, used to excite the X-ray tube, and the tube itself was estimated to be 160 ps full-width-at-half-maximum (FWHM). To select only the perovskite emission, a bandpass filter centered at 530 nm with 40 nm FWHM was used.
The scintillation decays were fitted with the convolution of the system IRF and the intrinsic scintillation decay function. Because a (semi-)prompt component was observed for all samples, we decided to model the intrinsic scintillation function as the sum of three exponential functions and a Dirac-delta function, in analogy to what was done in ref. 37 to model Cherenkov photons. Before opting for this model, others were tested using only exponential components (between two and five). However, these did not properly account for the ultra-fast decay component, which is of utmost importance for fast timing radiation detectors. We finally chose the model that allowed us to fit all the samples well enough, particularly in the fastest part of the decay, while maintaining the lowest number of components. In the fit procedure, the rise-time was fixed at 0 ps since it was well below the time resolution of the system. This allowed better stability of the fit for the decay part.
The detector time resolution (DTR) – also known as single time resolution (STR), as opposed to coincidence time resolution (CTR) which takes into account a pair of detectors – was measured using a novel experimental setup with low energy X-ray irradiation developed purposely for the characterization of low-stopping power scintillators.32,33 The setup includes a Hamamatsu pulsed X-ray tube, excited by a picosecond diode laser whose external trigger is used as the start signal for the DTR measurement. The stop signal instead is given by the output of the SiPM and is used for collecting the scintillation light emitted by the sample following the X-ray excitation. The DTR was evaluated as the FWHM of the time delay (time difference between start and stop) peak.
For these measurements, 3 × 3 mm2 plates were cut from the 5 cm discs to match the active area of the SiPM (3 × 3 mm2, S13360-3050CS from Hamamatsu), operated about 10 V above the breakdown voltage. The optical coupling was done with Meltmount glue (refractive index n = 1.58), and no reflective material was used to wrap the samples to avoid X-ray absorption from this material.
3 Results
3.1 Transmission
