
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decisive role of heavy-atom orientation for efficient enhancement of spin–orbit coupling in organic thermally activated delayed fluorescence emitters†
Michał Mońkaa,
Daria Grzywaczb,
Estera Hoffmana,
Vladyslav Ievtukhovb,
Karol Kozakiewiczb,
Radoslaw Rogowskia,
Aleksander Kubickia,
Beata Liberek b,
Piotr Bojarskia and
Illia E. Serdiuk*a
b,
Piotr Bojarskia and
Illia E. Serdiuk*a
aFaculty of Mathematics, Physics and Informatics, University of Gdańsk, Wita Stwosza 57, 80-308 Gdańsk, Poland. E-mail: illia.serdiuk@ug.edu.pl; Tel: +48 58 523 22 44
bFaculty of Chemistry, University of Gdańsk, Wita Stwosza 63, 80-308 Gdańsk, Poland
First published on 1st August 2022
Abstract
In view of the rapidly growing interest in the hybrid materials for heavy-metal-free optoelectronics, the research described here aimed to confirm the potential ability of abundant heavy atoms (HAs) in improving key parameters of organic emitters with thermally activated delayed fluorescence. Namely, the enhancement of reverse intersystem crossing (rISC) while keeping a reasonable value of fluorescence rate was investigated in red emitters with bromine atom(s) introduced into the ortho positions of the N,N-ditolylaniline donor fragment. The results of photophysical investigations and quantum chemical calculations indicate that selective acceleration of rISC by HAs without a substantial decrease of the fluorescence rate is possible. Molecular design principles of such hybrid materials, however, do not seem simple. In the investigated emitters, the oscillator strength of the S1–S0 transition which defines the fluorescence rate is not directly influenced by the bromine atoms, and remains similar or decreases weakly for the brominated emitters. The maximal enhancement of spin–orbit coupling (SOC) does not depend directly on the number of HAs either, but on their relative position and orientation in the emitter. The analysis of numerous rotational isomers of emitters revealed that SOC enhancement cannot be explained by either the internal or the external HA effect. Taking into account the lack of significant contribution of bromines in orbital and spin momentum of the T1–S1 transition, but yet significant SOC enhancement, we explain the observed phenomenon as a heavy-atom field effect (HAFE) which increases the total angular momentum. The most impressive SOC enhancement by the HAFE up to 60 times is observed when HAs align asymmetrically and are oriented towards the chromophore fragment with the largest orbital momentum change. Another important observation reveals that, in the case of symmetrical structures, the field of a heavy atom can be compensated by another one leading to almost zero SOC and 650-fold rISC inhibition. Such species should be avoided at the stage of molecular design planning.
Introduction
Hybrid organic emitters bearing cheap and abundant non-metallic heavy atoms (HAs) attract more and more attention lately.1–4 Such materials can be the Golden mean between all-organic materials and heavy-metal complexes or inorganic analogues. Potentially, the rational design of such hybrid materials can preserve the advantages of organic and heavy-metal materials to enable fast conversion of electric energy into light in a submicrosecond regime but avoid low efficiency of organics and high cost and environmental issues connected with the use of heavy metals.Nowadays, the application of various emitters in organic light emitting diodes (OLEDs) is realized via two main strategies which represent separate solutions for the problem of triplet harvesting under electric excitation. Widely commercialized generation of phosphorescent heavy-metal complexes converts all the electrically generated excitons to the triplet ones.5 Due to very high values of spin–orbit coupling (SOC) of the T1–S0 transition caused by the presence of a heavy-metal, such excitons are deactivated radiatively within 1–10 μs. Recently, various attempts were made to substitute such heavy-metal materials with organic analogues for application in such phosphorescence OLEDs.6–8 However, the best pure organic candidates phosphoresce in the second or millisecond time domain and/or with low quantum yields.9–11 Very recent approaches of incorporation of abundant non-metallic heavy atoms like selenium in organic materials provide submillisecond phosphorescence (385 μs) with a 20% quantum yield.6 These data indicate that we are still far away from efficient organic phosphors.
The second strategy for OLED emitters expected to be closer to commercialization utilizes the thermally activated delayed fluorescence (TADF) phenomenon.12 In TADF materials, the radiative deactivation of all excitons occurs from the singlet S1 state. Conversion of triplet excitons to singlet ones occurs via endothermic spin–flip transition named reverse intersystem crossing (rISC). It is enabled by the energetic proximity of the lowest excited singlet and triplet states in TADF emitters. Being “spin–forbidden”, rISC still depends on SOC. However, in contrast to the all-organic phosphorescent materials, some of their TADF analogues are capable of 100% quantum yields of luminescence within micro- and submicrosecond regimes.13 Taking into account the most recent achievements,14–17 development of organic and hybrid TADF materials seems to be the most promising direction for environment friendly multifunctional optoelectronics.
One of the most critical problems of organic TADF materials remains the slow rate of triplet harvesting.18,19 Acceleration of rISC while maintaining the fast fluorescence rate is supposed to be a solution for the minimization of the external quantum efficiency (EQE) roll-off under increasing current density and low stability of OLED devices in general. In light-atom organic materials due to weak SOC, efficient rISC can be achieved mainly via minimization of the energy gap between the S1 and T1 states (ΔEST): the lower the ΔEST value, the higher the rate constant of rISC (krISC). In general, small ΔEST requires strong spatial separation of the highest occupied (HOMO) and the lowest unoccupied molecular orbital (LUMO). For this reason an overwhelming number of TADF emitters are strong electron donor–acceptor (D–A) molecular systems whose S1 and T1 states have charge-transfer (CT) nature with a high transition dipole moment.
Another important parameter of a TADF emitter is its emissive rate. In donor–acceptor emitters, the latter is proportional to the oscillator strength of the 1CT-S0 transition (f1CT-S0). The above mentioned separation of HOMO and LUMO unavoidably leads to the decrease of f1CT-S0, or in terms of selection rules, the 1CT-S0 transition becomes forbidden. Low oscillator strength is clearly a negative factor for an OLED emitter, which elongates the conversion of excitation energy to light. Taking this into account, the relation of key parameters of D–A type TADF emitters can be expressed as:
| f1CT-S0 ∼ 1/krISC. | (1) |
Within the light-atom organic D–A materials, a solution to this dilemma is extremely challenging, because most of the structural changes sacrifice either the rISC rate and thus triplet harvesting efficiency or the 1CT-state radiative deactivation rate constant (kr) and thus fluorescence efficiency (Fig. 1). Fastly evolving hybrid materials based on organic emitters modified with HAs can potentially solve this dilemma. As correlation (1) is caused by different indirect electronic reasons, the presence of HAs can increase SOC, but hypothetically should not affect the fluorescence rate. However, in spite of the rapidly growing interest in such materials,14–17 very scarce knowledge on the mechanism of HA action on organic TADF emitters prevents their targeted rational molecular design. Taking into account the complex nature of TADF, the effect of a particular heavy atom on the nature of electronic states and other parameters connected with f1CT-S0(kr) and krISC(SOC) is highly unintuitive and should be extensively investigated.
