
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of composition engineering on charge carrier cooling in hybrid perovskites: computational insights†
Dibyajyoti
Ghosh
 *ab,
Carlos Mora
Perez
c,
Oleg
Prezhdo
c,
Wanyi
Nie
b,
Sergei
Tretiak
bd and
Amanda J.
Neukirch
*d
*ab,
Carlos Mora
Perez
c,
Oleg
Prezhdo
c,
Wanyi
Nie
b,
Sergei
Tretiak
bd and
Amanda J.
Neukirch
*d
aDept. Materials Science and Engineering (DMSE) and Dept. Chemistry Indian Institute of Technology, Delhi, Hauz Khas, New Delhi-110016, India. E-mail: dibyajyoti@iitd.ac.in
bCenter for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
cDepartment of Chemistry, University of Southern California, Los Angeles, California 90089, USA
dTheoretical Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: ajneukirch@lanl.gov
First published on 26th May 2022
Abstract
Mixed A-cation halide perovskites have emerged as one of the most promising materials for next-generation optoelectronic applications due to such factors as attractive charge carrier transport properties and enhanced stability under operating conditions. However, the influence of A-cation mixing on the excited state charge carrier dynamics and, particularly, on ultrafast hot-carrier relaxation processes, is yet to be studied in sufficient detail. We combine nonadiabatic molecular dynamics and time-domain density functional theory methods to establish the impact of formamidinium (FA)–cesium (Cs) mixing on the subpicosecond-scale hot-charge carrier cooling processes in FA1−xCsxPbI3 (x ≤ 0.5) materials. Our ab initio study illustrates that the partial substitution of organic FA species with inorganic Cs cations substantially extends the hot-electron and hot-hole relaxation times. Observed increases in the hot-carrier lifetimes indicate better performance of FA1−xCsxPbI3 compared to parent FAPbI3 in the field of hot carrier solar cells. The atomistic details of lattice dynamics reveal that FA–Cs cation mixing partially suppresses thermal fluctuations in the structure, weakening the carrier–phonon interaction under ambient conditions. Increased structural rigidity and weakened carrier–phonon interactions in turn lower the rates of intraband nonadiabatic transitions of hot-carriers and enhance their excited state lifetimes. The in-depth understanding of the relationship between the dynamic structure and carrier relaxation allows us to further propose rational design principles that can enhance the hot-carrier lifetimes in photoactive materials. The computational guides will help to realize photovoltaic devices that efficiently harvest hot-carriers and exhibit an improved power conversion performance compared to traditional single-junction solar cells.
Hot carrier solar cells (HCSCs) are attracting considerable attention due to their theoretical power conversion efficiency (PCE) as high as 66%, being much larger than the theoretical maximal PCE for traditional single-junction technologies.1–5 The proposed HCSCs efficiently extract the hot charge carriers that are generated from the high-energy incident photons. However, the efficient extraction of carriers with excess energy in semiconductors is a challenging task.6 The energy dissipation through intraband relaxation of hot electrons is generally a much faster process than their extraction rate. Suitable absorber materials for HCSCs have a slow cooling rate that allows the extraction of hot carriers through appropriate energy selective contacts. Recently, hybrid organic–inorganic halide perovskites have emerged as a promising class of materials for optoelectronics.7–14 In particular, elongated picosecond carrier cooling time, along with a long hot carrier diffusion length make these hybrid materials suitable for HCSC devices.15–20 The presence of a hot phonon bottleneck in these materials can significantly extend the carrier cooling time, up to several hundreds of picoseconds.19–22 Compared to traditional semiconductors, the carrier cooling mechanisms are substantially different in hybrid perovskites, primarily due to their highly dynamic ionic lattices, which give rise to strong coupling between photo-excited electrons and phonon modes.23,24 The chemical formula of three-dimensional hybrid halide perovskites is ABX3, where X is a halogen (iodine, bromine, or chlorine), B is a divalent metal such as lead, tin, and germanium, and A is an organic (methylammonium (MA) or formamidinium (FA)) or inorganic (cesium, Cs) monocationic atom.
Despite interesting initial reports, significant improvements are still required to realize halide perovskite-based HCSCs that take advantage of long-lived high carrier dynamics. In this regard, strategic compositional engineering has proven to be a powerful tool to fine-tune the hot carrier cooling rate in halide perovskites.25–27 Controlled mixing of halides (that are iodide and bromide) in MAPbBr3−xIx can modify the hot carrier lifetime, where iodide-based perovskites are suggested to be the best candidates.24,28 The broadened conduction band and increased electron–phonon interactions in bromide incorporated perovskites result in faster hot carrier cooling. Verma et al. further demonstrated that the addition of Sn in MAPb1−ySnyI3 slows down the cooling dynamics due to tuned carrier–carrier scattering rates and the widened gap between the optical and acoustic phonon branches.29 The critical role of A-cations in carrier cooling has also been studied extensively. In separate studies, Chen et al. and Hopper et al. found that the all-inorganic perovskite CsPbBr3 has a much slower hot carrier cooling rate than that in the hybrid bromide counterparts.30,31 This trend remains unchanged for nanocrystals and spin-coated films of APbBr3. A computational investigation by Madjet et al. further supports this A-cation dependent hot-carrier cooling but considered iodide perovskites.32 Despite superior characteristics in terms of hot carrier lifetime, all-inorganic halide perovskites suffer from structural instability due to spontaneous phase-transition under ambient conditions.33 Compositional engineering by mixing different A cations such as FA, MA, and Cs is a proven approach to enhance the phase-stability and thermostability of halide perovskites without compromising the power efficiency.8,10,34–36 Furthermore, Wang et al. have recently used ultrafast transient absorption spectroscopy to illustrate that incorporation of inorganic cations in hybrid halide perovskites enhances the hot carrier lifetime.25 However, the impact of A-cation mixing on hot carrier cooling has not been explored thoroughly yet.