CsPbBr3 samples capped with DDAB have generally better transparency, as visible in the photographs in Table 1 and then confirmed by transmission spectra shown in Fig. 1 (OA + OAm (a) DDAB (c)). CsPbBr3@PS (1%) capped with OA and OAm have 50% transmittance at 800 nm, while CsPbBr3@PS (1%) capped with DDAB has 56% transmittance. The difference is even more pronounced for higher filling factors, as can be observed in Fig. 1(a) and (c).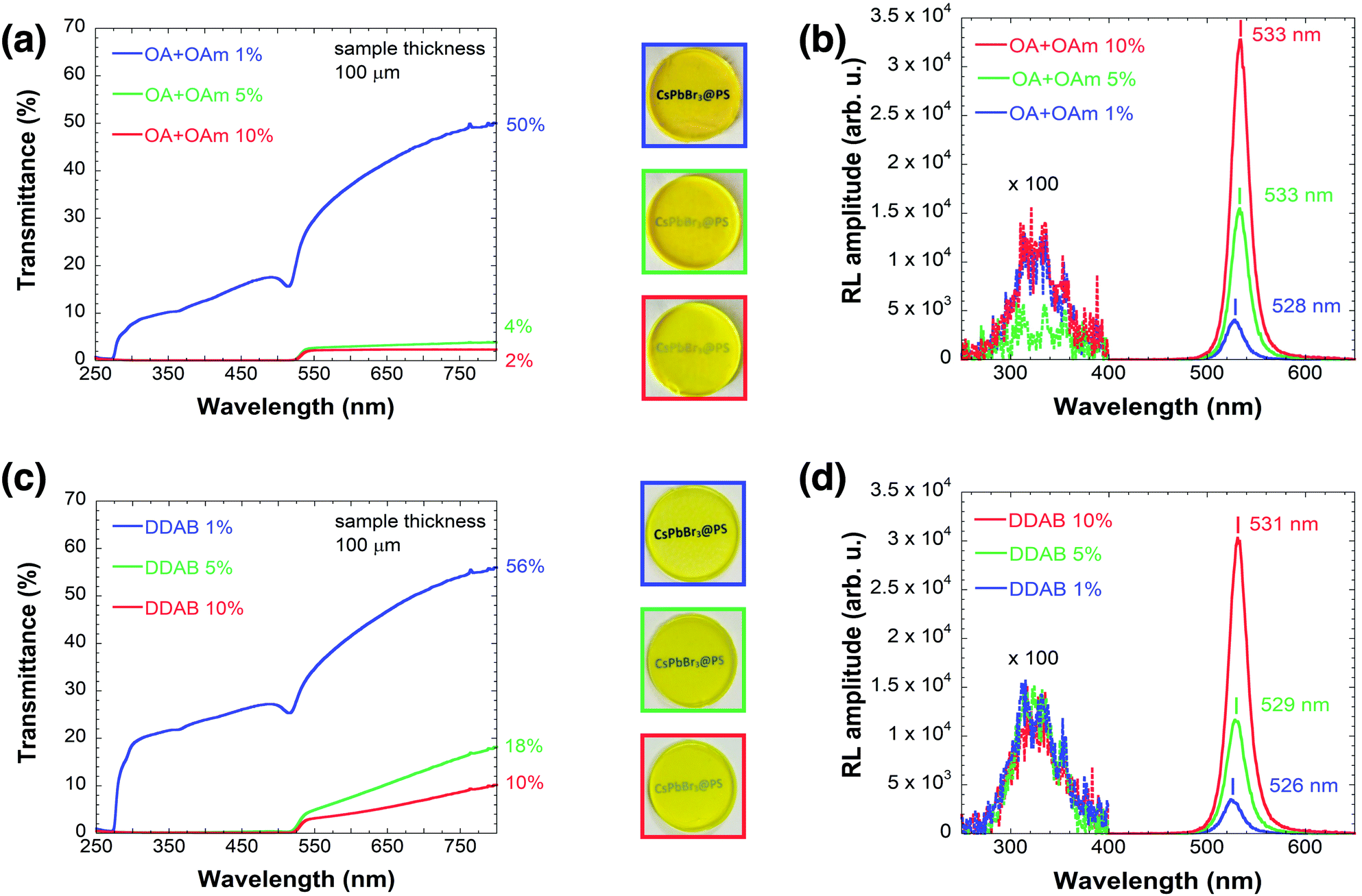 | ||
| Fig. 1 (a) Transmission spectra and (b) RL spectra of samples from the OA + OAm set, (c) transmission spectra and (d) RL spectra of samples from the DDAB set, with various filling factors. Blue, green and red lines represent 1%, 5% and 10% filling factors, respectively. The RL spectra were multiplied by the factor of 100 in the interval 250 –400 nm to reveal the weak polystyrene emission. | ||
From Fig. 1(a) and (c), it is also clear that samples with 1% filling factor have good transparency even at lower wavelengths than the absorption edge of CsPbBr3 (around 510–530 nm), unlike those with higher filling factors.
3.2 Radioluminescence
Fig. 1(b) and (d) show the RL spectra of the two sets of samples. The RL intensity – calculated by integrating the spectrum between 470 and 630 nm – of the OA + OAm 10% sample is 2× higher compared to the OA + OAm 5% sample, while the intensity of the DDAB 10% sample is 2.5× higher compared to the corresponding 5% sample.The positions of RL emission bands were obtained by fitting the curve around its maximum with a quadratic function to reduce the effect of noise. The errors were estimated by taking into account both the precision of the chosen fitting procedure and the systematic error of the setup. For more details and examples of such curves, see Fig. S3 in the ESI.†
The positions of RL emission bands of samples with higher filling factors are increasingly red-shifted from 528 ± 1 nm for 1% filling factor up to 533 ± 0.5 nm for the 10% filling factor using OA + OAm ligands and from 526 ± 1 nm (1% filling factor) to 531 ± 0.5 nm (10% filling factor) using the DDAB ligand. This red-shift is even more pronounced in the PL emission, as shown in Fig. S4 in the ESI.†
All samples have very weak emission centered at 325 nm, which originates from the polystyrene itself, as shown by the 100× magnification in Fig. 1(b) and (d).
The samples were stored under ambient conditions, and the RL spectra were recorded three times over the course of one month. No significant changes in the RL intensity were observed (see Fig. S5 in the ESI†).
