
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From phosphorescence to delayed fluorescence in one step: tuning photophysical properties by quaternisation of an sp2-hybridised nitrogen atom†
Anastasia
Klimash
 a,
Antonio
Prlj
bc,
Dmitry S.
Yufit
b,
Abhijit
Mallick
b,
Basile F. E.
Curchod
bc,
Paul R.
McGonigal
b,
Peter J.
Skabara
*a and
Marc K.
Etherington
*de
a,
Antonio
Prlj
bc,
Dmitry S.
Yufit
b,
Abhijit
Mallick
b,
Basile F. E.
Curchod
bc,
Paul R.
McGonigal
b,
Peter J.
Skabara
*a and
Marc K.
Etherington
*de
aWestCHEM, School of Chemistry, University of Glasgow, University Ave, Glasgow, G12 8QQ, UK. E-mail: Peter.Skabara@glasgow.ac.uk
bDepartment of Chemistry, Durham University, South Road, DH1 3LE, UK
cSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
dDepartment of Physics, Durham University, South Road, DH1 3LE, UK
eDepartment of Mathematics, Physics and Electrical Engineering, Northumbria University, Ellison Place, Newcastle upon Tyne, NE1 8ST, UK. E-mail: marc.k.etherington@northumbria.ac.uk
First published on 25th May 2022
Abstract
Control of the delayed emission of organic compounds is an important factor in the development of new display technology and for the emerging use of organic emitters in sensing and fluorescence microscopy. In particular, there is a need to understand how the phenomena of room-temperature phosphorescence and thermally activated delayed fluorescence intersect. Here, we show that delayed fluorescence can be activated in room temperature phosphorescence emitters by quaternising the sp2-hybridised heterocyclic nitrogens. Furthermore by judicious positioning of a carbazole donor in the meta- or para-position with respect to the ring nitrogen atom, structural and sterical influences combine to tune the origins of the delayed fluorescence from triplet–triplet annihilation to thermally activated delayed fluorescence. Crucially, the quaternisation of nitrogen provides us with the means to fine-tune singlet and triplet states in a predictable manner, uncover the intersection between phosphorescence and delayed fluorescence and tip the balance in favour of delayed fluorescence.
Introduction
For a relatively young field of materials science, organic electronics has attracted significant interest from researchers across the globe and thousands of novel semiconducting organic materials have already been reported.1–3 For many of these compounds the preparation involves complex multi-step syntheses that are time-consuming and require tedious purification methods. These issues provide limitations to the scale-up of the materials and their use in commercial products. Finding pathways for the preparation of highly efficient compounds that use readily available starting materials and simple synthetic routes would avoid these issues and be beneficial for the field.It is particularly tempting to discover candidates for use in organic light-emitting diodes (OLEDs) and other optoelectronic and photonic applications among already existing fluorescent materials and then control their properties by simple chemical modifications. Recently it was shown that the quaternisation of one or both nitrogen atoms of quinine, a well-known alkaloid naturally occurring in cinchona trees, leads to significant changes in its emission profile.4
This initial work provides a platform to explore the quaternisation of a range of amines with tertiary nitrogen atoms in their aromatic units and highlights a stepwise method with which to alter the photophysical properties of a compound. As a result, numerous readily available natural compounds containing heteroaromatic rings could be modified by this simple procedure and can potentially demonstrate a drastic change in their photophysical properties. Moreover, it might be a promising strategy for inducing phenomena such as thermally activated delayed fluorescence (TADF).5–8 Initially TADF was observed in Eosin-Y (hence also being known as E-type delayed fluorescence),9 a material that, similar to quinine, has a protonation site. Protonation and quaternisation have been shown4,10 to have a significant impact on the behaviour of the charge-transfer (CT) states—states that are crucial to TADF. Quaternisation, therefore, provides an attractive means to explore the control and modification of delayed fluorescence in quinine and similar compounds such as quinoline.