Our previous work reports extensive studies of the structural fluctuations and their impact on the charge carrier dynamics in halide perovskites including carrier cooling processes.37–40 Here, we investigate the influence of cation mixing on the hot carrier dynamics in FA1−xCsxPbI3 (x = 0, 0.25, 0.375, 0.5). To obtain a realistic time-dependent description of excited-state dynamics, we employ a combination of ab initio time-domain density functional theory (TD-DFT) and nonadiabatic molecular dynamics (NAMD). According to our study, the hot carrier cooling rate depends on the phonon-mediated excited-state dynamics that can be fine-tuned through A-cation mixing in halide perovskites. Detailed atomic-scale lattice dynamics provide insights on the excited-state carrier-phonon interactions, underpinning the complex carrier cooling mechanisms in these hybrid materials. These key results provide a materials design strategy to suppress the hot carrier cooling, which may benefit high-efficiency HCSCs and plasmonic photocatalytic processes.41,42
Here, we consider lead iodide perovskites where A cations are either 100% FA or its mixture with the inorganic Cs cation (Fig. 1). The FA1−xCsxPbI3 lattices have been modeled through partial substitution of FA with Cs where x ≤ 0.5. Previous reports indicate that the high concentration of Cs in FAPbI3 results in the poor crystallinity and formation of FA and Cs rich phases in the bulk, limiting the homogeneous A-cation mixing.34,36,43,44 Such inhomogeneous distribution of A-cations also negatively impacts the overall optoelectronic properties of these materials.10 Thus, the present study mostly focuses on the experimentally realized FA-dominant mixed cation lattices. Note that we particularly include the FA0.5Cs0.5PbI3 lattice in our study to determine the impact of high Cs concentration on excited carrier dynamics. Single crystal MA0.5Cs0.5PbBr3 has been synthesized recently and likely a similar process can be applied to produce a FA–Cs mixed system as well.45,46
 | ||
| Fig. 1 The DFT optimized structures of (a) FAPbI3, (b) FA0.75Cs0.25PbI3, and (c) FA0.5Cs0.5PbI3. A-cations (FA or Cs) occupy the cuboctahedral cage of Pb/I. The PbI6 octahedra are shown in grey. Note that these are for illustration purposes, and are not the simulation cells used for the study. Key: brown: lead, iodine: purple; hydrogen: white; carbon: cyan; nitrogen: blue; cesium: orange. | ||
As the concentration of the Cs cation increases in FA1−xCsxPbI3, the lattice volume per formula unit reduces, indicating an enhanced internal chemical pressure in these materials (see Fig. S1, ESI†). This lattice contraction occurs as the large-sized FA+ (ionic radii 2.53 Å) molecules are partially replaced by the much smaller Cs+ (ionic radii 1.67 Å) in mixed A-cation lattices.7 Thus, although the A-cations occupy the cuboctahedral cage of the BX3 framework and interact with the inorganic lattice through non-covalent interactions, its size does impact the overall lattice volume of halide perovskites.
First, we investigate the impact of different A-cation compositions on the electron cooling rates within a dynamic electronic landscape. The large magnitude of transient structural distortions at ambient conditions in these halide perovskites leads to significant fluctuations in their electronic energy levels, making the carrier cooling processes challenging to track and investigate. To tackle these issues, here we use a NAMD-based simulation technique that computes the nonadiabatic transitions among the energetically closely-spaced states and provides valuable insights into the carrier relaxation mechanisms.25,47–49 To have a proper comparison of cooling rates, we simulate the electron excitation from the VBM to higher states in the CBs with comparable excess energy. The initial excess energy of the hot electron is considered to be ≈ 0.55 eV for the parent and mixed A-cation perovskites. With this excess energy, the highest state that becomes populated with hot electrons is CBM + 7 for FA1−xCsxPbI3. With time, this hot electron population cools down and accumulates at the CB edge. To understand the trend of electron relaxation, we track the increase of cooled electron population at the CB edge over the simulation time. As the conduction band minimum (CBM) and CBM + 1 remain close to each other energetically (average energy difference being less than 0.1 eV) over simulation time, we define the total cooled population as the sum of the populations of these two states (see Fig. S2, ESI†). Spectroscopic techniques like the transient-absorption and optical-pump THz-probe based tools track the increased cooled electron population at the band edges of semiconductors after the above band gap excitation.26,50–53 The faster the increase in the population at the conduction band edge, the quicker the electron cooling happens in that material. Fig. 2a depicts that the hot carrier cooling process prominently depends on the composition of A-cations in FA1−xCsxPbI3. Overall, the hot electrons cool down at a slower rate in mixed A-cation perovskites compared to that in the pristine FAPbI3 material. After 4 ps of the electron excitation, 59% of the hot electron population cools down to the CB edge through the intraband relaxation process in FAPbI3. However, within the same time window only 40% of the hot electron population reaches the CB edge in FA0.5Cs0.5PbI3, indicating that most of the hot electronic population remains at higher energetic electronic states (Fig. 2a). We further track the change in excited electron population over time as plotted in Fig. 2b. This demonstrates the rapid depopulation of the most excited state and concomitant accumulation of partially relaxed electrons at relatively low-energy states at and near the conduction band edge. In these simulations, the most excited energy level that is the CBM + 7 state loses 75–62% of its initial population within 500 fs for all hybrid halide perovskites considered here. These results agree well with recent experimental reports suggesting a longer hot-carrier lifetime for FA–Cs mixed halide perovskites compared to that for FAPbI3.25Fig. 2a and b also illustrate that the electron cooling rate can be tuned by controlled mixing of FA and Cs cations. By partially substituting FA cations with inorganic Cs, the rate of hot electron cooling reduces considerably. To quantitatively calculate the carrier cooling time, we fit the decay curves in Fig. 2b with a function that is the sum of Gaussian and exponential functions (see details in the Methods section).49 The calculated electron cooling times with different A-cation concentrations in the FA1−xCsxPbI3 lattice are presented in the bar chart displayed in Fig. 2c. The sub-picosecond timescale for electron cooling in these iodide perovskites is in agreement with the experimentally reported values for similar materials.15–18 More importantly, the increased cooling time with cation-mixing demonstrates the possibility to control the intraband relaxation of hot electrons through compositional engineering.