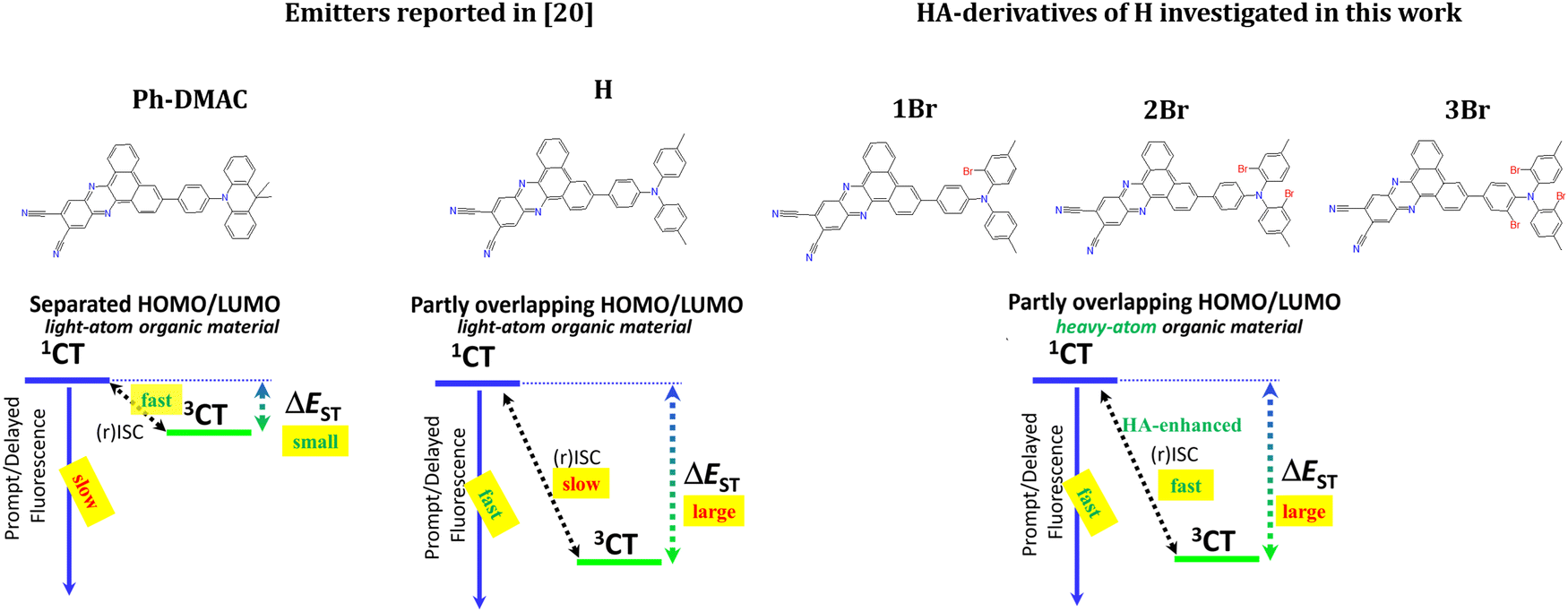 | ||
| Fig. 1 Canonic structures of the investigated emitters with a schematic representation of excited states and key processes leading to light emission. | ||
In this research, we aimed to find a molecular strategy for the HA-modification of an organic TADF emitter so that it could preserve its fluorescence rate but enhance the rISC rate. The most suitable molecular design strategy for such modifications seems to be the one compromising the ΔEST and f1CT-S0 values as reported recently.20 This strategy relies on a smaller separation of HOMO and LUMO in red and NIR emitters with a highly stabilized CT state. It utilizes a partly conjugated donor with a strong acceptor fragment20,21 and sacrifices the reduction of ΔEST for a higher f1CT-S0 value. The thus developed 11,12-dicyanodibenzo[a,c]phenazine emitter bearing a more planar N,N-ditolylaniline (DTA) donor (emitter H, Fig. 1) in comparison with a highly twisted 10-phenyl-9,9-dimethylacridan (Ph-DMAC) donor was reported to have a 12 times higher kr value. Simultaneously, the ΔEST increased almost 3.7 times causing a 30-fold drop in krISC. The authors also performed OLED tests, which confirmed that in spite of kr increase, rISC inhibition led to worse device work due to a much higher EQE roll-off. One should also note that the described emitters are one of the best representatives of red/NIR TADF emitters,22 and thus their further improvement seems to be very topical for OLED technology.
Taking into account the good fluorescent properties but unsatisfactory slow rISC, in this work, H was modified with heavy atoms. Bromine was selected as HAs thanks to its abundance, low cost and easiness of introduction into organic molecules. Next, possible positions of HAs in the emitter's structure were analyzed. According to our previous findings,23 one should avoid direct electronic interaction of HAs with the fragment responsible for the lowest excited triplet state of local character (3LE) to avoid intersystem crossing (ISC) enhancement via the 1CT→3LE(HA) channel, which is a negative factor regarding triplet harvesting efficiency. The reported phosphorescence investigations indicate that a triplet state localized on a triarylamino donor fragment has energy near 3.05 eV.24 Triplet energy of dibenzo[a,c]phenazine acceptor derivatives is below 2.50 eV,25 which makes it the lowest 3LE state in such kinds of emitters. For these reasons, one, two, and three bromine atoms were introduced into the donor, namely, at the ortho position of each of the benzene rings of the DTA fragment, providing emitters 1Br, 2Br, and 3Br, respectively (Fig. 1). Thorough analysis of the photophysical properties with the help of DFT calculations allowed us to reveal and explain rISC enhancement whilst maintaining a reasonable kr value. In the example of red TADF emitters, it was found that HAs in ortho positions of triarylamine donors enrich rotational isomerism of emitting species. The analysis of electronic features of such rotamers afforded valuable conclusions on the effect of relative positions of HAs on spin–orbit coupling and rISC.
Synthesis
3-(4-(Di-p-tolylamino)phenyl)dibenzo[a,c]phenazine-11,12-dicarbonitrile (H) was synthesized by the reaction of 3-(4-(di-p-tolylamino)phenyl)phenanthrene-9,10-dione (1) with 4,5-diaminophthalonitrile (Scheme 1), conducted according to the procedure described in the literature.20 Bromination of H with 1.1 eq. of NBS gave 1Br. Analogous bromination of H with 2.1 and 3.1 eq. of NBS was less selective. Therefore, we decided to brominate first substrate 1 with 2.1 and 3.1 eq. of NBS, which gave us 2 and 3, respectively. Finally, 2Br and 3Br were obtained by cyclization of 2 and 3 with 4,5-diaminophthalonitrile.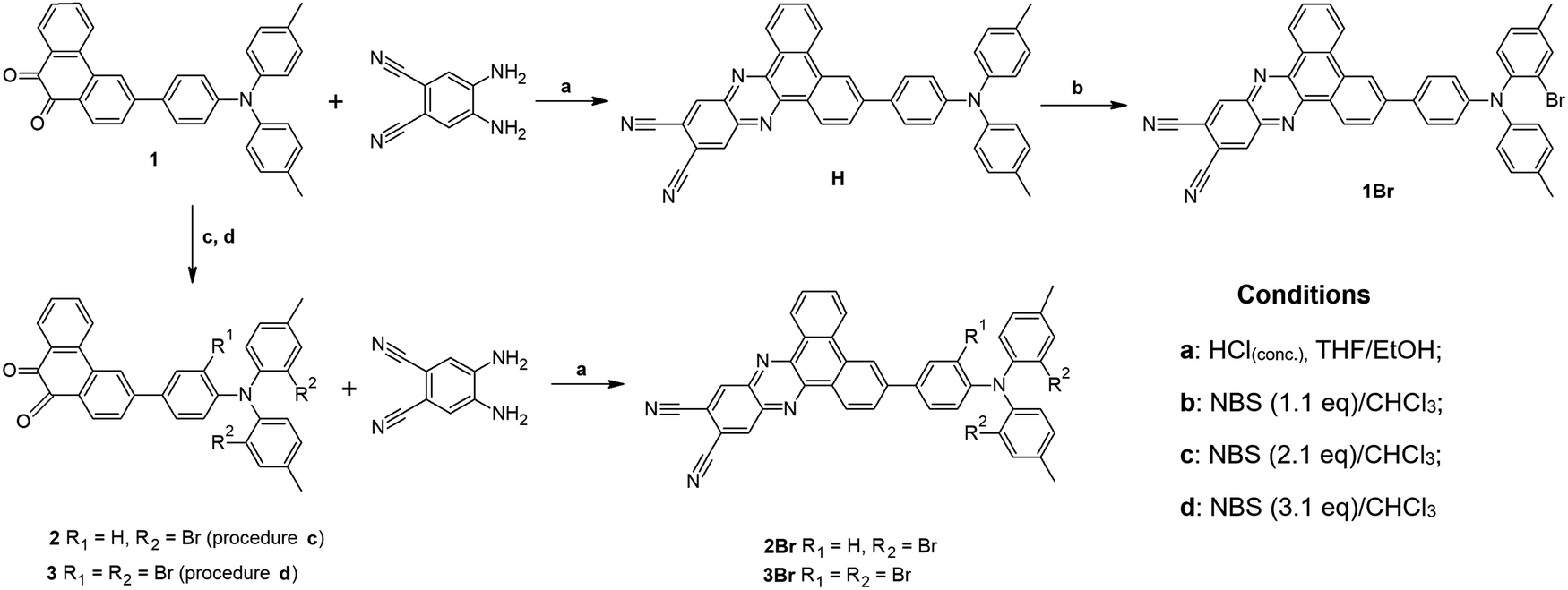 | ||
| Scheme 1 Synthetic scheme for the investigated emitters. | ||
Experimental section
Generals
Zeonex® (ZNX, Zeonex480R, density = 1.01 g cm−3), 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP, sublimated), reagents for synthesis, and solvents of respective grades for spectroscopy and synthesis were purchased and used without further purification.Synthesis and analysis
The 1H NMR spectra were recorded on a Bruker AVANCE III 500 (500.13/125.76 MHz) instrument, under standard experimental conditions in CDCl3 with internal Me4Si. The 13C NMR spectra were unavailable due to the low solubility of the investigated compounds. Positive-ion mode MALDITOF mass spectra were obtained using a Bruker Biflex III spectrometer with 2,5-dihydroxybenzoic acid matrixes. Purification by column chromatography was performed on silica gel (70–230 mesh).3-(4-(Di-p-tolylamino)phenyl)phenanthrene-9,10-dione (1) and 3-(4-(di-p-tolylamino)phenyl)dibenzo[a,c]phenazine-11,12-dicarbonitrile (H) were obtained according to the procedures described in the literature.20
General procedure for bromination of 1 and H
NBS (1.1 eq., 2.1 eq., or 3.1 eq., respectively) was added to 1 or H and dissolved in CHCl3 (8 mL). After stirring overnight at rt in the absence of light, the solution was concentrated and MeOH (5 mL) was added to the residue. The precipitated solid was filtered, washed and purified by silica gel column chromatography (eluent: CHCl3/n-hexane 1/5).3-(4-(Bis(2-bromo-4-methylphenyl)amino)phenyl)phenanthrene-9,10-dione (2)
Bromination of 1 (60 mg, 0.13 mmol) with 2.1 eq. of NBS (49 mg, 0.27 mmol) gave 2 (red solid, 70 mg, 84%). 1H NMR (500 MHz, CDCl3-d): δ ppm 2.36 (s, 6 H), 6.71 (d, J = 8.85 Hz, 2 H), 7.12 (dd, J= 7.93, 0.61 Hz, 2 H), 7.19 (d, J = 7.93 Hz, 2 H), 7.48 (td, J = 7.62, 0.61 Hz, 1 H), 7.50 (d, J = 1.53 Hz, 2 H), 7.56 (dt, J = 8.85, 2.14 Hz, 2 H), 7.65 (dd, J = 8.24, 1.53 Hz, 1 H), 7.72 (td, J = 7.33, 1.53 Hz, 1 H), 8.10 (d, J = 7.93 Hz, 1 H), 8.18 (d, J = 1.53 Hz, 1 H), 8.21 (dd, J = 7.93, 1.53 Hz, 1 H), 8.22 (d, J = 8.24 Hz, 1 H); MALDITOF-MS: m/z: calcd for C34H23Br2NO2 637.2, found 638.0 [M + 1]+.3-(4-(Bis(2-bromo-4-methylphenyl)amino)-3-bromophenyl)-phenanthrene-9,10-dione (3)