Compounds containing heterocyclic aromatic moieties with a sp2-hybridised ring nitrogen are commonplace and include molecules such as quinoline, pyridine and their derivatives. Quinoline has already been exploited as an acceptor motif in a TADF emitter. A carbazole group was introduced at the 8-position of the quinoline, and naphthyl moieties are situated in 2- and 4-positions.11 Pyridine, in its turn, was only used in TADF materials as a fragment of a larger electron-accepting moiety combined with either benzoyl linkers or triazine.12,13 There is a significant wealth of literature on the photophysical behaviour of quaternised pyridine derivatives. For instance, 4,4′-bipyridinium salts, also called viologens, exhibit electrochromism.14,15 The properties of these materials can be easily tuned chemically and electrochemically, which makes them good candidates to use in organic field-effect transistors and for energy storage.16–18 Similarly, quinolines change their properties significantly when quaternised. For example, Hancock et al. reported a quinoline-based fluorescent emitter where the luminescence was modulated by protonation of its nitrogen atoms by camphorsulfonic acid.19 Work on materials containing N-alkylated quinoline has demonstrated that such compounds also show second-order nonlinear optical responses with good characteristics for terahertz wave generation.20 Quinolinium salts, therefore, provide an easily accessible starting point to uncover new optical properties and have even been shown to exhibit aggregation-induced emission (AIE) behaviour,21–23 opening a pathway to non-doped OLED architectures.24
Here we present a series of quaternised quinoline-based donor–acceptor compounds and reveal that such compounds provide access to delayed fluorescence. There has been recent interest in the switching between room temperature phosphorescence (RTP) and TADF and the interplay between these phenomena.25–28 The alkylation step tunes the energy levels in such a fashion to induce either TADF or triplet–triplet annihilation (TTA) from two room temperature phosphorescence (RTP) materials. Additionally, the quinolinium-based materials presented in this work demonstrate AIE and mechanochromism (MC) meaning they have potential for both functional and photonic applications. There has been significant recent interest in the study of materials that simultaneously exhibit TADF, AIE and MC29–31 and while not the only TADF-active salts,32–34 to our knowledge, these compounds are the first organic salts that are prepared from non-TADF materials to show all three. To summarise, the modification of known N-heterocyclic chromophores by quaternisation provides rapid access to new materials with altered charge transfer and emission properties.
Results and discussion
Synthesis
To investigate the influence of quaternisation on the photophysical properties of quinoline-based donor–acceptor materials, we synthesised two new compounds, Q1 and Q2 (Scheme 1). Here, 3,6-di-tert-butylcarbazole (tBu-CBZ) was introduced as a donor in the 3- or 4-position of quinoline. The compounds were then methylated to prepare (Scheme 1) a series of quinoline salts MeQ1·X and MeQ2·X with triflate, iodide or tetrafluoroborate as the anion, X. | ||
| Scheme 1 General strategy for the preparation of the series of N-alkylated quinoline-based compounds MeQ1·X and MeQ2·X. (i) 3,6-Di-tert-butylcarbazole, XPhos, Pd2(dba)3, NaOtBu, PhMe, 110 °C, 18 h; (ii) MeOTf, CH2Cl2, 0 °C to rt, 10 h; (iii) MeI, PhMe, 100 °C, microwave, 3 h; (iv) NH4BF4, (MeO)3CH, 110 °C, microwave, 1 h. dba = dibenzylideneacetone; OTf = triflate. | ||
Both Q1 and Q2 were prepared via Buchwald–Hartwig coupling between 3,6-di-tert-butylcarbazole and 3-bromoquinoline (1) or 4-bromoquinoline (2), respectively. Compounds MeQ1·X and MeQ2·X were prepared by the methylation of Q1 and Q2, respectively, with corresponding methylating agents (Scheme 1). For the preparation of tetrafluoroborate salts MeQ1·BF4 and MeQ2·BF4, we followed the synthetic procedure previously reported by Kim et al.35 for the synthesis of imidazolium ionic liquids. We modified the procedure for use in a microwave to reduce reaction times. This method allows for direct alkylation of Q1 and Q2, without the need of an anion exchange step, and uses trimethyl orthoformate as a more environmentally friendly alternative to the standard N-methylating agents. This procedure will be further explored in our future work. General synthetic characterisation such as electrochemistry and thermal analysis can be found in the ESI† (Fig. S1–S20 and Tables S1, S2).
Photophysical characterisation
The emission and absorption for Q1 and Q2 were investigated in solutions of methylcyclohexane (MCH), toluene (PhMe) chloroform (CHCl3), 2-methyltetrahydrofuran (2-MeTHF), dichloromethane (DCM) and acetonitrile (MeCN) at 20 μM concentrations (see Fig. 1 and Fig. S21, ESI†).
 | ||
| Fig. 1 Steady-state photoluminescence (solid) and absorption (dash) spectra of (a) Q1, (b) Q2, (c) MeQ1·OTf and (d) MeQ2·OTf in a series of solvents. | ||
The absorption spectra for Q1 and Q2 in most solvents demonstrate two strong bands corresponding to π–π* transitions with several additional smaller peaks (Fig. 1 and Fig. S21, Table S3, ESI†). The two main bands are consistent with calculations, which indicate that the lower intense band of both compounds mainly correspond to a moderate CT transition, while the higher energy intense band comes from a LE state localised on carbazole (Table S8, ESI†).