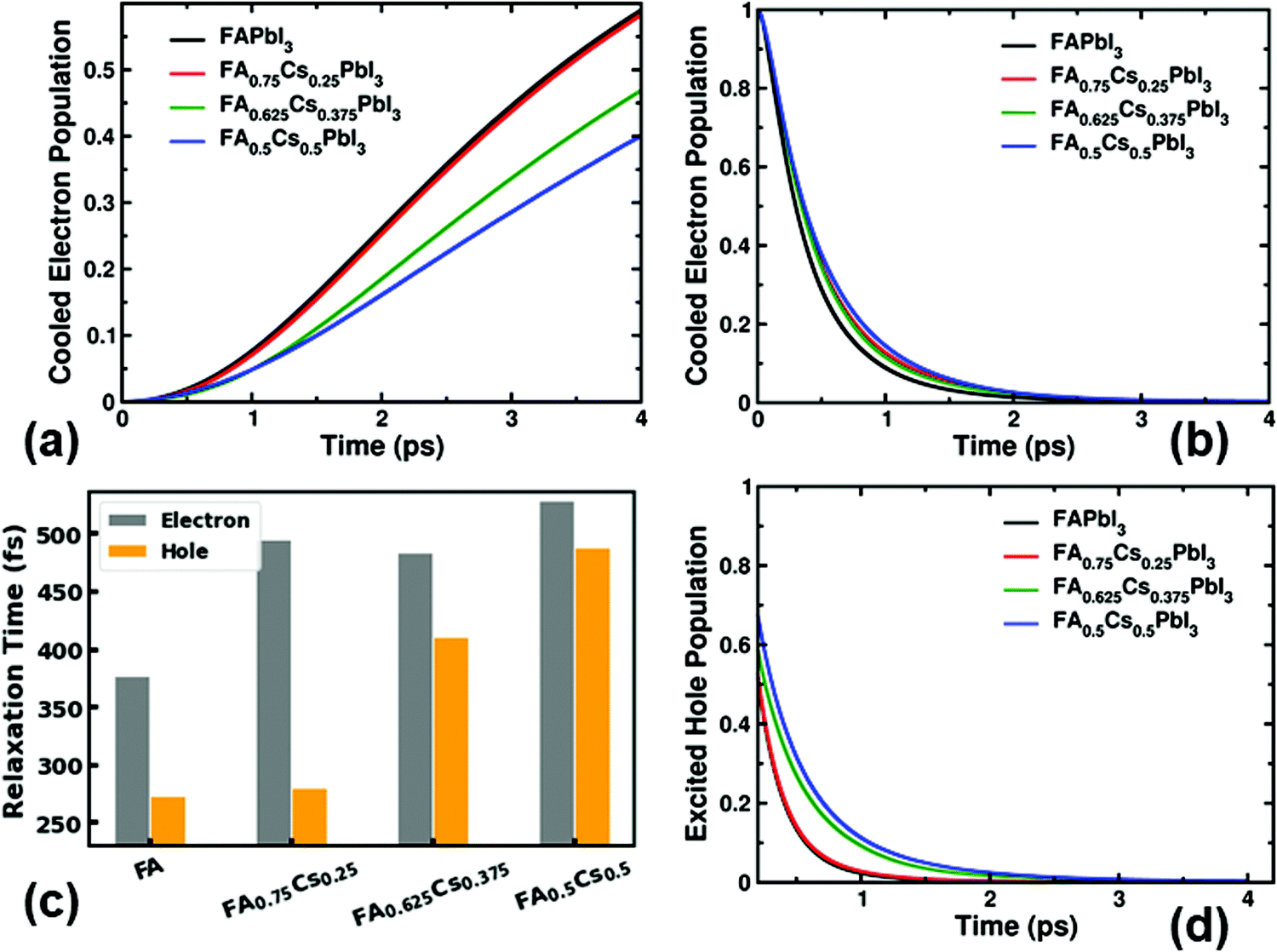 | ||
| Fig. 2 The impact of A-cation mixing on the carrier cooling rates in FA1−xCsxPbI3. (a) The increased population of conduction band edge states over time after the excitation. (b) The decay of electron population in the CBM + 7 state over time. (c) The electron and hole relaxation times for FA1−xCsxPbI3. (d) The population decay of the most excited hole state over time. The hot carrier relaxation processes can be tuned through A-cation mixing in lead iodide perovskites. | ||
We use a very similar computational framework to study the hot hole cooling process in these materials. An electron is excited from deep-states in the valence band to the CBM, leaving a hot hole in the VB. Like the electron, the hot hole initially (at t = 0 fs) remains in the VB state that is energetically below the VBM by ≈ 0.55 eV. We track the population decay of the hot hole in this VB state over the simulation time and evaluate the relaxation time for FA1−xCsxPbI3. The detailed time-resolved hot hole populations for all of the states involved are analyzed in Fig. S3 (ESI†). As shown in Fig. 2d, the hot hole relaxation time also substantially depends on the concentration of different A-cations in the lattice. Within all considered systems, FA0.5Cs0.5PbI3 has the longest relaxation time, which reduces with the concentration of Cs in FAPbI3. We conclude here that electron and hole cooling rates follow a similar trend with FA and Cs mixing in halide perovskites. The decreased hot-carrier cooling rates in the compositionally engineered perovskite can give rise to a longer hot carrier diffusion length, ultimately leading to improved hot-carrier extraction at perovskite/extraction layer interfaces. Note that the band gap of FA1−xCsxPbI3 widens with an increased concentration of Cs, however, such a change does not directly impact the carrier cooling processes that occur through intraband transitions.36
Cooling down only a fraction (only up to 59%) of the excited electron population within 4 ps (Fig. 2a) indicates that a considerable amount of transient hot electrons populate the electronic states above the conduction band edge. To determine the details of hot-electron distribution in the above-CBM states, we further inspect the population over time for all electronic states that are in the middle of two extreme energy bands (see Fig. S4, ESI†). The plotted transient population in individual electronic states show that the CBM + 3 state contains most of the electron density for all FA1−xCsxPbI3 systems that are investigated here. Thus, overall the CBM + 3 state creates a bottleneck to the intraband electron relaxation process which can be attributed to a larger energy gap below, and subsequently accumulates the excited carriers for a few picosecond timescale.