Bromination of 1 (60 mg, 0.13 mmol) with 3.1 eq. of NBS (70 mg, 0.40 mmol) gave 3 (orange solid, 79 mg, 85%). 1H NMR (500 MHz, CDCl3-d): δ ppm 2.34 (s, 6 H), 6.77 (d, J = 8.54 Hz, 1 H), 6.80 (d, J = 8.24 Hz, 1 H), 6.93 (d, J = 8.24 Hz, 1 H), 7.05 (2 × d, J = 7.93 Hz, 2 H), 7.45 (2 × s, 2 H), 7.51 (t, J = 7.63/7.33 Hz, 1 H), 7.52 (dd, J = 8.24 Hz, J = 2.14 Hz, 1 H), 7.64 (d, J = 8.24 Hz, 1 H), 7.75 (t, J = 7.63/7.47 Hz, 1 H), 7.93 (d, J = 2.14 Hz, 1 H), 8.12 (d, J = 8.24 Hz, 1 H), 8.17 (s, 1 H), 8.22 (d, J = 7.63 Hz, 1 H), 8.25 (d, J = 7.94 Hz, 1 H); MALDITOF-MS: m/z: calcd for C34H22Br3NO2 715.2, found 716.1 [M + 1]+.3-(4-((2-Bromo-4-methylphenyl)(p-tolyl)amino)phenyl)dibenzo[a,c]phenazine-11,12-dicarbonitrile (1Br)
Bromination of H (42 mg, 0.07 mmol) with 1.1 eq. of NBS (14 mg, 0.08 mmol) led to 1Br (red solid, 40 mg, 84%). 1H NMR (500 MHz, CDCl3-d): δ ppm 2.35–2.40 (4 × s, 6 H), 7.03 (d, J = 8.85 Hz, 1 H), 7.05–7.15 (m, 5 H), 7.17–7.23 (m, 2 H), 7.52 (dd, J = 10.38, 1.83 Hz, 1 H), 7.67 (d, J = 8.54 Hz, 1 H); 7.68 (d, J = 8.85 Hz, 1 H); 7.80 (tdd, J = 7.94, 2.75, 1.53 Hz, 1 H), 7.91 (tdd, J = 7.63, 2.75, 1.53 Hz, 1 H), 7.98 (dt, J = 8.54, 1.53 Hz, 1 H), 8.64 (dd, J = 8.09, 3.36 Hz, 1 H), 8.71 (bs, 1 H), 8.76 (dd, J = 1.83, 0.61 Hz, 1 H); 8.77 (t, J = 0.61 Hz, 1 H); 9.32 (dd, J = 8.24, 3.66 Hz, 1 H), 9.34 (dt, J = 8.24, 1.52 Hz, 1 H); MALDITOF-MS: m/z: calcd for C42H26Br1N5 679.2, found 680.2 [M + 1]+.General procedure for the synthesis of 2Br and 3Br
4,5-Diaminophthalonitrile (1.2 eq.) was added to 2 or 3 and dissolved in THF/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 5 mL) with HCl(conc) (10 μL). After stirring overnight at rt the solution was concentrated and purified by silica gel column chromatography (eluent: CHCl3/n-hexane 1/3).
:1; 5 mL) with HCl(conc) (10 μL). After stirring overnight at rt the solution was concentrated and purified by silica gel column chromatography (eluent: CHCl3/n-hexane 1/3).
3-(4-(Bis(2-bromo-4-methylphenyl)amino)phenyl)dibenzo[a,c]phenazine-11,12-dicarbonitrile (2Br)
Reaction of 4,5-diaminophthalonitrile (12 mg, 0.076 mmol) with 2 (40 mg, 0.063 mmol) gave 2Br (red solid, 40 mg, 83%). 1H NMR (500 MHz, CDCl3-d); 2.37 (s, 6 H), 6.79 (dt, J = 8.85, 2.14 Hz, 2 H), 7.13 (ddd, J= 8.24, 2.14, 0.61 Hz, 2 H), 7.19 (d, J = 8.24 Hz, 2 H), 7.51 (dd, J = 1.83, 0.61 Hz, 2 H), 7.69 (dt, J = 8.85, 2.14 Hz, 2 H), 7.80 (td, J= 7.02, 2.14 Hz, 1 H), 7.91 (td, J= 7.32, 1.53 Hz, 1 H), 7.99 (dd, J = 8.54, 1.53 Hz, 1 H), 8.65 (d, J= 7.94 Hz, 1 H), 8.73 (d, J = 1.53 Hz, 1 H), 8.79 (dd, J= 3.66, 0.61 Hz, 2 H), 9.33 (d, J = 8.24 Hz, 1 H), 9.36 (dd, J = 7.93, 1.53 Hz, 1 H); MALDITOF-MS: m/z calcd for C42H25Br2N5 759.2, found 759.2 [M]+.Synthesis of 3-(4-(bis(2-bromo-4-methylphenyl)amino)phenyl)dibenzo[a,c]phenazine-11,12-dicarbo-nitrile (3Br)
Reaction of 4,5-diaminophthalonitrile (10 mg, 0.067 mmol) with 3 (40 mg, 0.056 mmol) gave 3Br (yellow solid, 38 mg, 81%). 1H NMR (500 MHz, CDCl3-d); 2.37 (s, 6 H), 6.83 (d, J = 8.24 Hz, 1 H), 6.87 (d, J = 7.96 Hz, 1 H), 7.01 (d, J = 8.24 Hz, 1 H), 7.09 (dd, J = 7.96 Hz, J = 1.37 Hz, 2 H), 7.49 (bs, 2 H), 7.68 (dd, J = 8.51, 2.20 Hz, 1 H), 7.85 (t, J = 7.69 Hz, 1 H), 7.96 (ddd, J = 8.51 Hz, J = 7.14 Hz, J = 1.37 Hz, 1 H), 8.00 (dd, J = 8.51, 1.65 Hz, 1 H), 8.09 (d, J = 1.92 Hz, 1 H), 8.69 (d, J = 8.24 Hz, 1 H), 8.74 (d, J = 1.37 Hz, 1 H), 8.82 (d, J = 1.1 Hz, 1 H), 8.82 (m, 1 H), 9.38 (dd, J = 7.96 Hz, J = 1.1 Hz, 1 H), 9.39 (d, J = 8.51 Hz, 1 H); MALDITOF-MS: m/z: calcd for C42H24Br3N5 837.2, found 838.1 [M + 1]+.The NMR spectra of the obtained compounds can be found in the ESI.†
Sample preparation for photophysical measurements
Films in ZNX and CBP were prepared by the spin-coating method; the final mass fractions of dispersed emitters were ca. 0.1% and 6%, 10%.Photoluminescence measurements
UV-Vis absorption spectra were recorded using a Shimadzu UV-1900 spectrophotometer. Steady-state photoluminescence spectra were recorded using an FS5 spectrofluorometer (Edinburgh Instruments) using front-face excitation geometry. Absolute PL quantum yields (PLQYs) were measured using an integrating sphere (Quantaurus C11347-11, Hamamatsu). Time-resolved measurements were performed at different temperatures using a customized system26 consisting of a pulsed YAG:Nd laser (PL2251A, EKSPLA) coupled with an optical parametric generator (PG 401/SH) as the excitation light source and 2501S grating spectrometer (Bruker Optics) combined with a streak camera system (C4334-01 Hamamatsu) as the detection unit. The system was equipped with a double-stage high vacuum pump (T-Station 85 Edwards) coupled with a closed-cycle helium cryostat (APD DE-202) and a temperature controller (LakeShore 336). To reduce scattering, reflections and secondary order artifacts, a set of various high performance optical bandpass (BP) and longpass (LP) filters were used, in the excitation path 325/50BP or 375/50BP (CWL/FWHM), depending on the selected excitation wavelength, together with LP filters: 375LP or 350LP (Edmund Optics). In order to build PL intensity decay profiles, streak camera images were integrated over a constant specified wavelength interval. Radiative rate constants kr and (reverse) intersystem crossing rates k(r)ISC were calculated using procedures described in detail previously and in Section S4 (ESI†).23Quantum chemical calculations
Quantum chemical calculations were conducted at the DFT/TD-DFT level of theory using the Gaussian 16 program package.27 The B3LYP functional was used with the LAN2LDZ basis set.28 Unspecific solvent effect (toluene) was included at the level of the Polarized Continuum Model (PCM).First, for each compound, unconstrained geometry optimizations were performed for the ground (S0), excited singlet (S1) and excited triplet electronic (T1, T2) states. Convergence of all geometry optimizations was verified by the vibrational analysis, and no negative frequencies were observed; therefore, the calculated minima correspond to “true” stationary points. The nature of electronically excited states was determined by the analysis of molecular orbitals. Spin–orbit coupling constants were computed using the ORCA 4.2 software package29 with B3LYP functional and DEF2-TZVP basis set with included relativistic zero-order regular approximation (ZORA).