Q1 is a blue emitter and, as expected for donor–acceptor compounds, it undergoes a bathochromic shift of up to 0.35 eV (from MCH to MeCN) in its photoluminescence onset with increasing solvent polarity. This is indicative of the CT character of the emission (Fig. 1 and Fig. S21, Table S4, ESI†). The emission peak of Q2 in MCH is slightly red-shifted compared to Q1 but the onsets are similar. Q2 also exhibits similar bathochromic shifts in its photoluminescence onset (0.45 eV from MCH to MeCN) with increasing solvent polarity (Table S4, ESI†).
Interestingly, upon methylation the compounds become almost non-emissive in solution and, therefore, only weak emission spectra could be recorded in solution for MeQ1·OTf and MeQ2·OTf (see Fig. 1 and Fig. S22, ESI†). This indicates the presence of nonradiative decay channels quenching the emission in solution.
The methylated compounds are insoluble in MCH, so the spectra were recorded in 2-MeTHF, DCM, CHCl3 and MeCN. Similar to Q1, the absorption spectra of MeQ1·OTf exhibits several absorption peaks between 4.28 eV (290 nm) and 3.71 eV (334 nm). A new CT band emerges at a longer wavelength (around 2.76 eV (450 nm) in 2-MeTHF and MeCN, Fig. 1 and Fig. S22, Table S3, ESI†). The last band demonstrates a red shift of almost 0.29 eV in DCM and CHCl3 (peak at 2.47 eV (502 nm)) compared to the 2-MeTHF and MeCN solutions. MeQ2·OTf demonstrates very similar behaviour. It exhibits a new CT band around 2.76 eV (450 nm) in 2-MeTHF and MeCN that is similarly red-shifted in DCM (0.16 eV to 2.60 eV (477 nm)) and CHCl3 (0.18 eV to 2.58 eV (480 nm)) (Fig. 1 and Fig. S22, Table S3, ESI†). Strong CT character of the lowest excited singlet state is also predicted by calculations for both compounds (i.e. cations in vacuum, see Table S8, ESI†).
All MeQ1+ and MeQ2+-based salts demonstrate very similar absorption and emission profiles, so most of the measurements were performed only for MeQ1·OTf and MeQ2·OTf compounds. The salts only become emissive in an aggregated state, indicative of their AIE character (see Fig. 2). Aggregation is known to restrict and inhibit deleterious nonradiative decay channels, simply by limiting molecular motions which lead to such deactivation processes.36,37 It is also possible that in solid state the molecules are predominantly adopting a more emissive conformation.
 | ||
| Fig. 2 Photoluminescence spectra of (a) MeQ1·OTf and (b) MeQ2·OTf in THF/hexane mixtures with different hexane fractions (fHex). The excitation wavelength is 450 nm. | ||
Due to the non-emissive nature of the salts in well-dissolved solutions, we confirmed their AIE behaviour using solvent mixtures of tetrahydrofuran (THF) and hexane (see Fig. 2). As the compounds are insoluble in hexane, the addition of hexane to the THF solutions causes the compounds to form aggregates and activate their AIE. The intensity of the emission increases for both MeQ1·OTf and MeQ2·OTf with the increase in volume fraction of hexane (fhex).
| Compound | Emission peaka/eV | PLQYab/% | ΔES–Tac/eV |
|---|---|---|---|
| a In 1 wt% PMMA with neat film values in brackets. b Measured in air. c Experimentally obtained from the onsets of the prompt fluorescence and phosphorescence spectra. | |||
| Q1 | 3.03 | 20 | 0.57 |
| Q2 | 2.99 | 19 | 0.57 |
| MeQ1·OTf | 2.12 (2.16) | 4 (13) | 0.14 (0.28) |
| MeQ2·OTf | 2.10 (2.13) | 13 (34) | 0.06 (0.11) |
Absorption spectra of materials Q1 and Q2 in PMMA films (Fig. S23 and Table S5, ESI†) are very similar to those recorded in solution (Fig. S21 and Table S3, ESI†) and both feature the two main bands consistent with the calculations (Table S8, ESI†). As with the solution measurements, several smaller peaks are also observed and are detailed in Table S5 (ESI†). Both Q1 and Q2 emit light in the blue region of the spectrum with peaks at 3.05 eV (407 nm) and 3.01 eV (412 nm), respectively (Fig. S23 and Table S5, ESI†). The photoluminescence quantum yields (PLQYs) of the two materials in PMMA are approximately 20% (Table 1).