We next investigate the energy dissipation of excited electrons and holes that can be tracked through time-resolved photoluminescence experiments. Fig. 3a depicts that even after 4 ps of excitation, the electrons retain 0.12–0.19 eV excess energy (about 5–8 times that of room-temperature, kBT) representing 23–36% of the initial excitation energy. Likewise, excited holes also possess considerable excess energy after a 4 ps timescale as shown in Fig. 3b. The presence of such persistent warm carrier concentration in hybrid and inorganic halide perovskites has been broadly studied by different experimental techniques.54–56 Similar to the hot carrier population loss, the average energy dissipation is fastest in FAPbI3, and it becomes slower as Cs replaces the FA cations in the mixed-cation lattices. The persistent excess energies for electrons and holes monotonically increases with an increase of x in FA1−xCsxPbI3 from 0 to 0.5 (Fig. 3a and b). Thus, like carrier populations, the energy dissipation of excited carriers distinctly depends on the type and relative concentration of A-site cations in hybrid perovskites.
 | ||
| Fig. 3 The excess energy dissipation associated with hot carrier cooling in FA1−xCsxPbI3. The energy dissipation over time during (a) electron cooling in the conduction band and (b) hole cooling in the valence band. The energy dissipation rates depend on the A-cation composition of these perovskites. | ||
We now attempt to rationalize the atomistic origin for the A-cation dependence of hot-carrier relaxation in hybrid perovskites. The partial density of states for FA1−xCsxPbI3 (x = 0, 0.5) over AIMD trajectories illustrates that the electronic states of A-site cations (that are FA or Cs) considerably contribute to the total density of states far away (in excess of 2 eV at least) from the band edge states (Fig. S5, ESI†). Thus, even after including highly dynamic electronic landscapes and involving energy states that are ≈ 0.55 eV away from the band edges, we do not find any direct electronic contributions from the A-site cations into relevant states. Stated differently, the A-site cations are electronically inert during the hot carrier relaxation processes that occur after above-band gap excitation. This overall electronic structure of halide perovskites under ambient conditions agrees well with previous reports.37,57–59
For mechanically soft halide perovskite lattices, the dynamic coupling between the electronic excited states and lattice vibrations significantly impacts the energy and charge dynamics through non-radiative channels.38,40,48,60 Under the vibronic coupling, the inelastic carrier–phonon interactions dominantly take part in energy exchange between electronic and vibrational subsystems. During the carrier cooling process, the hot electrons and holes dissipate their excess energy to the vibrational degrees of freedom.48,53 In this regard, the time-averaged nonadiabatic couplings (NACs) between electronic states measure the carrier–phonon interaction strengths at ambient conditions.47,61 Generally, the higher the NAC values, the stronger the interactions between electronic and vibrational degrees of freedom. These strong interactions give rise to the fast phonon-assisted nonadiabatic transitions among the electronic states involved and cause rapid decay of hot-carrier populations. So the energy dissipation during hot carrier cooling is expected to be faster for the material with higher NAC strengths.32,48,62,63Fig. 4a and b display the absolute time-averaged NAC values involving the states ranging from VBM − 7 to CBM + 7 for FAPbI3 and FA0.5Cs0.5PbI3. For all other materials, NAC values near band edge states are shown in Fig. S6, ESI.† As the NAC values are considerably higher only along the sub-diagonal lines, we conclude that the nonadiabatic transitions during hot-carrier relaxation predominantly occur between energetically adjacent electronic states. There can be multiphonon processes through which higher energy states can directly couple to the much lower energy states, however, such couplings are found to be weak in these materials, a minor contribution of these processes to carrier relaxation.
 | ||
| Fig. 4 The time-averaged NAC values for (a) FAPbI3 and (b) FA0.5Cs0.5PbI3. The electronic states near the valence and conduction band edges have been considered for plotting. i = 0 and i = 1 represent the VBM and CBM states, respectively. The scale of heatmap applied for both (a) and (b) is given in the right column of (b). (c) Variation in the arithmetic means of NAC values for valence (VBM to VBM − 7) and conduction (CBM to CBM + 7) states with A-cation composition in Fa/Cs lead iodide perovskites. Overall the NAC strengths for both types of states reduce with increased Cs incorporation. | ||
We further calculate the arithmetic means of NAC values for valence (VBM to VBM − 7) and conduction (CBM to CBM + 7) states, separately (Fig. 4c). Irrespective of the A-cation concentration in hybrid perovskites, the mean NAC values for intraband transitions involving valence states, are much higher compared to those with conduction states. This overall trend of mean NAC justifies the faster relaxation rates and shorter hot-carrier lifetime for holes in these hybrid materials.27,32,48,49
Considering NACs for conduction states, we find that FAPbI3 has the highest averaged NACs for overall intraband transitions (Fig. 4c). These coupling strengths reduce with the increased Cs concentration in mixed-A cation perovskites. The incorporation of Cs cations decreases the averaged NAC values for conduction bands by ≈ 8–13% compared to the parent FAPbI3. While tracking the hot-hole relaxation process over time, a similar trend is apparent for FA1−xCsxPbI3. The mean NAC for the valence states reduces by ≈ 9–28% when FA cations are partially replaced with Cs+. These results unambiguously illustrate the relationship between the NACs and carrier lifetime: the weaker carrier–phonon interactions are concomitant to the longer hot-carrier lifetimes in FA1−xCsxPbI3. Thus, simulations involving the nonadiabatic processes are necessary to analyse the hot-carrier relaxation processes in halide perovskites.