3.3 Decay time under X-ray excitation
Fig. 2 shows an example of a measured scintillation decay together with its fit. The fitted scintillation decays of all six samples can be found in the ESI† (see Fig. S6 and S7). The results are summarized in Table 2. For all samples, we observe an ultra-fast decay time component (modeled with a Dirac-delta function) of about 20% weight and another sub-ns decay time component, also with around 20% contribution. Those ultra-fast decay time components are of utmost importance to boost the timing capability of the material. For samples belonging to the OA + OAm set, it can be observed that the slowest decay component (τd3) and its corresponding weight (R3) progressively increase together with the filling factor, at the expense of the fastest exponential decay component (τd1). In contrast, for the DDAB samples, no clear trend is observed.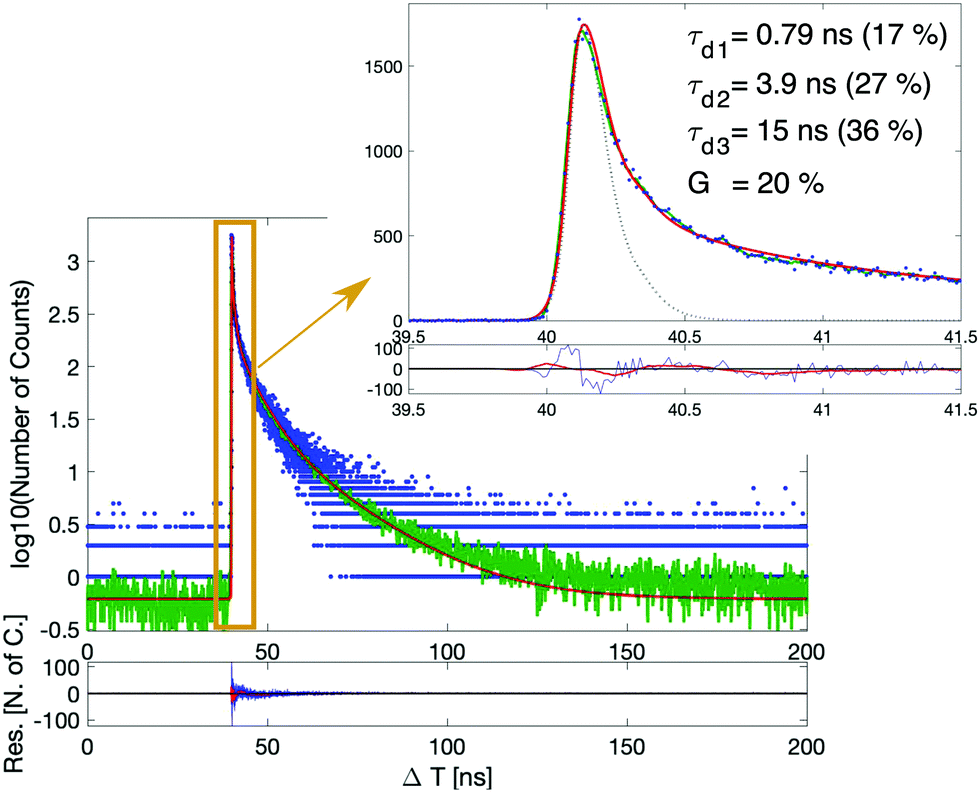 | ||
| Fig. 2 Scintillation decay of the CsPbBr3 10% sample capped with DDAB. The scintillation decay is shown in the semi-logarithmic scale over the whole range (≈150 ns). The blue dots are the measured data points, the green line is their average, and the red curve is the fit function. Inset: Details of the ultra-fast component in the linear scale. The dotted gray line represents the system IRF. | ||
The effective decay time (τd,eff), defined as the weighted harmonic mean of the exponential components, was chosen as the figure of merit to summarize the three exponential decay components in only one parameter, simplifying the comparison between the samples.21 The obtained values are reported in the last column of Table 2 and confirm what was previously observed. For this calculation, re-normalization was applied to omit the almost prompt contribution for better comparison. For a final evaluation, both parameters τd,eff and G, the weight of the prompt component, need to be considered simultaneously.
| Sample | τ d1 [ns] | R 1 [%] | τ d2 [ns] | R 2 [%] | τ d3 [ns] | R 3 [%] | G [%] | τ d,eff [ns] | |
|---|---|---|---|---|---|---|---|---|---|
a The fit function was normalized so that the weights of the four components add up to one (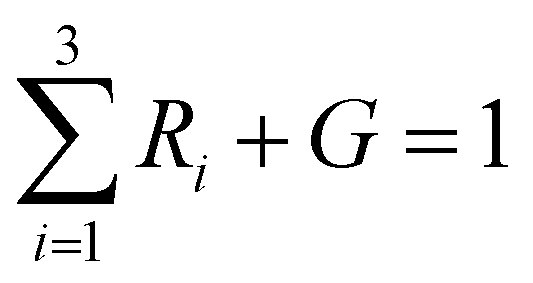 ), but the effective decay time was calculated with the re-normalized ratio: ), but the effective decay time was calculated with the re-normalized ratio: 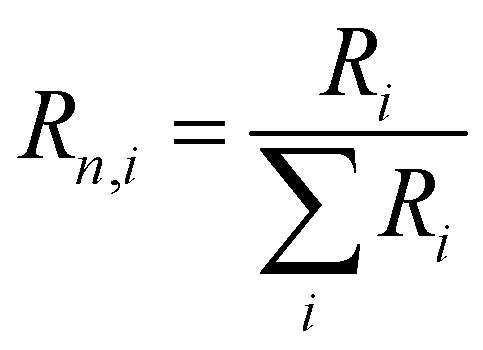 and and 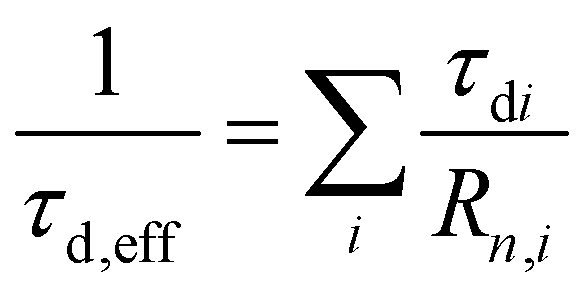 . .