The absorption spectra for the MeQ1 salts are nearly identical, independent of the nature of the counter-ion (Fig. S24 and Table S5, ESI†). MeQ2 derivatives demonstrate similar behaviour (Fig. S24 and Table S5, ESI†). All six salts are yellow emitters with photoluminescence peaks between 2.15 eV (578 nm) and 2.10 eV (590 nm) (Fig. S24 and Table S5, ESI†). The methylation of the aromatic quinoline unit has therefore had a significant effect on the colour of the photoluminescence. This observation is consistent with the behaviour observed upon methylating quinine,4 although with a more drastic energy shift.
For compounds MeQ1·OTf and MeQ2·OTf, PLQYs were recorded as both 1 wt% PMMA dispersions and as neat films. The emission is largely quenched in the PMMA matrix for both MeQ1·OTf (13% in neat film and 4% in PMMA) and MeQ2·OTf (34% in neat film and 13% in PMMA) (see Table 1). This is due to the aggregation-induced nature of the emission of the materials. The restriction of molecular motion increases from solution to PMMA matrix to neat film. This inhibits the nonradiative decay, hence the increase of PLQY. The more than twofold increase of PLQY for MeQ2·OTf compared to MeQ1·OTf is in correlation with the theoretical findings indicating a higher oscillator strength for the S1 of MeQ2+ (0.263) compared to MeQ1+ (0.051, Table S8, ESI†), which is also apparent from the relative intensity of the CT band in the absorption spectra (Fig. 1 and Fig. S24, ESI†).
All salts exhibit mechanochromism, i.e. their colours change after mechanical force is applied. Due to the quenching of luminescence in MeQ1·I and MeQ2·I in the solid-state, mechanochromism was only investigated for the OTf and BF4 salts. Initially all four materials are yellow emitters that, after grinding, exhibit shifts in their photoluminescence to the red region (Fig. S25, S26 and Table S6, ESI†). The MeQ1+ salts, MeQ1·OTf and MeQ1·BF4, demonstrate much smaller PL peak shifts of 0.11 eV and 0.08 eV, compared to MeQ2·OTf (0.19 eV) and MeQ2·BF4 (0.24 eV). This suggests that when tBu-CBZ is in the 4-position of the quinoline ring, the material exhibits more significant changes in the packing after grinding than when tBu-CBZ is in the 3-position.
 | ||
| Fig. 3 Time-resolved photoluminescence decays for (a and b) MeQ1·OTf and (c and d) MeQ2·OTf in neat film at RT and 80 K. | ||
To give further comparison between the starting materials Q1 and Q2 and the salts, time-resolved photoluminescence measurements were also performed on 1 wt% PMMA films (see Fig. 4 and Fig. S29–S32, ESI†). These measurements confirm the room temperature phosphorescence (RTP) nature of Q1 and Q2 and demonstrate that upon alkylation, the RTP is tuned to delayed fluorescence. In a simple, single synthetic step, we have converted RTP materials into delayed fluorescence emitters with AIE behaviour.