Results and discussion
Absorption spectra
The effect of bromine substitution on the ground state (S0) of emitters can be analyzed from the absorption spectra (Fig. 2A). In solutions, the long wavelength absorption maximum responsible for the S0–1CT transition shifts gradually to higher energies with the number of introduced bromines. There are two main factors which can cause such an effect: (i) the electron-withdrawing feature of bromine as a halogen, which reduces the donor strength and thus increases the energy of the CT state and (ii) the increase of the dihedral angle between the donor and acceptor fragment due to the large size of bromine atoms in the ortho position, leading to further HOMO–LUMO separation and reduction of conjugation between molecular fragments. The important consequence of the ii factor is the decrease of oscillator strength fS0–1CT. This is not the case for 1Br and 2Br, as determined oscillator strength is almost equal or increases as compared to H (Table 1).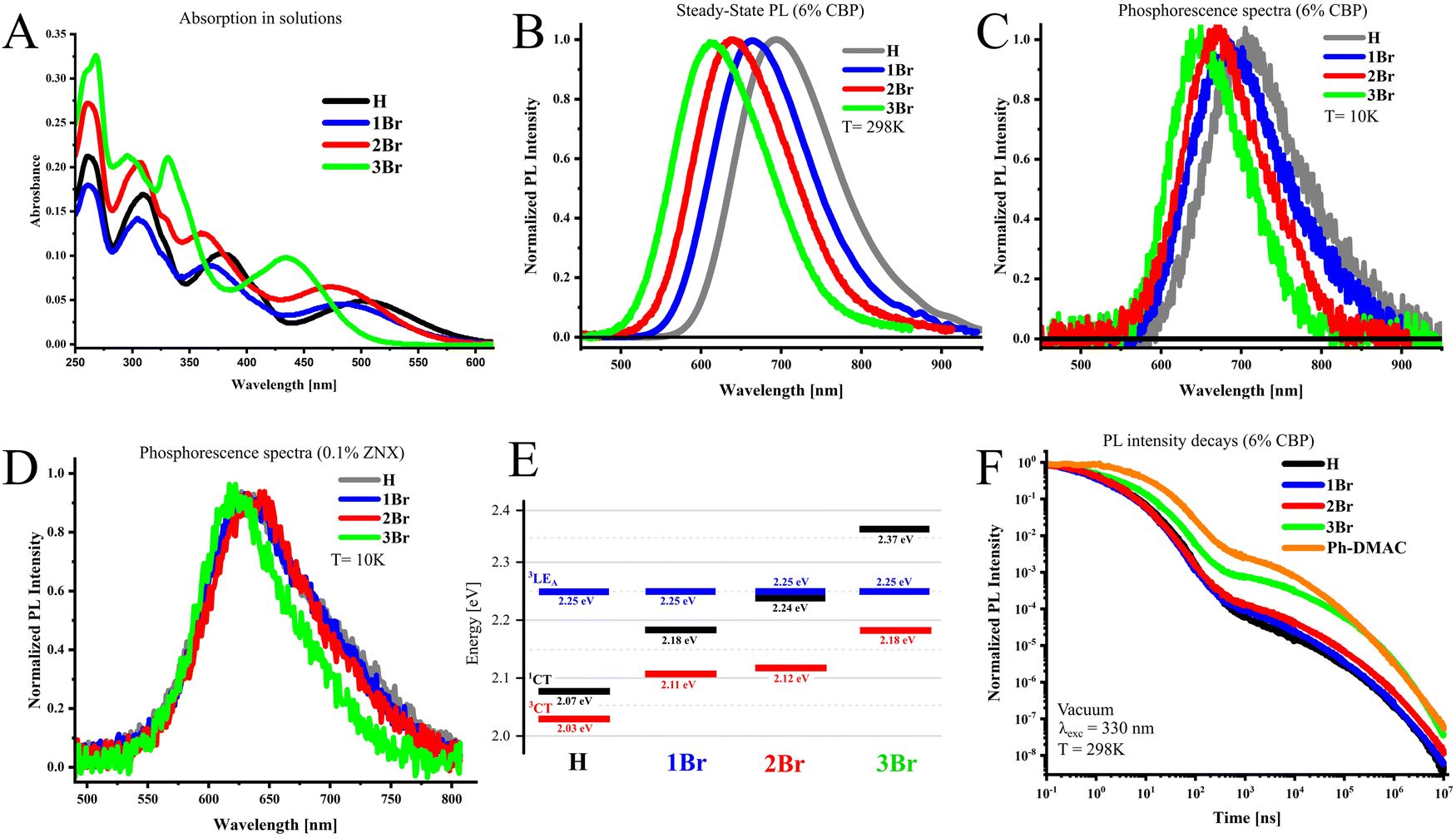 | ||
| Fig. 2 Absorption spectra in dichloromethane solutions (A); steady-state PL spectra of the investigated compounds in 6% CBP measured under excitation wavelength λexc = 330 nm (B); phosphorescence spectra in 6% CBP (T = 10 K, 20 ms delay after excitation pulse) (C); phosphorescence spectra in 0.1% ZNX (T = 10 K, 20 ms delay) (D); alignment of excited states in respective compounds determined from steady-state PL and phosphorescence onsets (E); and PL intensity decays of the investigated compounds in 6% CBP (F). | ||
| Cmpd | λ abs | f S0-1CT | |
|---|---|---|---|
| [nm] | Experimental | Calculateda | |
| a Calculated fS0-1CT is a statistical mean value for various rotamers discussed further; calculated by taking into account the probability of each rotamer predicted by Boltzmann distribution at 298 K. | |||
| H | 502 | 0.214 | 0.107 |
| 1Br | 483 | 0.206 | 0.131 |
| 2Br | 474 | 0.289 | 0.216 |
| 3Br | 435 | 0.450 | 0.118 |
This indicates that the electron-withdrawing feature of bromine has a key influence on the CT absorption band. Compound 3Br exhibits the highest blue shift together with an imaginary increase of the f value. The latter is explained by a strong overlap of the CT absorption with the next band near 400 nm, which disables experimental determination of fS0–1CT. As discussed further, DFT calculations confirm close fS0–1CT values for all compounds.