|
|||||||||
| OA + OAm | 1% | 0.76 ± 0.02 | 24 ± 4 | 3.0 ± 0.3 | 27 ± 3 | 11 ± 1 | 31 ± 7 | 18 ± 2 | 1.9 ± 0.2 |
| 5% | 0.68 ± 0.02 | 18 ± 3 | 3.4 ± 0.3 | 30 ± 4 | 18 ± 2 | 28 ± 5 | 24 ± 3 | 2.0 ± 0.2 | |
| 10% | 0.69 ± 0.02 | 14 ± 2 | 4.1 ± 0.4 | 28 ± 4 | 26 ± 3 | 42 ± 7 | 16 ± 2 | 2.9 ± 0.3 | |
| DDAB | 1% | 0.92 ± 0.03 | 18 ± 3 | 3.9 ± 0.4 | 29 ± 4 | 21 ± 3 | 37 ± 7 | 16 ± 2 | 2.9 ± 0.3 |
| 5% | 0.79 ± 0.02 | 17 ± 3 | 3.5 ± 0.3 | 35 ± 4 | 18 ± 2 | 28 ± 5 | 20 ± 2 | 2.4 ± 0.2 | |
| 10% | 0.79 ± 0.02 | 17 ± 3 | 3.9 ± 0.4 | 27 ± 3 | 15 ± 2 | 36 ± 6 | 20 ± 2 | 2.6 ± 0.2 | |
3.4 Time resolution under X-ray excitation
Time resolution results are presented in Table 3. One can observe that overall, the samples capped with DDAB showed slightly better timing capability. The time delay distribution of all samples can be observed in the ESI† (see Fig. S8).| Filling factor | DTR (FWHM) [ps] | |
|---|---|---|
| OA + OAm | DDAB | |
| 1% | 305 ± 9 | 308 ± 9 |
| 5% | 330 ± 10 | 309 ± 9 |
| 10% | 319 ± 9 | 295 ± 8 |
| LYSO:Ce | 695 ± 21 | |
| EJ232 | 332 ± 10 | |
To give a better idea about the timing performance of this material, a comparison with two well-known scintillators – LYSO:Ce and EJ232 plastic scintillators – with similar geometry (3 × 3 × 0.2 mm3) is shown as well in Table 3. LYSO:Ce was chosen as the state-of-the-art inorganic scintillator for TOF-PET: it has high density and high effective atomic number (7.1 g cm−3 and 66, respectively), hence good stopping power and photofraction, the intrinsic light yield of about 40 ph keV−1 and the effective decay time of about 40 ns.21 On the other hand, EJ232 was chosen as an ultra-fast plastic scintillator: it shows similar properties to the BC422 plastic scintillator (effective decay-time of ≈1.7 ns, rise time of ≈35 ps, intrinsic light yield of about 10 ph keV−121) but it can be produced in very thin layers with a better surface state. For the aforementioned reasons, EJ232 is one of the favorite materials in preliminary studies27,28 about heterostructures.
The respective time delay distribution is shown in Fig. 3 in comparison to the best performing CsPbBr3 sample (10% filling factor, DDAB set). All CsPbBr3 samples perform twofold better than LYSO:Ce and similarly or slightly better than EJ232. This is explained by the scintillation decay kinetics of the considered samples (1.5 ns effective decay time for CsPbBr3 samples and EJ232 instead of 40 ns for LYSO) and by the low irradiation energy.
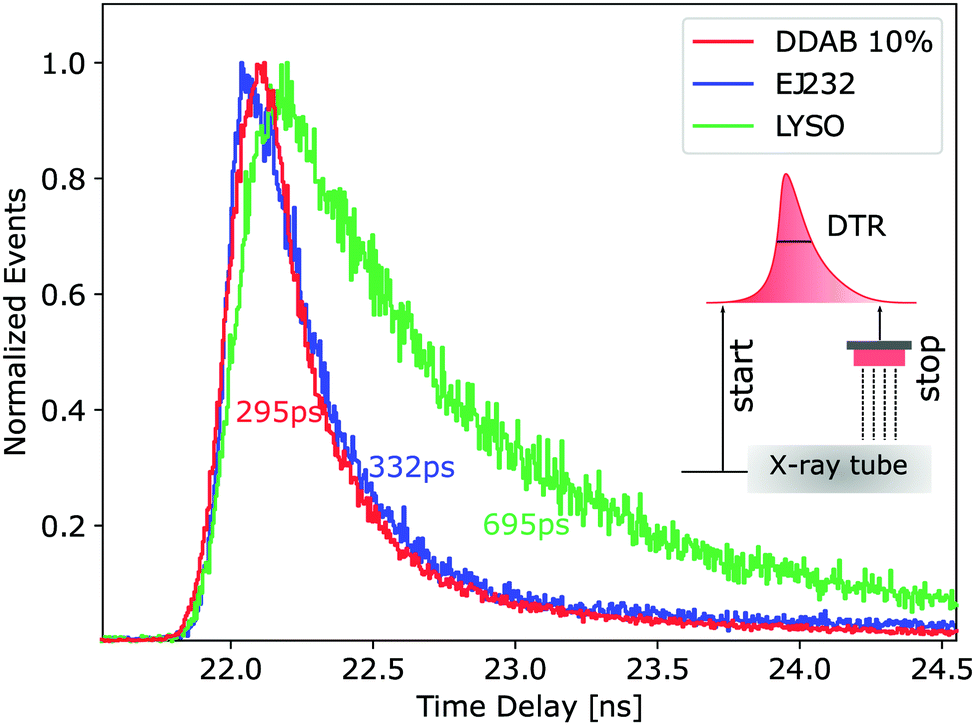 | ||
| Fig. 3 Comparison of the time delay distribution for the CsPbBr3 10% sample capped with DDAB (red) and standard scintillators EJ232 (blue) and LYSO (green). Inset: Sketch of the measurement condition of the pulsed X-ray tube exciting the tested sample and the evaluation of the DTR from the time difference of start and stop signals. | ||
4 Discussion
Two sets of CsPbBr3@PS samples with various nanocrystal filling factors were prepared. One set used CsPbBr3 nanocrystals capped with OA + OAm (OA + OAm set), and the second set used CsPbBr3 capped with DDAB after the ligand exchange procedure (DDAB set). First, free nanocrystals were characterized (XRPD, PL and RL spectra), then transparency, RL emission spectra, decay times, and time resolution of fabricated nanocomposites of CsPbBr3 in polystyrene of both sets were determined.4.1 Basic optical characterization
Generally, samples from the DDAB set exhibited superior transparency compared to those from the OA + OAm set. Together with the fact that a 10% filling factor was achieved only in this set (cf. the CsPbBr3 residue in the 10% OA + OAm sample Petri dish in Table 1), we also conclude that for the embedding, the DDAB ligand is a better choice than the standard OA + OAm combination. Because even 10% filling factor could not be achieved completely in the OA + OAm set, we did not explore higher filling factors in this study.The better transparency of the DDAB set is probably a result of its better surface passivation capability. By careful analysis of the scintillation decay components in Table 2, we concluded that it effectively suppresses aggregation, because no trend of gradually slower decay times was observed in this set. In contrast, in the OA + OAm set, we observed this trend in the third exponential decay component τd3. We reason that when larger crystals are formed by aggregation, the scintillation response is slowed down due to the quantum confinement effect.