 | ||
| Fig. 4 Photoluminescence decay for (a and b) MeQ1·OTf and (c and d) MeQ2·Otf in 1 wt% PMMA films at RT and 80 K. | ||
Q1 and Q2 show clear room temperature phosphorescence in 1 wt% PMMA film. Radiative decay from triplet states of Q1 and Q2 is favoured due to the large energy gap between the lowest triplet and the lowest singlet states (Table S8, ESI†). Calculations also predict sufficiently short phosphorescence lifetimes of 0.86 s and 1.35 s, respectively, for Q1 and Q2 giving additional confirmation that RTP is taking place in these compounds. Quaternisation significantly tunes the singlet and triplet levels – in methylated compounds the singlet–triplet energy gap (ΔES–T) is reduced, opening up the possibility for delayed fluorescence due to reverse intersystem crossing from a triplet to singlet state. This change from RTP to delayed fluorescence can be quantitatively explained by considering the energetics of the two compounds. The ΔES–T gaps for MeQ1·OTf and MeQ2·OTf in both neat films and 1 wt% PMMA film can be estimated from measuring the S1 and T1 energies for both samples from the onset of the high energy-edge of the fluorescence and phosphorescence peaks, respectively (Table S7, ESI† and Fig. 5). For the neat films the first singlet excited states S1 are nearly isoenergetic for MeQ1·OTf and MeQ2·OTf (2.51 eV), while the energy of the T1 triplet state is 0.17 eV lower for the MeQ1·OTf (2.23 eV) compared to MeQ2·OTf (2.40 eV). The ΔES–T is consequently much smaller for MeQ2·OTf (0.11 eV) compared to MeQ1·OTf (0.28 eV). Theoretically, the computed ΔES–T (based on ground-state minimum energy geometries in vacuum) are 0.25 and 0.17 eV for MeQ1+ and MeQ2+, respectively. However, we note that estimates of excitation energies from these experimental fluorescence and phosphorescence onsets, or calculations based on fixed ground state geometries, assume that the ΔES–T can be obtained as in Scheme 2a. However, if there is a difference in geometry between the S1 and T1, then the experimental values from measurement of the onset relate instead to Scheme 2b and thus ΔES–T cannot be obtained directly unless the energy gap due to the change in geometry is also known  . As an alternative, we optimised the S1 and
. As an alternative, we optimised the S1 and 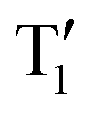 states of both compounds theoretically as in Scheme 2b. The downside of this approach is that it still corresponds to the situation in vacuum – the molecules have more flexibility as compared to the solid state. Nevertheless, we found that S1 and
states of both compounds theoretically as in Scheme 2b. The downside of this approach is that it still corresponds to the situation in vacuum – the molecules have more flexibility as compared to the solid state. Nevertheless, we found that S1 and  excited state minima are nearly degenerate for MeQ2+, while in the case of MeQ1+ the absolute energy of the S1 minimum is 0.14 eV above the
excited state minima are nearly degenerate for MeQ2+, while in the case of MeQ1+ the absolute energy of the S1 minimum is 0.14 eV above the 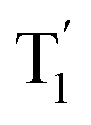 minimum (Table S10, ESI†). Once again, our results imply that the intersystem crossing may be more easily achieved in MeQ2+ than MeQ1+ compounds.