Photoluminescence spectra and alignment of excited states
The photoluminescence (PL) features of emitters were investigated in the amorphous film media, namely, 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP), widely used as a host material for OLEDs. In CBP films, the PL maximum of the investigated emitters shifts from red-NIR to orange-red with the increasing number of bromine atoms in the donor fragment (Fig. 2B, Table 2). In 6% w/w films, according to the PL onset values (Fig. S1A, ESI†), the 1CT state energy increases from 2.07 eV in H by 0.11 eV (1Br, 2.18 eV) and by 0.17 eV (2Br, 2.24 eV), when the first and second bromine atoms are introduced into the side phenyl rings (Table S1, ESI†). Similarly to absorption, the appearance of third bromine in the linker phenyl ring (3Br) causes the strongest 1CT-energy increase by 0.30 eV, reaching 2.37 eV. PL quantum yields vary from moderate high 61% in 3Br to almost absolute in 2Br. The phosphorescence spectra measured at 10 K with a 20 ms delay after the excitation pulse exhibit similar dependence, but the absolute shifts are smaller (Fig. 2C, Table 2). The energy of the 3CT-state derived from phosphorescence onsets (Fig. S1B, ESI†) increases thus by 0.06 eV (1Br), 0.08 eV (2Br), and 0.13 eV (3Br) compared to H. The described spectral measurements indicate that ΔEST increases from 0.04 eV in H to 0.08 eV (1Br) and 0.13 eV (2Br) and reaches maximal 0.20 eV for 3Br. At a higher doping concentration (10%), spectral behavior is generally similar (Fig. S1C and D, ESI†); however, PL spectra shift 5–10 nm to the red which results in a slight decrease of ΔEST by 10–30 meV (Table S1, ESI†).| w X/CBP (%) | PLQY | λ PL | λ Ph | ΔEST | τ PF | τ DF | k r | k ISC | k rISC | |
|---|---|---|---|---|---|---|---|---|---|---|
| [%] | [nm] | [nm] | [eV] | [ns] | [μs] | [107] s−1 | [107] s−1 | [104] s−1 | ||
| H | 6 | 83 | 692 | 706 | 0.04 | 16.7 | 646 | 3.2 | 2.1 | 0.25 |
| 10 | 51 | 704 | 709 | 0.03 | 18.8 | 356 | 2.7 | 2.0 | 0.50 | |
| 1Br | 6 | 76 | 665 | 684 | 0.08 | 15.5 | 422 | 2.6 | 3.0 | 0.48 |
| 10 | 77 | 671 | 690 | 0.06 | 15.2 | 296 | 2.9 | 2.9 | 0.60 | |
| 2Br | 6 | 98 | 641 | 669 | 0.13 | 14.8 | 781 | 1.6 | 5.5 | 0.50 |
| 10 | 97 | 647 | 672 | 0.10 | 15.0 | 442 | 2.1 | 3.9 | 0.50 | |
| 3Br | 6 | 61 | 602 | 648 | 0.20 | 27.2 | 403 | 0.51 | 5.0 | 1.9 |
| 10 | 71 | 606 | 633 | 0.19 | 23.3 | 267 | 0.58 | 3.5 | 2.0 | |
| Ph-DMAC | 6 | 71 | 600 | 601 | 0.09 | 39.4 | 272 | 0.39 | 2.0 | 1.7 |
To estimate the energy of the higher triplet state, phosphorescence measurements were performed in nonpolar Zeonex® (ZNX) polymer, where the CT states are destabilized. In ZNX films, the phosphorescence onset is almost identical for all emitters and provides the energy of a triplet state near 2.25 eV (Fig. 2D and Fig. S2, ESI†). Under such conditions, phosphorescence most likely occurs from the triplet state with a dominating locally-excited character (3LE). As substitution in the donor does not affect its energy, such a 3LE-state is localized on the acceptor fragment identical to all emitters, which is supported by the phosphorescence spectra of the acceptor molecule (Fig. S2, ESI†). Thus estimated alignment of the lowest excited states is summarized in Fig. 2E, and will be further used for the discussion of photophysical properties.
PL decays and rates of photophysical processes
The analysis of time-resolved emission spectra and PL decay curves evidences that all emitters dispersed in the CBP host exhibit TADF (Fig. 2F and Fig. S3A–D, ESI†). In the region of prompt fluorescence (PF), H and emitters with bromines in the side rings of the DTA donor (1Br and 2Br) fluoresce with a similar lifetime τPF close to 15 ns (Table 2). The kr values seem to depend on the doping ratio more than on the bromine contamination. At 6% and 10% doping with 1Br, kr slightly decrease and increase, respectively, by around 15%. In the case of 2Br in 6% films, kr decreases twice, but at a higher doping ratio, kr is only 20% lower as compared to H. In 3Br, PF lifetime increases to 27 ns, which is mainly caused by an almost fivefold kr decrease. 2Br and 3Br exhibit a ca. twice faster ISC rate than the rest of the emitters. This is the result of the energetic closeness of the 1CT and 3LE states (Fig. 2E), as the latter state plays an important role in ISC as was revealed previously.23,30As compared to H, the delayed fluorescence lifetime (τDF) of 1Br and 3Br shortens ca. 1.5-fold. This indicates that the presence of one and/or three heavy atoms in the TDA donor helps to convert triplet excitons to light more than 1.5 times faster. This becomes possible because of 2 and 8 times faster rISC, respectively, as for 6% CBP films. Importantly, in 3Br, rISC enhancement outranges the decrease of the kr value.
Surprisingly, the τDF value of 2Br is higher than that of H, because of the more efficient enhancement of ISC than rISC. As compared to 1Br, the introduction of a second Br atom has either negligible (6% doping) or even negative (10% doping) effects on the rISC rate (Fig. S4 (ESI†), Table 2). Apparently, in the investigated emitters, the HA-effect is not additive by the number of bromines, hence their relative position should play an important role. Such a peculiar rISC behavior is further explained with the help of quantum chemical calculations.
From the point of view of the geometry–property relationship, it is interesting to compare 3Br with Ph-DMAC (Fig. 1, Fig. S1E and F, ESI†). Due to the large size of three bromine atoms, the donor fragment of 3Br can have a more twisted structure than 1Br and 2Br, but a less rigid one than 9,9-dimethylacridan in Ph-DMAC. Under the same conditions, as compared to Ph-DMAC, 3Br exhibits an almost 1.5-fold higher kr value and a slightly faster rISC rate, whilst ΔEST is more than twice higher. Taking into account the exponential dependence of krISC on −ΔEST from the Marcus–Hush formula (eqn (S1), ESI†) this indicates ca. a hundred times higher SOC in 3Br. This is an important conclusion for further design of 3Br analogues with lower ΔEST towards fast rISC emitters.
Rotational isomerism and calculations of the rISC rates
Due to the large size of bromine at the ortho positions of benzene rings in the DTA donor, rotations of these rings have increased the energy barrier. This leads to various rotational isomers which can coexist at room temperature in a medium of low viscosity. In amorphous films, which are the media of interest for OLED emitters, high viscosity disables the rotations of large molecular fragments and thus such rotational isomers become individual photophysical species with restricted donor fragment geometries. In the investigated emitters there are three types of dihedral angles which provide rotational isomerism (Fig. 3). First is the dihedral angle θL between the plane of the dibenzo[a,c]phenazine acceptor and linker phenyl ring. In all the investigated rotamers, the optimal value θL is near +35° or –35° (Table 3), where the sign defines whether the donor fragment is above or beyond the acceptor plane (relatively to the molecular orientation depicted in Fig. 3). Two next dihedral angles between the linker phenyl ring and side rings A and B are depicted as θA and θB, respectively.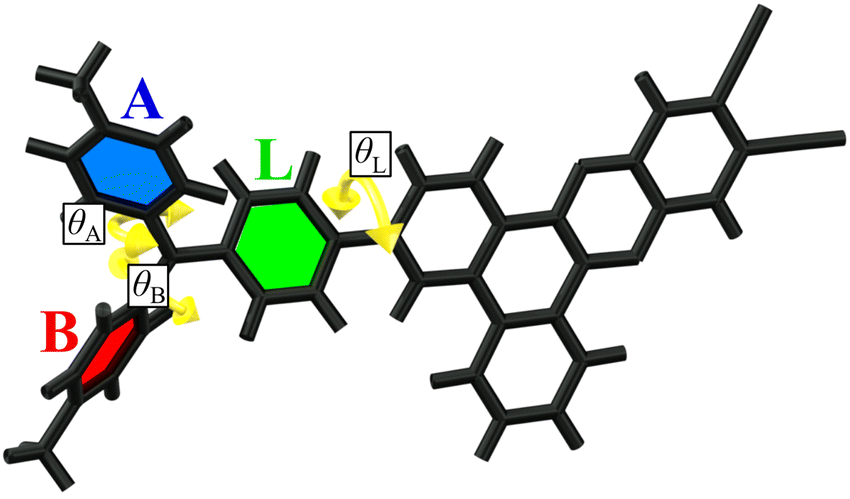 | ||