However, because the transparency of the samples from the DDAB set still decreases slightly with the increasing filling factor, we expect that clustering occurs to some extent in this set. We distinguish the simple clustering – i.e. nanocrystals forming larger clusters but preserving their shape and size – from the real aggregation – i.e. nanocrystals forming bigger particles with their neighbors. Naturally, the aggregation can be understood as an extreme case of simple clustering (first, nanocrystals undergo the clustering process and then the aggregation may occur). As a result of this phenomenon, we observe a gradual speed-up of scintillation response in the first exponential decay component τd1. This is probably caused by increased self-absorption and subsequent luminescence quenching within those clusters (a similar trend is also observed in the OA + OAm set).
Both phenomena could have caused the observed red-shifts in the RL spectra; both the formation of larger nanocrystals by aggregation and the increased self-absorption within the clusters can result in this shift. As this article is mainly focused on the timing capability of the produced samples, a more in-depth discussion of all the above-mentioned phenomena and observed red-shifts in the RL spectra can be found in the ESI† (Section S3).
4.2 Timing performance
Timing performance of the prepared nanocomposite samples is the crucial characteristic for the target application. TCSPC measurements revealed the ultra-fast scintillation decay kinetics of these samples: in all of them, we observed a (semi-)prompt component, which has been modeled with a Dirac-delta function, and a second sub-nanosecond component (between 700 and 900 ps). Summing up the contributions, almost 50% of light is emitted within the first nanosecond. This contributes to an increase in the initial photon time density, which, taking into account both scintillation decay kinetics and intrinsic light yield, is the main contribution to time resolution.21 The presence of the ultrafast components can be explained by luminescence quenching or the formation of biexcitons.38–40The ultra-fast decay kinetics of these samples explains the two-fold better time resolution of CsPbBr3@PS compared to LYSO at 10 keV. This is a significant result because LYSO is a scintillator that is currently used in commercial TOF-PET scanners. Our nanocomposite is therefore a viable candidate for applications in fast timing detectors.
Because LYSO is a high density crystal, we also compared the timing performance of CsPbBr3@PS with that of the EJ232 plastic scintillator, which is one of the favorite options as a fast material for heterostructured scintillators which we aim to substitute with CsPbBr3@PS. CsPbBr3@PS proved to have even faster decay kinetics than EJ232 (similar effective decay time but with a more significant contribution from the prompt component, see Table 2), nevertheless, at this moment, the time resolution improvement with respect to EJ232 is not significant. This is probably due to less efficient energy conversion as a result of still too low filling factor. Despite the similar density and the atomic number of the two samples, the polymer matrix of CsPbBr3@PS does not contribute to the overall scintillation intensity significantly. Therefore, scintillating photons are produced almost exclusively when X-rays interact directly with nanocrystals, and with filling factors between 1 and 10%, the probability for this happening is quite low. Such a not-optimized energy conversion further results in a lower number of produced photons and typically the timing capability scales with the inverse square root of the light yield.