minimum (Table S10, ESI†). Once again, our results imply that the intersystem crossing may be more easily achieved in MeQ2+ than MeQ1+ compounds.
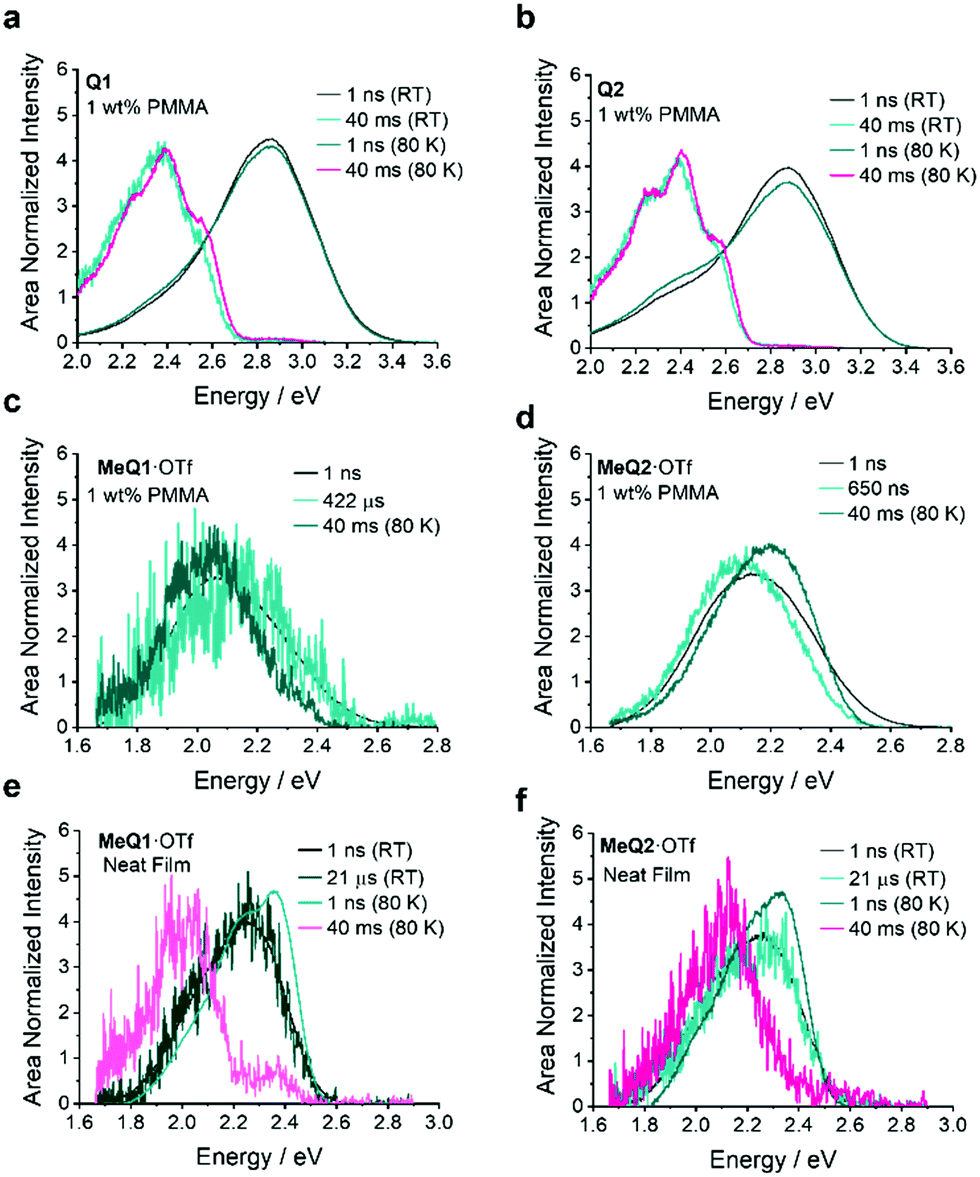 | ||
| Fig. 5 Time resolved spectra at RT and 80 K for demonstrating the RTP, TADF and TTA characters of (a) Q1, (b) Q2, (c and e) MeQ1·OTf and (d and f) MeQ2·OTf. | ||
 | ||
| Scheme 2 Relaxation of the lowest singlet and triplet excited states via fluorescence (F) and phosphorescence (P), respectively, according to (a) idealised Jablonski scheme where geometry changes are not taken into account and (b) realistic scheme where S1 and T1 optimal geometries differ. | ||
While we have demonstrated that the singlet and triplet states change energy between the original compounds and the salts, explaining why the changes are different between MeQ1 and MeQ2 is more complex. The different response to quaternisation for these two compounds is quite a surprising outcome as one would expect MeQ2·OTf, with its tBu-CBZ group in the para-position, to have a better conjugation between the donor and acceptor moieties and thus a larger overlap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels compared to MeQ1·OTf, where the communication between the quinoline nitrogen and the tBu-CBZ unit should be partially interrupted by virtue of meta-position. The fact that MeQ2·OTf is a better TADF emitter, compared to MeQ1·OTf, might indicate that other factors can influence the magnitude of the ΔES–T gap. It is also possible that in the excited state the dihedral angle between the donor and acceptor moieties is larger for MeQ2·OTf than it is MeQ1·OTf, which leads to a better separation of HOMO and LUMO for the former. Indeed, theoretical excited-state optimisations performed in vacuum predict a significantly larger dihedral angle for the S1 and  minimum geometries of MeQ2+, as compared to the S1 and
minimum geometries of MeQ2+, as compared to the S1 and 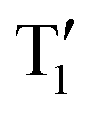 states of MeQ1+ (Table S10, ESI†).
states of MeQ1+ (Table S10, ESI†).
As a result of these different changes and, therefore, different energy gaps for MeQ1+ and MeQ2+, we must consider the origin of the delayed fluorescence. Having imparted the salts with delayed fluorescence it is now possible to determine its origin by measuring the dependence of fluorescence intensity (IF) on laser fluence (Eex). TTA demonstrates quadratic power dependence at lower excitation dose with the slope n being 2 for IF ∼ Enex that turns into 1 at high triplet densities.38,39 For TADF materials the dependence is always linear, i.e., n = 1. To confirm the nature of delayed fluorescence for the quaternised compounds, we performed laser fluence measurements on the neat films of the OTf salts. The estimated slope for the MeQ1·OTf salt is 1.29, which indicates that the emission has bimolecular contribution and most likely originates from TTA (Fig. S33, ESI†). For MeQ2·OTf, the power dependence is linear, thus the fluorescence originates from TADF (Fig. S33, ESI†). We can also compare the estimated reverse intersystem crossing rates for MeQ1·OTf and MeQ2·OTf (Table S7, ESI†) showing that MeQ2·OTf has an order of magnitude higher rISC rate consistent with its smaller ΔES–T, larger TADF contribution to the delayed fluorescence and overall better performance as an emitter.