| Fig. 3 Structure of the investigated emitters with three types of dihedral angles (θA, θB and θL) highlighted which provide rotational isomerism. | ||
| Unit | H-1 | H-2 | 1Br-exo | 1Br-endo | 2Br-syn | 2Br-anti | 3Br-c3V | 3Br-A | 3Br-B | 3Br-C | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| θ L, θA, θB – dihedral angles between phenyl rings of the donor fragment (Fig. 3). ΔG(S1), ΔG(T1) – Gibbs free energies of S1 and T1 electronic states, respectively. S1–S0, T1–S0, T2–S0 – predicted energies of respective vertical transitions. f1CT-S0 – oscillator strength of the S1–S0 transition. SOCT1–S1, SOCT2–S1 – SOC constants for T1–S1 and T2–S1 transitions, respectively. ΔE3CT–1CT – energy gap between 3CT and 1CT states. λ3CT–1CT – reorganization energy. Ea – activation energy; χ3CT,χ3LE – relative population of 3CT and 3LE states at room temperature predicted by Boltzmann distribution (Section S2, ESI). | |||||||||||
| θ L | [°] | 27.9 | −28.8 | −23.1 | −22.5 | 17.8 | 17.6 | 19.0 | 19.5 | 17.8 | 18.1 |
| θ A | [°] | −64.6 | −65.1 | −69.1 | −74.9 | −63.5 | −68.5 | 76.6 | 76.5 | −77.5 | −71.0 |
| θ B | [°] | −64.8 | −65.2 | 55.3 | −74.9 | 77.3 | −68.4 | −78.0 | 61.7 | −73.6 | 61.4 |
| ΔG(S1) | [a.u.] | −1889.647927 | −1889.647680 | −1902.208629 | −1902.206972 | −1914.764446 | −1914.765059 | −1927.32269 | −1927.324124 | −1927.322466 | −1927.321232 |
| ΔG(T1) | [a.u.] | −1889.650547 | −1889.649878 | −1902.211800 | −1902.211480 | −1914.773697 | −1914.774187 | −1927.33009 | −1927.328483 | −1927.330665 | −1927.331147 |
| S1–S0 | [eV] | 1.38 | 1.37 | 1.58 | 1.59 | 1.74 | 1.77 | 1.84 | 1.77 | 1.85 | 1.80 |
| S1–S0 | [nm] | 899 | 907 | 787 | 779 | 713 | 702 | 674 | 702 | 670 | 687 |
| f CT-S0 | 0.228 | 0.209 | 0.331 | 0.335 | 0.438 | 0.461 | 0.407 | 0.340 | 0.450 | 0.39 | |
| T1–S0 | [eV] | 1.26 | 1.26 | 1.38 | 1.38 | 1.45 | 1.43 | 1.55 | 1.55 | 1.53 | 1.54 |
| T1–S0 | [nm] | 980 | 984 | 896 | 892 | 854 | 864 | 797 | 800 | 810 | 805 |
| T2–S0 | [eV] | 1.92 | 1.91 | 1.96 | 1.96 | 2.01 | 2.03 | 2.00 | 1.96 | 2.02 | 1.99 |
| T2–S0 | [nm] | 645 | 646 | 632 | 631 | 616 | 610 | 619 | 630 | 613 | 622 |
| SOCT1–S1 | [cm−1] | 0.06 | 0.08 | 0.32 | 0.44 | 0.58 | 0.04 | 0.58 | 0.88 | 2.3 | 3.45 |
| SOCT2–S1 | [cm−1] | 0.23 | 0.17 | 0.58 | 0.74 | 0.40 | 0.13 | 0.39 | 0.65 | 0.87 | 3.08 |
| ΔE3CT-1CT | [eV] | 0.03 | 0.03 | 0.11 | 0.12 | 0.10 | 0.15 | 0.20 | 0.14 | 0.24 | 0.18 |
| λ 3CT–1CT | [eV] | 0.34 | 0.33 | 0.38 | 0.38 | 0.39 | 0.39 | 0.41 | 0.41 | 0.41 | 0.41 |
| E a | [eV] | 0.18 | 0.17 | 0.23 | 0.23 | 0.23 | 0.25 | 0.29 | 0.25 | 0.32 | 0.28 |
| k 3CT–1CT·χ3CT | [104 s−1] | 0.16 | 0.30 | 0.56 | 0.80 | 1.8 | 2.8 × 10−3 | 0.14 | 1.54 | 0.93 | 8.0 |
| k 3LE–1CT·χ3LE | [104 s−1] | 0.032 | 9 × 10−3 | 1 × 10−3 | 1 × 10−3 | 1 × 10−3 | 1 × 10−3 | <1 × 10−3 | <1 × 10−3 | <1 × 10−3 | 3 × 10−3 |
Due to a large number of structural and electronic parameters, and very poor knowledge of the mechanism of their influence on spin–flip transitions, geometry optimizations and calculations of excited state properties were performed for all possible rotamers. The analysis of electronic properties included radiative deactivation 1CT–S0 and the parameters of 3CT→1CT transition: SOC constants, ΔEST, activation energy (Ea), which takes into account the internal reorganization energy, and finally calculation of the 3CT→1CT rate constants (k3CT–1CT) using the Marcus–Hush equation ((S1), ESI†). To obtain statistically weighted values of f1CT-S0 and k3CT-1CT, the respective values of all rotamers were added taking into account the contribution of each rotamer provided by relative energies of rotamers and Boltzmann distribution law (S2) (ESI†). As was reported previously, in donor–acceptor type TADF emitters including their HA-derivatives, the rotational30 and vibrational23 isomerism has key importance for efficient rISC via the 3CT→1CT pathway.
According to the experimental data, the kr values show complex behavior depending on the number of bromine atoms and the emitter/CBP doping ratio indicating the specific role of intermolecular interactions in amorphous films (Table 2). Regarding the aim of this study, the important observation is that H, 1Br, and 2Br show rather similar kr values in the 2–3 × 107 s−1 range. Neither statistically weighted calculated f1CT-S0 values (Table 1) nor f1CT-S0 of individual rotamers (Fig. S5, S6, Table S2, ESI†) show a substantial decrease with the growing number of bromines in the emitter.
The statistically weighted k3CT–1CT correlate very well with the experimental krISC values (Fig. 4), which confirms that various rotamers are responsible for the photophysical behavior of emitters in films. This also enables the analysis of 3CT→1CT transition parameters in key rotamers as discussed below. We should note that the contribution of the second triplet state in rISC was found to be negligible even in 3Br, where the 3LE-state is relatively close to the 3CT one (Fig. 2E). The statistically weighted k3LE–1CT values do not exceed 1 × 103 s−1, which is only 5% of the k3CT–1CT one (Fig. 4 and Section S2, ESI†). In other compounds, the 3LE→1CT transition is slower. One can conclude that such a slow rISC pathway can only compete in rotamers with the slowest 3CT→1CT transition such as 2Br-anti ones (Table 3), but even in the latter case, k3LE–1CT is three times lower than k3CT–1CT.
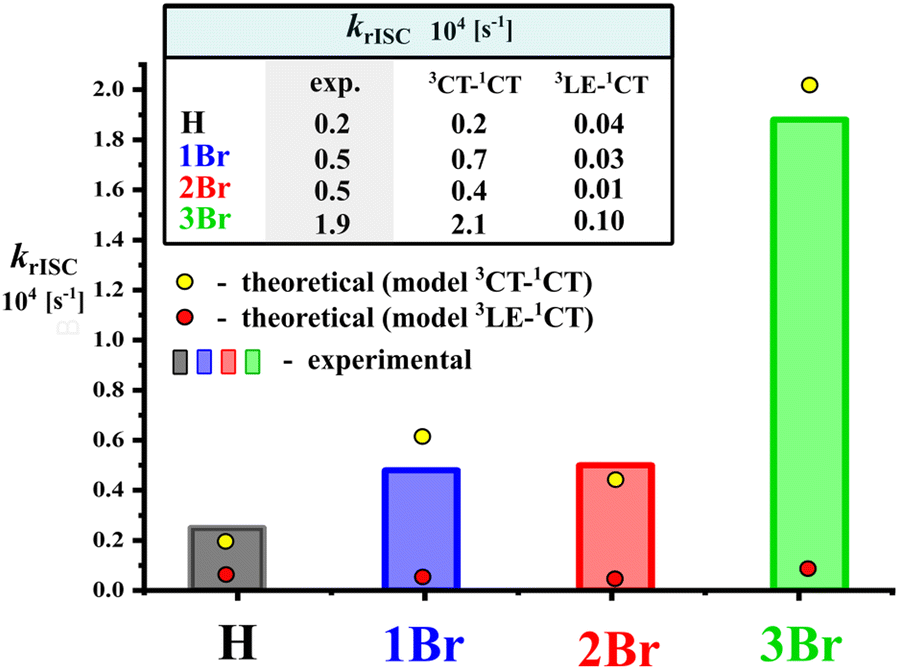 | ||
| Fig. 4 Comparison of experimental krISC with calculated statistically weighted k3CT–1CT and k3LE–1CT. | ||
In H, two rotamers with different θL (H-1 and H-2 in Table 3, Table S2, Fig. S5 and S7, ESI†) have different SOC constants: 0.06 and 0.08 cm−1, in spite of the absence of HAs and minor differences in the electronic parameters including ΔEST and Ea (Fig. S5, ESI†). Different positions of linker phenyl provide different orientations of donor versus acceptor; in the rotamer H-2 with θL = –29°, the donor fragment is more twisted versus the acceptor, providing larger change in the orbital moment ΔL during the 3CT→1CT transition (Fig. S7, ESI†). One should note that the change of orbital moment is typically small as for transition between states of very close nature. In spite of minor changes in ΔL and SOC, the calculated k3CT–1CT values of H rotamers differ by 2 times.