Finally, we explore the atomistic origin of the reduced NAC strengths for FA1−xCsxPbI3 lattices with increased Cs concentration. The NAC strengths for intraband transition closely depend on the thermal motions of atoms that contribute to the wavefunctions of the involved electronic states.32,40,48,64 Generally, a more dynamic lattice gives rise to higher NAC values due to the stronger carrier–phonon interactions. The inorganic Pb and I atoms predominantly contribute to the electronic states that are involved in the carrier cooling processes (see the pDOS plots in Fig. S7, ESI†). Therefore, we primarily focus on the root-mean square fluctuations (RMSF) that measure the extent of atomic displacements due to thermal fluctuations in the inorganic framework. The RMSF values for the inorganic Pb–I lattice in Fig. 5 suggest that the atomic displacements reduce as the FA molecules are replaced by Cs+ in the mixed-A cation perovskites. The replacement of 50% FA with Cs cations results in 18% reduction in the RMSF of the inorganic framework. Stated differently, the Cs incorporation enhances the rigidity of the inorganic sublattice by partially suppressing the structural fluctuations at ambient conditions. Here, we also note that the RMSF of A-cations reduces with Cs concentration, demonstrating constrained cation dynamics (Fig. 5). The partially suppressed A-cation dynamics in FA1−xCsxPbI3 also weakens the dynamic coupling with the Pb–I lattice, effectively reducing its RMSF values as shown in Fig. 5. We conclude that the weakened NAC strengths originate from the reduced mobility of inorganic sites and A-cations in the FA1−xCsxPbI3.
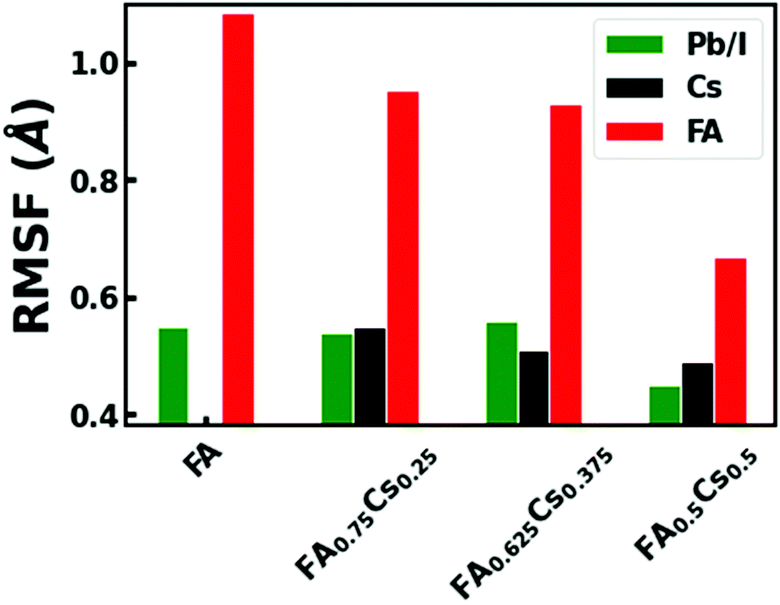 | ||
| Fig. 5 The extent of structural fluctuations with A-cation mixing in halide perovskites. The root mean square fluctuations (RMSF) for inorganic framework (Pb/I), FA, and Cs cations for FA1−xCsxPbI3 at 300 K. | ||
Our analyses identify two main sources of the reduced structural motions in the Cs+ incorporated mixed A-cation perovskites: (1) an increased internal chemical pressure in the lattice, and (2) partially suppressed rotational dynamics of FA molecules.12,35,36,65,66 As discussed in previous studies, the replacement of larger sized FA+ (2.53 Å) cations with smaller Cs+ (1.67 Å) ions causes contraction of the unit cell volume.12,36 These contracted perovskite cells limit the spatial freedom of the Pb and I atoms to displace under ambient conditions and mostly lock the corner-shared octahedra in a tilted conformation.35 Considering other factors, we realize that light-weight atoms (C, N, and H) of FA+ have much larger displacement than inorganic framework and Cs cations. Due to this molecular cation, the entire organic–inorganic perovskite lattice becomes dynamically fluctuating under ambient conditions. However, the lattice contraction confines the motion of FA molecules by shrinking the cuboctahedral cage volume. The reduced RMSF values of FA in these mixed A-cation lattices compared to pristine FAPbI3 support the restricted motions of organic molecules. Reduction in FA cation dynamics consequently increases the non-covalent hydrogen bonding interactions between the molecule and iodide of the Pb–I framework. Strengthened non-covalent interactions between organic and inorganic sublattices help to partially mitigate the thermal fluctuations in Pb/I lattice-sites, reducing their overall RMSF values. Finally, we find that less dynamic mixed A-cation lattices enhance the hot carrier cooling time due to weak nonadiabatic transition probability. Thus, even though the A-cations do not participate electronically in the carrier cooling processes, their indirect impact through lattice dynamics significantly influences these phenomena. Our study unambiguously depicts that rational modification of halide perovskite compositions can provide a powerful tool to manipulate ultrafast carrier dynamics.