Thus, through a single-step synthesis we have converted one RTP compound into a TADF emitter and a very similar RTP compound into an emitter that exhibits a mixture of TADF and TTA. This demonstrates the level of control that can be exerted over the energetic states of organic chromophores utilising a combination of quaternisation and structural and steric properties (Fig. S40, ESI†).
 | ||
| Fig. 6 The X-ray crystal structures of Q2, MeQ2·OTf and MeQ1·OTf. SR = steric repulsion. | ||
In the crystalline state, the minor differences in the conformations of the cations do not affect the lengths of the N–C bonds between the planar donor and acceptor fragments, which is very similar in all studied molecules. The fact that the dihedral angles between the donor and acceptor moieties do not change significantly with the methylation, while the luminescence type changes from phosphorescence to delayed fluorescence, demonstrates that achieving high twist between the donor and acceptor unit is not sufficient for the separation of HOMO and LUMO and consequent reduction of the ΔES–T gap. Instead, tuning the relative strength of donor and acceptor plays a much more important role. In this case, the methylation of the N-atom on the quinoline unit makes it more electron-deficient, changing the energy gap and inducing delayed fluorescence.
The molecules of Q2 in the crystalline state are linked by both CH⋯N and π⋯π intermolecular interactions (Fig. 7 and Fig. S37, ESI†). On the contrary, π-stacking between the aromatic rings are suppressed in MeQ2·OTf and MeQ1·OTf on account of the triflate counterion increasing the spacing between each cation (Fig. 7 and Fig. S38, S39, ESI†). This indicates a significant influence of inter-ionic electrostatic interactions on packing motifs in crystal structures of the studied salts. These interactions could also explain why the salts exhibit AIE and mechanochromism. The packing of MeQ2·OTf also lacks strong direction-specific interactions between the MeQ2+ units.
 | ||
| Fig. 7 Face-to-face aromatic intermolecular interactions in Q2 and their absence in MeQ2·OTf from X-ray crystal packing. | ||
Conclusions
To conclude, the materials presented in this work can be prepared via a simple two-step synthesis involving a relatively quick purification process to convert RTP emitters into delayed fluorescence, and in particular, TADF emitters. This provides a simple pathway to expand the library of TADF emitters by enhancing already existing molecules without the need to design new molecular motifs from scratch. In total, six new quinoline-based salts were synthesised. Apart from TTA and TADF, the salts demonstrate additional functional behaviour including AIE and mechanochromism.The tuning of the energy levels by alkylation has been rationalised via calculations and experimentally verified through time-resolved photoluminescence measurements. Interestingly while both materials exhibit delayed fluorescence and despite a very similar structure, MeQ1·OTf shows a mixture of TADF and TTA while MeQ2·OTf is a solely TADF emitter.
Author contributions
A. K.: conceptualisation, formal analysis, investigation, methodology, project administration, resources, visualisation, writing – original draft, writing – review & editing; A. P.: conceptualisation, investigation, formal analysis, software, visualisation, writing – original draft, writing – review & editing; D. S. Y.: investigation, visualisation, writing – original draft, writing – review & editing; A. M.: investigation, writing – review & editing; B. F. E. C.: conceptualisation, formal analysis, software, supervision, writing – review & editing; P. R. M.: conceptualisation, writing – review & editing; P. S.: conceptualisation, supervision, funding acquisition, writing – review & editing; M. K. E.: conceptualisation, formal analysis, investigation, methodology, project administration, resources, supervision, visualisation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement H2020-MSCA-ITN2015/674990 project ‘‘EXCILIGHT’’. A. K. also thanks the EPSRC for funding (EP/T013710/1). M. K. E. thanks the Royal Society of Chemistry (R20-1668) for support. The authors also thank Andrew P. Monkman for use of his facilities for the spectroscopic work.Notes and references
- Y. C. Liu, C. S. Li, Z. J. Ren, S. K. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29 Search PubMed.