In 1Br, the highly asymmetric donor fragment enables 16 rotamers. The calculated f1CT-S0 values are very similar for all rotamers (Fig. S5, ESI†). As compared to H, the calculated ΔEST values are increased more than 3 times, and for all rotamers of 1Br they exceed 100 meV. The presence of a heavy atom causes more than a fivefold SOC constant rise up to 0.31–0.45 cm−1, depending on the rotamer structure. The highest SOC is observed when the bromine atom in the donor fragment is oriented towards the acceptor (8 rotamers are attributed to 1Br-endo group in Fig. 5 and Fig. S5, ESI†), whilst the opposite direction provides the lowest value (8 rotamers, 1Br-exo). SOC rise is, however, partially compensated by the ΔEST increase, which provides more than 3 times rISC acceleration as compared to H. As neither experimental kr nor calculated f1CT-S0 values of 1Br are substantially lower than those of H, one can conclude that the introduction of one bromine atom has a positive effect on triplet harvesting in such emitters.
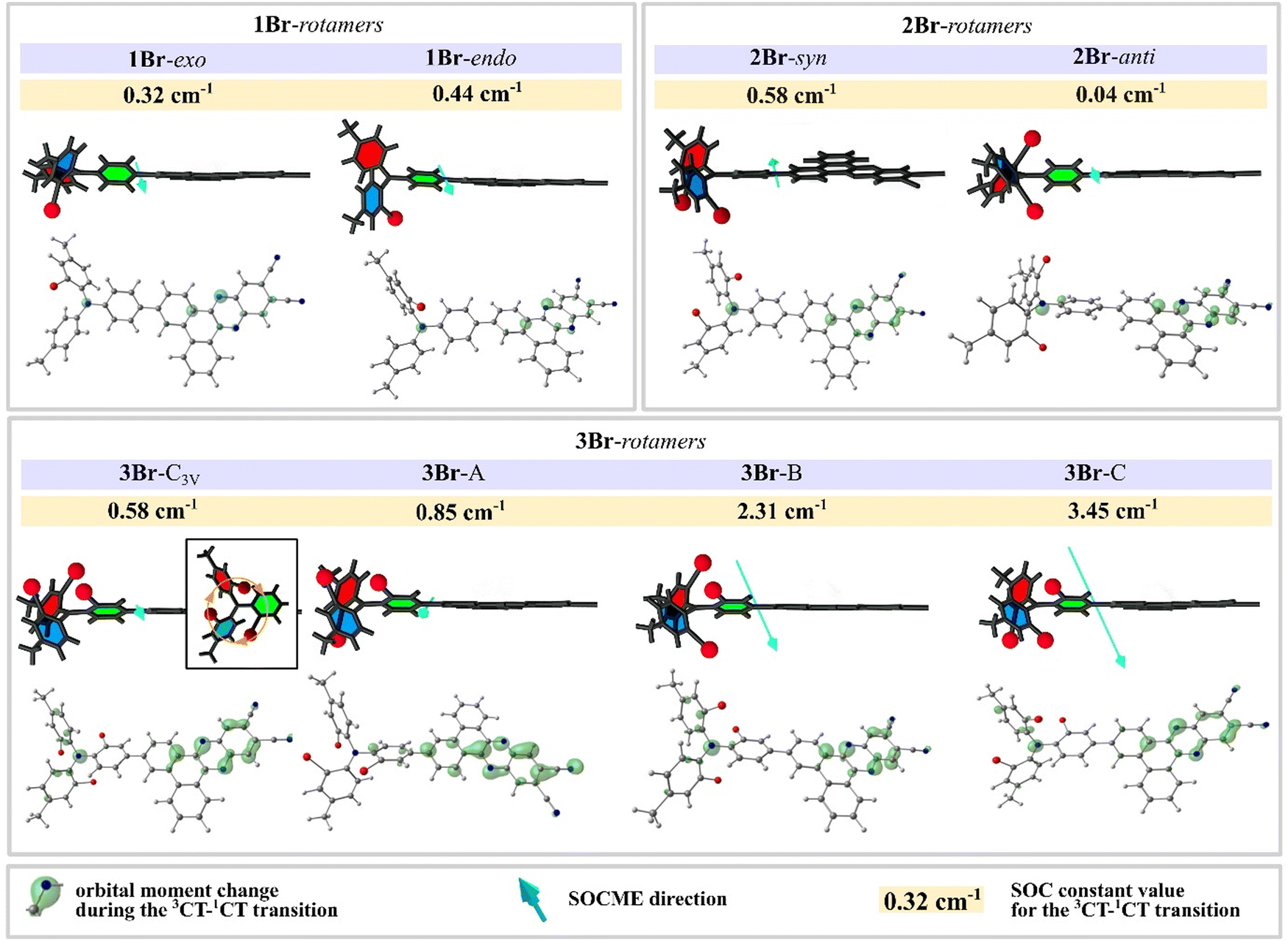 | ||
| Fig. 5 Optimized structures of key rotamers (see Fig. S5 and S6, ESI†) of 1Br, 2Br, and 3Br with SOCME value, SOCME direction (turquoise arrows), and ΔL (green clouds, contour value 0.02); illustrations show the simplified orientation of heavy atoms relative to the acceptor plane. | ||
Regarding the relative position of two bromine atoms, 16 rotamers of 2Br can be divided into two main groups: syn and anti. The expected strong heavy-atom effect is realized only in the syn rotamers with both bromines on the same side of the acceptor plane (Fig. 5 and Fig. S5 (ESI†), Table 3). In such HA configurations, the increased SOC constants reach 0.60 cm−1. In the anti rotamers, bromines on the different sides of the acceptor plane cause surprisingly low SOC constants, dropping below 0.05 cm−1, resembling those for an HA-free emitter H. In the result, the k3CT–1CT of the most efficient syn rotamer is 650 higher than that of the least efficient anti one. These observations indicate that the HA effect can be zeroed when heavy atoms are situated in a symmetric way or at the opposite sides of the donor fragment.
3Br exhibits the most complex isomerism. Regarding the relative position of three bromine atoms, its 32 rotamers can be divided into four groups. The rotamers in which bromines and the nitrogen atom form a symmetric pyramid-like structure of C3V symmetry (3Br-c3V in Fig. 5) exhibit the lowest SOC values below 0.70 cm−1 (Fig. S6 (ESI†), Table 3 and Table S2, ESI†). When such a symmetry is broken by the rotation of A or B rings so that side bromines occur in the anti configuration (3Br-A on Fig. 5 and Fig. S6, ESI†), the SOC increases slightly up to 0.9 cm−1. As an exception, another group of anti rotamers with bromine atoms of the side rings oriented towards the acceptor fragment exhibit a high SOC reaching 2.40 cm−1 (3Br-B). In such rotamers, in contrast to anti rotamers of 2Br, the symmetry of the anti configuration is broken by the bromine in the linker phenyl of 3Br. The most impressive SOC values reaching 3.50 cm−1 are observed in the case when the C3V symmetry is broken via rotation of the linker phenyl ring, so that bromines in the side phenyl rings remain in the syn configuration (3Br-C). Such a dependence on the relative positions of bromine atoms supports the conclusion that the asymmetric configuration of HAs favors the maximal increase of SOC. From the point of view of rISC efficiency, a negative factor of all 3Br rotamers is high ΔEST which ranges from 140 to 240 meV. Remarkably, in spite of such a high ΔEST value of 180 meV, high SOC in the most efficient rotamers 3Br-C enables k3CT–1CT above 8 × 104 s−1 outranging more than 25 times that in H.
The increase of SOC within an isolated atom or molecular system is usually explained by the increase of the orbital and/or spin transition moment, because these two quantum values define the change of the total angular momentum and thus the spin–orbit coupling.6 Thorough analysis of electronic parameters led us to the conclusion that, regarding total angular momentum, the 3CT→1CT transition in various rotamers bearing heavy atom(s) remains very similar. None of the calculated electronic parameters by itself can explain the drastic change of SOC and rISC rates. Neither calculated changes in the NTO orbitals of 3CT→1CT transitions nor partial contributions of electronic densities on the bromine atom below (Fig. S8 and S9, ESI†) reveal a substantial change in the orbital transition moment. Triplet spin density distribution (TSDD, Fig. S10, ESI†) maps remain similar and the contribution of bromine atom(s) is negligible too, which indicates that the change of spin transition moment is very similar for all isomers. Therefore, the observed heavy-atom effect cannot be regarded as the internal one. On the other hand, the notion “external heavy-atom effect” cannot be applied either, as heavy atoms are covalently bonded to the chromophore fragment.
In spite of the absence of direct influence of heavy atom(s) on the electronic spin–flip transition, their presence and orientation change immensely the SOC matrix elements and thus the direction and value of total angular momentum. The observed phenomenon can be regarded as the heavy-atom field effect (HAFE). Such a field interacts with the electron density during the electronic transition enhancing the change of spin. The peculiarity of the observed HAFE phenomenon, namely the inactivity of HAs in either 1CT-S0 or 3CT→1CT transitions as one can conclude on the basis of NTO analysis (Fig. S8, ESI†) can be explained by the electron-withdrawing effect of bromine which disables its participation in the charge-transfer transition in the role of the donor.