In this work, we combine time-domain DFT and NAMD modeling techniques to explore the hot carrier cooling process in FA–Cs cation mixed halide perovskites at ambient conditions. Our simulations demonstrate that the ultrafast hot-carrier relaxation can be suppressed by substituting FA cations with inorganic Cs. However, the carrier relaxation processes directly involve only the electronic states that are predominantly delocalized only over the inorganic framework of Pb/I. This apparent discrepancy emphasizes the importance of lattice dynamics, modulating electron–phonon interactions at ambient conditions. By analyzing the nonadiabatic transitions during carrier relaxation processes, we identify that organic FA molecules induce large structural fluctuations in FAPbI3 causing strong nonadiabatic coupling among the electronic states. The light atoms of FA are dynamically more active and consequently couple strongly with the Pb/I framework. An intentional substitution of FA molecules with inorganic Cs considerably suppresses the thermal fluctuations in the FA1−xCsxPbI3 which is evident from our RMSF analyses. In turn, the reduced lattice dynamics in mixed A-cation perovskites weakens the electron–phonon interaction strength under ambient conditions. Consequently, the hot carrier relaxation processes become relatively slower, enhancing the carrier lifetime in these comparatively rigid lattices. Several factors such as size mismatch induced chemical pressure in the lattice, reduced concentration of light atoms in the A-site, and stronger non-covalent interactions between the A-cation and Pb/I framework cause the increased rigidity of the FA1−xCsxPbI3 lattice. Our study provides in-depth analyses of the dynamic structure elucidating hot carrier relaxation in mixed A-cation perovskites, and suggests synthetic routes to fine-tune the excited state carrier dynamics in these materials. Some feasible strategies that can be considered to enhance the hot-carrier lifetime are (1) incorporating elemental A-cations with a smaller size than Cs (such as Rb), (2) strengthening non-covalent interactions between organic–inorganic sublattices through substituting I with Br/Cl, and (3) choosing appropriate A-cations that can form stronger non-covalent interactions with the Pb/X (X = halogens) framework through hydrogen bonding.67,68
Methods
We perform density functional theory (DFT) calculations and ab initio molecular dynamics (AIMD) simulations using the Vienna Ab Initio Simulation Package (VASP).69,70 All simulations are done with the projected augmented wave (PAW) method and consider a plane-wave basis set with a cut-off energy of 420 eV.71 As the approximated exchange and correlation interactions, we use the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof functional (PBE) form.72 To model the simulation cells with varying A-cation composition, we set up a 2 × 2 × 2 supercell that contains 8 formula units of APbI3 (A = FA/Cs). We conjugate the gradient algorithm to relax the geometries of FA1−xCsxPbI3 at 0 K until all the interatomic forces become smaller than 0.01 eV Å−1. In the structural optimization we do not impose any constraints and allowed the cell size to change during the simulation. We use the DFT-D3 correction method of Grimme et al. to include the dispersion interactions for all the simulations.73 The optimization and self-consistent field calculations use k-point mesh as 3 × 3 × 3 and 6 × 6 × 6, respectively.The mixed quantum-classical non-adiabatic molecular dynamics (NAMD) simulations with the decoherence-induced surface hopping (DISH) technique have been adapted to investigate the nonadiabatic transitions of hot carriers.47,74,75 The NAMD simulations use the framework of time-domain density functional theory (DFT) as implemented in the PYthon eXtension for Ab Initio Dynamics (PYXAID) code.49,61,76 Within this methodology we consider the electrons as the quantum mechanical particle and nuclei as the classical entity. Here, the back-reaction of the electronic degrees of freedom on the nuclear ones has been neglected as halide perovskites are solid and assumed to have very similar nuclear geometries under different electronic states. Several groups including us have applied this computational approach for studying the electron–hole recombination, and hot-carrier cooling processes in halide perovskites.32,37,38,60,62,77 For generating dynamical trajectories, we perform AIMD simulations considering a 2 × 2 × 2 Monkhorst–Pack k-point mesh and a time step of 1 fs. We begin with 0 K DFT optimized geometries and heat those up to 300 K using repeated velocity rescaling for 4 ps. To ensure the thermal equilibrium in these materials, we further generate an AIMD trajectory of another 4 ps under the canonical ensemble (NVT). Finally, trajectories of 15 ps have been simulated using the microcanonical ensemble (NVE). While complete 15 ps trajectories are used for analyzing the structural dynamics in the lattice, we consider the first 5 ps of these trajectories for NAMD simulations. In these simulations every fifth geometry throughout the 5 ps trajectory (total 1000) has been considered as the starting geometries. For each starting geometry, 1000 stochastic realizations of the DISH process have been simulated to evaluate the ultrafast intraband carrier cooling in these materials.61,76 The current NAMD simulations capture the carrier relaxation along the Γ-point of the Brillouin zone only. To calculate the carrier cooling time, we fit the population decay of the highest excited state with the following fitting function,27,32,78
| f (t) =A·exp(− t/τ1) + (1 − A)exp(− (t/τ2)2) | (1) |
The lifetime of hot electrons and holes is
| τ =A·τ1+ (1 − A)τ2 | (2) |
Note that the inclusion of spin–orbit coupling (SOC) significantly impacts the quantitative magnitude of the carrier cooling time, however, this increases the computational cost as well.49,77 We do not consider SOC effects here as our main interest is to analyze the relative trend of carrier cooling rate with the change in A-cation composition in these halide perovskites. Furthermore, the composition of the inorganic framework (Pb/I) which is more sensitive to the SOC effects and dominantly participate in the intraband carrier relaxation, remains unchanged for all FA1−xCsxPbI3 lattices. Thus, it can be assumed that the impact of SOC on the carrier relaxation rates in these perovskites is similar and its absence will not fundamentally alter the reported trends.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work at Los Alamos National Laboratory (LANL) was supported by the LANL LDRD program. This work was done in part at the Center for Nonlinear Studies (CNLS) and the Center for Integrated Nanotechnologies (CINT), a U.S. Department of Energy and Office of Basic Energy Sciences user facility, at LANL. This research used resources provided by the LANL Institutional Computing Program. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy (Contract No. 89233218NCA000001). O. V. P. acknowledges support of the U.S. Department of Energy (grant no. DE-SC0014429).References
- R. T. Ross and A. J. Nozik, Efficiency of hot-carrier solar energy converters, J. Appl. Phys., 1982, 53, 3813–3818 CrossRef CAS.