- H. Nakanotani, Y. Tsuchiya and C. Adachi, Chem. Lett., 2021, 50, 938–948 CrossRef CAS.
- A. T. Turley, A. Danos, A. Prlj, A. P. Monkman, B. F. E. Curchod, P. R. McGonigal and M. K. Etherington, Chem. Sci., 2020, 11, 6990–6995 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- J. Qian and A. M. Brouwer, Phys. Chem. Chem. Phys., 2010, 12, 12562–12569 RSC.
- I. Bhattacharjee, N. Acharya and D. Ray, Chem. Commun., 2019, 55, 1899–1902 RSC.
- J. Pandidurai, J. Jayakumar, N. Senthilkumar and C. H. Cheng, J. Mater. Chem. C, 2019, 7, 13104–13110 RSC.
- S. J. Yoon, H. J. Lee, K. H. Lee and J. Y. Lee, J. Mater. Chem. C, 2020, 8, 7485–7491 RSC.
- C. J. Schoot, J. J. Ponjee, H. T. van Dam, R. A. van Doorn and P. T. Bolwijn, Appl. Phys. Lett., 1973, 23, 64–65 CrossRef CAS.
- P. M. S. Monk, D. R. Rosseinsky and R. J. Mortimer, Electrochromic Materials and Devices, ed. P. M. S. Monk, D. R. Rosseinsky and R. J. Mortimer, 2015, ch 3, pp. 57–90 Search PubMed.
- J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benitez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard, 3rd and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS PubMed.
- E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 2002, 86, 5524–5527 CrossRef.
- L. Michaelis, Chem. Rev., 1935, 16, 243–286 CrossRef CAS.
- J. M. Hancock and S. A. Jenekhe, Macromolecules, 2008, 41, 6864–6867 CrossRef CAS.
- S. I. Kim, B. J. Kang, C. U. Jeong, M. H. Shin, W. T. Kim, M. Jazbinsek, W. Yoon, H. Yun, D. Kim, F. Rotermund and O. P. Kwon, Adv. Opt. Mater., 2019, 7, 1801495 CrossRef.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- K. Tanabe, D. Kodama, M. Hasegawa and T. Kato, Chem. Lett., 2014, 43, 184–186 CrossRef CAS.
- P. Data, M. Okazaki, S. Minakata and Y. Takeda, J. Mater. Chem. C, 2019, 7, 6616–6621 RSC.
- T. Hosono, N. O. Decarli, P. Z. Crocomo, T. Goya, L. E. de Sousa, N. Tohnai, S. Minakata, P. de Silva, P. Data and Y. Takeda, J. Mater. Chem. C, 2022, 10, 4905–4913 Search PubMed.
- L. Zhan, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 17651–17655 CrossRef CAS PubMed.
- N. A. Kukhta, R. Huang, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Phys. Chem. C, 2019, 123, 26536–26546 CrossRef CAS.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC.
- S. Xu, T. Liu, Y. Mu, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- M. K. Etherington, Front. Chem., 2020, 8, 716 CrossRef CAS PubMed.
- M. Y. Wong, M.-G. La-Placa, A. Pertegas, H. J. Bolink and E. Zysman-Colman, J. Mater. Chem. C, 2017, 5, 1699–1705 Search PubMed.
- M. Y. Wong, G. J. Hedley, G. Xie, L. S. Kölln, I. D. W. Samuel, A. Pertegás, H. J. Bolink and E. Zysman-Colman, Chem. Mater., 2015, 27, 6535–6542 CrossRef CAS.
- Z. Xu, D. Hean, C. Climent, D. Casanova and M. O. Wolf, Mater. Adv., 2021, 2, 5777–5784 RSC.
- D. J. Kim, K. H. Oh and J. K. Park, Green Chem., 2014, 16, 4098–4101 RSC.
- R. Crespo-Otero, Q. Li and L. Blancafort, Chem. – Asian J., 2019, 14, 700–714 CrossRef CAS PubMed.
- H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS PubMed.
- F. B. Dias, Philos. Trans. R. Soc., A, 2015, 373, 20140447 CrossRef PubMed.
- V. Jankus, E. W. Snedden, D. W. Bright, V. L. Whittle, J. A. G. Williams and A. Monkman, Adv. Funct. Mater., 2013, 23, 384–393 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2149620–2149624. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc01737g |
| This journal is © The Royal Society of Chemistry 2022 |