Most likely, the HAFE has a direction defined by the orientation of heavy atoms. In fact, the analysis of the SOC matrix elements within the geometry coordinates of each rotamer (Fig. 5) indicates that maximal strengths of the HAFE and thus the highest SOC are achieved in two cases. The first one with the most substantial growth of SOC matrix elements is observed when HAs’ relative alignment is asymmetrical (compare 3Br-c3v and other rotamers of 3Br, Fig. 5) and the field of one heavy atom cannot be compensated by another one (compare syn and anti2Br rotamers). The second case is when heavy atoms are orientated towards the change of electronic density during the 3CT→1CT transition (compare endo with exo1Br rotamers, and 2Br-syn with 2Br-anti, Fig. 5). In fact, the symmetric orientation of bromines in 3Br-C3V and especially 2Br-anti rotamers results in self-compensation or even zeroing of the HA effect.
To the best of our knowledge, neither observation of the HAFE nor the possibility of self-compensation of the HA effect was reported before. The latter peculiarity is not intuitive for materials with more than one HA, and can lead to an imaginary lack of the HA effect or even the opposite result of SOC decrease. The possibility of formation of rotamers with zero HAFE during synthesis or sample preparation evidences another challenge for hybrid material engineers to analyze and eliminate such species at the stage of molecular design planning to achieve efficient rISC.
Conclusions
The results of the above discussed experimental and computational investigations evidence that hybrid organic materials bearing abundant heavy atoms in fact can solve the dilemma of all-organic TADF materials given by eqn 1. In the investigated example, selective acceleration of the 3CT→1CT rISC pathway without a drastic decrease of the S1–S0 oscillator strength is possible in a partially conjugated D–A type emitter, when heavy atoms do not self-compensate the created field and are oriented towards the fragment transition orbital moment. The most representative examples are various rotamers of 2Br and 3Br with similar oscillator strength values but very different SOC and rISC rates (Fig. S5 and S6, ESI†). This is a clear evidence that in contrast to pure organic emitters such hybrid molecular systems disobey the inverse relationship of f1CT-S0 and krISC. This is supported by the following:(1) the f1CT-S0 values measured for absorption as well as calculated ones, which indicate that bromine in the role of a heavy atom does not decrease the rate of radiative deactivation via S1–S0 transition;
(2) photophysical features of 1Br in 10% ww. film, which in comparison to H has twice higher krISC and only 1.2 times smaller kr;
(3) different rotamers of 2Br and 3Br, which have extremely different krISC rates, but close f1CT-S0 ones.
On the one hand, the applied bromine is a good choice for the role of a heavy atom as it can be easily introduced by common and powerful synthetic methods. However, its pronounced electron-withdrawing influence on the donor fragment reduces the charge-transfer strength leading to the blue-shift of TADF and increase of ΔEST. Both these factors are undesired for red emitters, whilst the latter one is negative for all TADF materials in general. Nevertheless, thanks to high SOC, in 3Br with a 200 meV energy gap almost disabling TADF, the rISC rate is the highest one. Another negative factor of the increase of 1CT energy especially in 2Br and 3Br is the decrease of the 1CT→3LE energy gap. This leads to the acceleration of ISC, and thus faster conversion of singlet states to triplet ones.
The analysis of various rotational isomers of the investigated emitters evidences that further improvement of hybrid organic emitters with the HAFE can be probably achieved via the stereoselective control of the position of HAs in the molecular structure. In the example of 3Br, selective synthesis or isolation of 3Br-C rotamers could provide a further fourfold rISC enhancement without the decrease of the fluorescence rate, making such an emitter more attractive commercially.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support within the LIDER XI grant LIDER/47/0190/L-11/19/NCBR/2020 (M. M., D. G., E. H., K. K., and I. E. S) and CHEMFIZ program WND-POWR.03.02.00-00-I059/16 (M. M.) of National Centre for Research and Development (NCBR), Poland is gratefully acknowledged. Quantum chemical calculations were performed on the computers of the Wroclaw Centre for Networking and Supercomputing (WCSS), Poland.References
- S. H. Mir, L. A. Nagahara, T. Thundat, P. Mokarian-Tabari, H. Furukawa and A. Khosla, J. Electrochem. Soc., 2018, 165, 3137–3156 CrossRef.
- V. Trifiletti, C. Asker, G. Tseberlidis, S. Riva, K. Zhao, W. Tang, S. Binetti and O. Fenwic, Front. Electron., 2021, 2, 758603 CrossRef.
- T. Hua, L. Zhan, N. Li, Z. Huang, X. Cao, Z. Xiao, S. Gong, C. Zhou, C. Zhong and C. Yang, Chem. Eng. J., 2021, 426, 131169 CrossRef CAS.
- Y. Xiang, Y. Zhao, N. Xu, S. Gong, F. Ni, K. Wu, J. Luo, G. Xie, Z.-H. Lu and C. Yang, J. Mater. Chem. C, 2017, 5, 12204–12210 RSC.
- E. Longhi and L. De Cola, Iridium(III) Complexes for OLED Application, Iridium(III) in Optoelectronic and Photonics Applications, John Wiley & Sons, Ltd, Chichester, UK, 2017, p. 205 Search PubMed.
- W. Shao, H. Jiang, R. Ansari, P. M. Zimmerman and J. Kim, Chem. Sci., 2022, 13, 789–797 RSC.
- M. Godumala, A. V. Kumar and R. Chandrasekar, J. Mater. Chem. C, 2021, 9, 14115–14132 RSC.
- H. F. Higginbotham, M. Okazaki, P. de Silva, S. Minakata, Y. Takeda and P. Data, ACS Appl. Mater. Interfaces, 2021, 13, 2899–2907 CrossRef CAS PubMed.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed.
- A. Lv, W. Ye, X. Jiang, N. Gan, H. Shi, W. Yao, H. Ma, Z. An and W. Huang, J. Phys. Chem. Lett., 2019, 10, 1037–1042 CrossRef CAS PubMed.
- Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X. Yin, Y. He, X. Wang, Z. Wu, E. Pang, J. Xu and J. Wang, Front. Chem., 2020, 8, 725 CrossRef CAS PubMed.
- T. Huang, X. Song, M. Cai, D. Zhang and L. Duan, Mater. Today Energy, 2021, 21, 100705 CrossRef CAS.
- Y. Ren, Y. Wada, K. Suzuki, Y. Kusakabe, J. Geldsetzer and H. Kaji, Appl. Phys. Express, 2021, 14, 071003 CrossRef CAS.
- D. Song, Y. Yu, L. Yue, D. Zhong, Y. Zhang, X. Yang, Y. Sun, G. Zhou and Z. Wu, J. Mater. Chem. C, 2019, 7, 11953 RSC.
- T. Hua, L. Zhan, N. Li, Z. Huang, X. Cao, Z. Xiao, S. Gong, C. Zhou, C. Zhong and C. Yang, Chem. Eng. Sci., 2021, 426, 131169 CrossRef CAS.
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- Y. Zhang and S. R. Forrest, Phys. Rev. Lett., 2012, 108, 267404 CrossRef PubMed.
- R. Furue, K. Matsuo, Y. Ashikari, H. Ooka, N. Amanokura and T. Yasuda, Adv. Opt. Mater., 2018, 6, 1701147 CrossRef.
- Y.-L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hanisch, S. Reineke, J. Fan and L.-S. Liao, Adv. Mater., 2019, 31, 1902368 CrossRef CAS PubMed.
- J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 6, 1800255 CrossRef.
- M. Mońka, I. E. Serdiuk, K. Kozakiewicz, E. Hoffman, J. Szumilas, A. Kubicki, S. Y. Park and P. Bojarski, J. Mater. Chem. C, 2022, 10, 7925–7934 RSC.
- R. D. Burkhart and N. I. John, J. Phys. Chem., 1991, 95, 7189–7196 CrossRef CAS.
- U. Balijapalli, Y.-T. Lee, B. S. B. Karunathilaka, G. Tumen-Ulzii, M. Auffray, Y. Tsuchiya, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 19364–19373 CrossRef CAS PubMed.
- A. A. Kubicki, P. Bojarski, M. Grinberg, M. Sadownik and B. Kukliński, Opt. Commun., 2006, 269, 275–280 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- I. E. Serdiuk, M. Mońka, K. Kozakiewicz, B. Liberek, P. Bojarski and S. Y. Park, J. Phys. Chem. B, 2021, 125, 2696–2706 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: additional results of photophysical investigations, computationally predicted electronic parameters and calculated rISC rate constants for various rotamers, NMR spectra. See DOI: https://doi.org/10.1039/d2tc01729f |
| This journal is © The Royal Society of Chemistry 2022 |