- P. Würfel, Solar energy conversion with hot electrons from impact ionisation, Sol. Energy Mater. Sol. Cells, 1997, 46, 43–52 CrossRef.
- M. A. Green, Third generation photovoltaics: Ultra-high conversion efficiency at low cost, Prog. Photovoltaics Res. Appl., 2001, 9, 123–135 CrossRef CAS.
- D. Ferry, S. Goodnick, V. Whiteside and I. Sellers, Challenges, myths, and opportunities in hot carrier solar cells, J. Appl. Phys., 2020, 128, 220903 CrossRef CAS.
- D. König, K. Casalenuovo, Y. Takeda, G. Conibeer, J. Guillemoles, R. Patterson, L. Huang and M. Green, Hot carrier solar cells: Principles, materials and design, Phys. E, 2010, 42, 2862–2866 CrossRef.
- Y. Takeda, T. Ito, T. Motohiro, D. König, S. Shrestha and G. Conibeer, Hot carrier solar cells operating under practical conditions, J. Appl. Phys., 2009, 105, 074905 CrossRef.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, K. N. Mohammad, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Cesium-containing Triple Cation Perovskite Solar Cells: Improved Stability, Reproducibility and High Efficiency, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, A Mixed-cation Lead Mixed-halide Perovskite Absorber for Tandem Solar Cells, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- R. G. Niemann, L. Gouda, J. Hu, S. Tirosh, R. Gottesman, P. J. Cameron and A. Zaban, Cs+ incorporation into CH3NH3PbI3 perovskite: substitution limit and stability enhancement, J. Mater. Chem. A, 2016, 4, 17819–17827 RSC.
- W. Rehman, D. P. McMeekin, J. B. Patel, R. L. Milot, M. B. Johnston, H. J. Snaith and L. M. Herz, Photovoltaic Mixed-cation Lead Mixed-halide Perovskites: Links between Crystallinity, Photo-stability and Electronic properties, Energy Environ. Sci., 2017, 10, 361–369 RSC.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Formamidinium and Cesium Hybridization for Photo-and Moisture-stable Perovskite Solar Cell, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- R. Prasanna, A. Gold-Parker, T. Leijtens, B. Conings, A. Babayigit, H.-G. Boyen, M. F. Toney and M. D. McGehee, Band Gap Tuning via Lattice Contraction and Octahedral Tilting in Perovskite Materials for Photovoltaics, J. Am. Chem. Soc., 2017, 139, 11117–11124 CrossRef CAS PubMed.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Methylammonium-free, high-performance and stable perovskite solar cells on a planar architecture, Science, 2018, 3583 Search PubMed.
- A. Y. Alsalloum, B. Turedi, K. Almasabi, X. Zheng, R. Naphade, S. D. Stranks, O. F. Mohammed and O. M. Bakr, 22.8%-Efficient single-crystal mixed-cation inverted perovskite solar cells with a near-optimal bandgap, Energy Environ. Sci., 2021, 14, 2263–2268 RSC.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Long-range balanced electron-and hole-transport lengths in organic-inorganic CH3NH3PbI3, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- H. Zhu, K. Miyata, Y. Fu, J. Wang, P. P. Joshi, D. Niesner, K. W. Williams, S. Jin and X.-Y. Zhu, Screening in crystalline liquids protects energetic carriers in hybrid perovskites, Science, 2016, 353, 1409–1413 CrossRef CAS PubMed.
- Z. Guo, Y. Wan, M. Yang, J. Snaider, K. Zhu and L. Huang, Long-range hot-carrier transport in hybrid perovskites visualized by ultrafast microscopy, Science, 2017, 356, 59–62 CrossRef CAS PubMed.
- H.-H. Fang, S. Adjokatse, S. Shao, J. Even and M. A. Loi, Long-lived hot-carrier light emission and large blue shift in formamidinium tin triiodide perovskites, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- Y. Yang, D. P. Ostrowski, R. M. France, K. Zhu, J. Van De Lagemaat, J. M. Luther and M. C. Beard, Observation of a hot-phonon bottleneck in lead-iodide perovskites, Nat. Photonics, 2016, 10, 53–59 CrossRef CAS.
- J. Yang and X. Wen, et al., Acoustic-optical phonon up-conversion and hot-phonon bottleneck in lead-halide perovskites, Nat. Commun., 2017, 8, 1–9 CrossRef PubMed.
- J. Fu, Q. Xu, G. Han, B. Wu, C. H. A. Huan, M. L. Leek and T. C. Sum, Hot carrier cooling mechanisms in halide perovskites, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- S.-T. Ha, C. Shen, J. Zhang and Q. Xiong, Laser cooling of organic-inorganic lead halide perovskites, Nat. Photonics, 2016, 10, 115–121 CrossRef CAS.