Comparing the time resolution of all of the synthesized samples, no significant difference was observed with the increasing filling factor, for either of the two sets. These results can give some insight into the effective light output of these samples with the increasing filling factor. From the optical measurements, we saw that samples with higher filling factors show not only higher PL and RL but also significantly lower transmittance. The absence of a clear trend in DTR with the filling factor suggests that these two effects balance each other. This also suggests that an optimum could exist, and it will be investigated in future studies with a denser scan of filling factors. Moreover, future experiments will be also dedicated to achieving higher filling factors while maintaining as good a transparency of the nanocomposites as possible.
Comparing the time resolution of the two sets, samples from the DDAB set performed slightly better, in agreement with previously discussed results on the optical properties of these samples.
4.3 Applications and outlook
From an application point of view, even a time resolution in the order of 300 ps will reduce the amount of scattered photons for TOF-CT. While in this study a pulsed X-ray tube with a median energy of 10 keV was used, for X-ray and CT examinations, typically higher energies are used (20–120 keV). To put the measured time resolution into perspective, already at 90 keV sub-100 ps (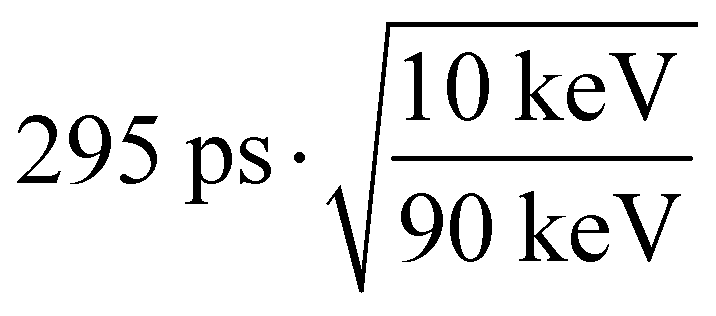 ), time resolution can be expected. Such a value allows most of the scattered photons for TOF-CT to be removed and thus results in better image quality32 or a lower required dose for the same examination.
), time resolution can be expected. Such a value allows most of the scattered photons for TOF-CT to be removed and thus results in better image quality32 or a lower required dose for the same examination.
A corresponding calculation can be done for TOF-PET working with 511 keV gamma-rays. Selecting at the 340 keV Compton edge, a coincidence time resolution of 35 ps FWHM is reported with a bulk plastic scintillator which is similar to the used EJ232.41 Such simple approximations highlight the promising R&D avenue of lead halide perovskite nanocrystals, in line with the ongoing 10 ps challenge for TOF-PET.14
All arguments above must be considered in light of the fact that in this manuscript, we have only presented preliminary measurements for TOF-CT and TOF-PET with certainly not optimized samples. There are several paths to improve the performance and applicability. For instance, by a careful choice of the surface ligands and the embedding procedure, 60% filling factor for Cd(Zn)S/ZnS core–shell nanocrystals was reported while keeping the nanocomposite monolith transparent.42 Moreover, the introduction of a wavelength-shifter to the matrix can be used to suppress the self-absorption, as successfully shown in the just mentioned study. Increasing the filling factor while enhancing the transparency automatically leads to a higher number of detected photons and thus an improvement in the timing capabilities. Another important question, in particular for TOF-PET and high energy physics detectors, is how to ensure high stopping power and sufficient energy deposition in the nanomaterials. One approach is to integrate it into a composite structure27 with heavy inorganic scintillators like BGO43 or GAGG:Ce,Mg.41,44
5 Summary and conclusion
In this work, we presented the fabrication and complete characterization of CsPbBr3 nanocrystals with two different surface ligands (OA + OAm and DDAB) embedded in polystyrene with various particle filling factors, up to 10%. The characterization spanned from optical properties (transmission, PL, RL) to timing properties under X-ray irradiation (scintillation decay kinetics and time resolution).The optical characterization highlighted a drop in transmission with an increasing filling factor. This effect was less pronounced in CsPbBr3 samples capped with DDAB, which, compared to OA + OAm, is known to better passivate the surface of nanocrystals.
Decay time measurements in TCSPC revealed the presence of a (semi-)prompt component – modeled with a Dirac-delta function – contributing by 20% and three exponential decay components. The scintillation decay kinetics of CsPbBr3 samples capped with DDAB were not affected by the increasing concentration of nanoparticles, while for samples capped with OA + OAm, an increase in the value of the slowest component itself and its weight was observed, at the expense of the fastest exponential component. Consequently, we can conclude that the DDAB ligand better prevents nanocrystals from aggregating.
The presented time resolution measurements confirm the applicability of this material to be used in radiation detector systems where fast timing is required. Already with non-optimized CsPbBr3@PS, a more than twofold better timing capability with respect to LYSO:Ce was achieved at low X-ray energy.
This study represents a promising starting point for the optimization of CsPbBr3 nanocrystals embedded in polymer matrices toward their use in time-based radiation detector systems.
Author contributions
Kateřina Děcká: conceptualization, formal analysis, investigation, validation, visualization, methodology, writing – original draft, writing – review & editing. Fiammetta Pagano: conceptualization, formal analysis, investigation, validation, visualization, methodology, software, writing – original draft, writing – review & editing. Isabel Frank: formal analysis, investigation, validation, visualization, writing – review & editing. Nicolaus Kratochwil: methodology, supervision, writing – original draft, writing – review & editing. Eva Mihóková: funding, resources, project administration, methodology, supervision, writing – review & editing. Etiennette Auffray: conceptualization, funding, resources, project administration, supervision, writing – review & editing. Václav Čuba: conceptualization, funding, resources, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was carried out in the frame of Crystal Clear Collaboration. This research was funded by the Czech Science Foundation, grant number GA20-06374S, Operational Programme Research, Development and Education financed by European Structural and Investment Funds and the Czech Ministry of Education, Youth and Sports (Project No. SOLID21 CZ.02.1.01/0.0/0.0/16_019/0000760) and by the Grant Agency of the Czech Technical University in Prague, grant number SGS20/185/OHK4/3T/14. Further support was provided from the CERN Budget for Knowledge Transfer to Medical Applications and from the European Union's Horizon 2020 Research and Innovation programme under Grant Agreement No 101004761 (AIDAinnova). The authors express their gratitude to Jan Král for introducing the ligand exchange procedure.Notes and references
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- Y. Wang, T. Zhang, M. Kan and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 12345–12348 CrossRef CAS PubMed.
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem., Int. Ed., 2015, 127, 15644–15648 CrossRef.
- Y. Zhang, R. Sun, X. Ou, K. Fu, Q. Chen, Y. Ding, L.-J. Xu, L. Liu, Y. Han and A. V. Malko, et al. , ACS Nano, 2019, 13, 2520–2525 CrossRef CAS PubMed.
- M. Gandini, I. Villa, M. Beretta, C. Gotti, M. Imran, F. Carulli, E. Fantuzzi, M. Sassi, M. Zaffalon and C. Brofferio, et al. , Nat. Nanotechnol., 2020, 15, 462–468 CrossRef CAS PubMed.
- V. Mykhaylyk, H. Kraus, V. Kapustianyk, H. Kim, P. Mercere, M. Rudko, P. Da Silva, O. Antonyak and M. Dendebera, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- O. D. Moseley, T. A. Doherty, R. Parmee, M. Anaya and S. D. Stranks, J. Mater. Chem. C, 2021, 9, 11588–11604 RSC.
- H. Yang, H. Li, R. Yuan, J. Chen, J. Zhao, S. Wang, Y. Liu, Q. Li and Z. Zhang, J. Mater. Chem. C, 2021, 9, 7905–7909 RSC.
- K. Tomanová, V. Čuba, M. G. Brik, E. Mihóková, R. Martinez Turtos, P. Lecoq, E. Auffray and M. Nikl, APL Mater., 2019, 7, 011104 CrossRef.
- A. Benaglia, S. Gundacker, P. Lecoq, M. Lucchini, A. Para, K. Pauwels and E. Auffray, Nucl. Instrum. Methods Phys. Res., Sect. A, 2016, 830, 30–35 CrossRef CAS.
- M. T. Lucchini, W. Chung, S. C. Eno, Y. Lai, L. Lucchini, M. Nguyen and C. G. Tully, J. Instrum., 2020, 15, P11005 CrossRef CAS.
- D. R. Schaart, G. Schramm, J. Nuyts and S. Surti, IEEE Trans. Radiat. Plasma Med. Sci., 2021, 5, 598–618 Search PubMed.
- P. Lecoq, C. Morel, J. O. Prior, D. Visvikis, S. Gundacker, E. Auffray, P. Križan, R. M. Turtos, D. Thers and E. Charbon, et al. , Phys. Med. Biol., 2020, 65, 21RM01 CrossRef CAS PubMed.
- J. Rossignol, R. M. Turtos, S. Gundacker, D. Gaudreault, E. Auffray, P. Lecoq, Y. Bérubé-Lauzière and R. Fontaine, Phys. Med. Biol., 2020, 65, 085013 CrossRef CAS PubMed.
- N. Kratochwil, S. Gundacker and E. Auffray, Phys. Med. Biol., 2021, 66, 195001 CrossRef CAS PubMed.
- S. I. Omelkov, V. Nagirnyi, S. Gundacker, D. A. Spassky, E. Auffray, P. Lecoq and M. Kirm, J. Lumin., 2018, 198, 260–271 CrossRef CAS.
- S. Gundacker, R. Pots, A. Nepomnyashchikh, E. Radzhabov, R. Shendrik, S. Omelkov, M. Kirm, F. Acerbi, M. Capasso and G. Paternoster, et al. , Phys. Med. Biol., 2021, 66, 114002 CrossRef PubMed.
- K. Tomanová, A. Suchá, E. Mihóková, L. Procházková, I. Jakubec, R. M. Turtos, S. Gundacker, E. Auffray and V. Čuba, IEEE Trans. Nucl. Sci., 2020, 67, 933–938 Search PubMed.
- R. Turtos, S. Gundacker, S. Omelkov, E. Auffray and P. Lecoq, J. Lumin., 2019, 215, 116613 CrossRef CAS.
- S. Gundacker, R. M. Turtos, N. Kratochwil, R. H. Pots, M. Paganoni, P. Lecoq and E. Auffray, Phys. Med. Biol., 2020, 65, 025001 CrossRef CAS PubMed.
- Z. Zhu, Q. Sun, Z. Zhang, J. Dai, G. Xing, S. Li, X. Huang and W. Huang, J. Mater. Chem. C, 2018, 6, 10121–10137 RSC.
- Z. Li, L. Kong, S. Huang and L. Li, Angew. Chem., Int. Ed., 2017, 129, 8246–8250 CrossRef.
- C. Wang, H. Lin, Z. Zhang, Z. Qiu, H. Yang, Y. Cheng, J. Xu, X. Xiang, L. Zhang and Y. Wang, J. Eur. Ceram. Soc., 2020, 40, 2234–2238 CrossRef CAS.
- Y. Tong, Q. Wang, H. Yang, X. Liu, E. Mei, X. Liang, Z. Zhang and W. Xiang, Photonics Res., 2021, 9, 2369–2380 CrossRef.
- R. M. Turtos, S. Gundacker, E. Auffray and P. Lecoq, Phys. Med. Biol., 2019, 64, 185018 CrossRef CAS PubMed.
- F. Pagano, N. Kratochwil, M. Salomoni, M. Pizzichemi, M. Paganoni and E. Auffray, Phys. Med. Biol., 2022, 67, 135010 CrossRef PubMed.
- G. Konstantinou, P. Lecoq, J. M. Benlloch and A. J. Gonzalez, IEEE Trans. Radiat. Plasma Med. Sci., 2021, 6, 510–516 Search PubMed.
- F. Maddalena, A. Xie, X. Y. Chin, R. Begum, M. E. Witkowski, M. Makowski, B. Mahler, W. Drozdowski, S. V. Springham and R. S. Rawat, et al. , J. Phys. Chem. C, 2021, 125, 14082–14088 CrossRef CAS.
- Eljen technology EJ232 datasheet, https://eljentechnology.com/products/plastic-scintillators/ej-232-ej-232q, 2021.
- S. Vinogradov, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 912, 149–153 CrossRef CAS.
- F. Pagano, N. Kratochwil, M. Salomoni, I. Frank, S. Gundacker, M. Pizzichemi, M. Paganoni and E. Auffray, IEEE Nuclear Science Symposium and Medical Imaging Conferences (20th October), Yokohama, Japan (remote), 2021.
- F. Pagano, N. Kratochwil, M. Salomoni, I. Frank, S. Gundacker, M. Pizzichemi, M. Paganoni and E. Auffray, Front. Phys., 2022 Search PubMed , submitted.
- L. Zhang, W. Liang, L. Xu, M. Zhu, X. Wang, J. Su, L. Li, N. Liu, Z. Zhang and Y. Gao, Chem. Eng. J., 2021, 417, 129349 CrossRef CAS.
- M. Imran, P. Ijaz, L. Goldoni, D. Maggioni, U. Petralanda, M. Prato, G. Almeida, I. Infante and L. Manna, ACS Energy Lett., 2019, 4, 819–824 CrossRef CAS.
- L. Bollinger and G. E. Thomas, Rev. Sci. Instrum., 1961, 32, 1044–1050 CrossRef CAS.
- S. Gundacker, R. Turtos, E. Auffray and P. Lecoq, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 891, 42–52 CrossRef CAS.
- N. Yarita, H. Tahara, T. Ihara, T. Kawawaki, R. Sato, M. Saruyama, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2017, 8, 1413–1418 CrossRef CAS PubMed.
- M. N. Ashner, K. E. Shulenberger, F. Krieg, E. R. Powers, M. V. Kovalenko, M. G. Bawendi and W. A. Tisdale, ACS Energy Lett., 2019, 4, 2639–2645 CrossRef CAS.
- R. Turtos, S. Gundacker, S. Omelkov, B. Mahler, A. Khan, J. Saaring, Z. Meng, A. Vasilev, C. Dujardin and M. Kirm, et al. , npj 2D Mater. Appl., 2019, 3, 1–10 CrossRef CAS.
- K. Děcká, J. Král, F. Hájek, P. Průša, V. Babin, E. Mihóková and V. Čuba, Nanomaterials, 2021, 12, 14 CrossRef PubMed.
- C. Liu, Z. Li, T. J. Hajagos, D. Kishpaugh, D. Y. Chen and Q. Pei, ACS Nano, 2017, 11, 6422–6430 CrossRef CAS PubMed.
- N. Kratochwil, E. Auffray and S. Gundacker, IEEE Trans. Radiat. Plasma Med. Sci., 2020, 5, 619–629 Search PubMed.
- L. Martinazzoli, N. Kratochwil, S. Gundacker and E. Auffray, Nucl. Instrum. Methods Phys. Res., Sect. A, 2021, 1000, 165231 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc02060b |
| This journal is © The Royal Society of Chemistry 2022 |