- M. Li, J. Fu, Q. Xu and T. C. Sum, Slow Hot-Carrier Cooling in Halide Perovskites: Prospects for Hot-Carrier Solar Cells, Adv. Mater., 2019, 31, 1802486 CrossRef CAS PubMed.
- S. Kahmann and M. A. Loi, Hot carrier solar cells and the potential of perovskites for breaking the Shockley-Queisser limit, J. Mater. Chem. C, 2019, 7, 2471–2486 RSC.
- T. Wang and L. Jin, et al., Protecting hot carriers by tuning hybrid perovskite structures with alkali cations, Sci. Adv., 2020, 6, eabb1336 CrossRef CAS PubMed.
- N. K. Tailor, S. Mishra, T. Sharma, A. K. De and S. Satapathi, Cation-Dependent Hot Carrier Cooling in the Lead-Free Bismuth Halide A3Bi2I9 (A= FA, MA, and Cs) Perovskite, J. Phys. Chem. C, 2021, 125, 9891–9898 CrossRef CAS.
- L. Tyler Mix and D. Ghosh, et al., Hot Carrier Cooling and Recombination Dynamics of Chlorine-Doped Hybrid Perovskite Single Crystals, J. Phys. Chem. Lett., 2020, 11, 8430–8436 CrossRef PubMed.
- T. Ghosh, S. Aharon, L. Etgar and S. Ruhman, Free carrier emergence and onset of electron-phonon coupling in methylammonium lead halide perovskite films, J. Am. Chem. Soc., 2017, 139, 18262–18270 CrossRef CAS PubMed.
- S. D. Verma, Q. Gu, A. Sadhanala, V. Venugopalan and A. Rao, Slow Carrier Cooling in Hybrid Pb-Sn Halide Perovskites, ACS Energy Lett., 2019, 4, 736–740 CrossRef CAS.
- J. Chen, M. E. Messing, K. Zheng and T. Pullerits, Cation-dependent hot carrier cooling in halide perovskite nanocrystals, J. Am. Chem. Soc., 2019, 141, 3532–3540 CrossRef CAS PubMed.
- T. R. Hopper, A. Gorodetsky, J. M. Frost, C. Muller, R. Lovrincic and A. A. Bakulin, Ultrafast intraband spectroscopy of hot-carrier cooling in lead-halide perovskites, ACS Energy Lett., 2018, 3, 2199–2205 CrossRef CAS PubMed.
- M. E. Madjet, G. R. Berdiyorov, F. El-Mellouhi, F. H. Alharbi, A. V. Akimov and S. Kais, Cation effect on hot carrier cooling in halide perovskite materials, J. Phys. Chem. Lett., 2017, 8, 4439–4445 CrossRef CAS PubMed.
- D. B. Straus, S. Guo, A. M. Abeykoon and R. J. Cava, Understanding the Instability of the Halide Perovskite CsPbI3 through Temperature-Dependent Structural Analysis, Adv. Mater., 2020, 32, 2001069 CrossRef CAS PubMed.
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Grätzel, S. M. Zakeeruddin, U. Röthlisberger and M. Grätzel, Entropic Stabilization of Mixed A-cation ABX3 Metal Halide Perovskites for High Performance Perovskite Solar Cells, Energy Environ. Sci., 2016, 9, 656–662 RSC.
- D. Ghosh, P. Walsh Atkins, M. S. Islam, A. B. Walker and C. Eames, Good Vibrations: Locking of Octahedral Tilting in Mixed-Cation Iodide Perovskites for Solar Cells, ACS Energy Lett., 2017, 2, 2424–2429 CrossRef CAS.
- D. Ghosh, A. R. Smith, A. B. Walker and M. S. Islam, Mixed A-cation perovskites for solar cells: atomic-scale insights into structural distortion, hydrogen bonding, and electronic properties, Chem. Mater., 2018, 30, 5194–5204 CrossRef CAS.
- D. Ghosh, D. Acharya, L. Zhou, W. Nie, O. V. Prezhdo, S. Tretiak and A. J. Neukirch, Lattice Expansion in Hybrid Perovskites: Effect on Optoelectronic Properties and Charge Carrier Dynamics, J. Phys. Chem. Lett., 2019, 10, 5000–5007 CrossRef CAS PubMed.
- D. Ghosh, A. J. Neukirch and S. Tretiak, Optoelectronic Properties of Two-Dimensional Bromide Perovskites: Influences of Spacer Cations, J. Phys. Chem. Lett., 2020, 11, 2955–2964 CrossRef CAS PubMed.
- Y. Wang, L. Pedesseau, C. Katan, J. Even, O. V. Prezhdo, S. Tretiak, D. Ghosh and A. J. Neukirch, Nonadiabatic molecular dynamics analysis of hybrid Dion-Jacobson 2D leads iodide perovskites, Appl. Phys. Lett., 2021, 119, 201102 CrossRef CAS.
- D. Ghosh, D. Acharya, L. Pedesseau, C. Katan, J. Even, S. Tretiak and A. J. Neukirch, Charge carrier dynamics in two-dimensional hybrid perovskites: Dion-Jacobson vs. Ruddlesden-Popper phases, J. Mater. Chem. A, 2020, 8, 22009–22022 RSC.
- L. Zhou and D. F. Swearer, et al., Quantifying hot carrier and thermal contributions in plasmonic photocatalysis, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- L. Zhou and M. Lou, et al., Hot carrier multiplication in plasmonic photocatalysis, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022109118 CrossRef CAS PubMed.
- A. Boziki, D. J. Kubicki, A. Mishra, S. Meloni, L. Emsley, M. Grätzel and U. Rothlisberger, Atomistic Origins of the Limited Phase Stability of Cs + -Rich FA x Cs (1-x) PbI3 Mixtures, Chem. Mater., 2020, 32, 2605–2614 CrossRef CAS.
- G. M. Dalpian, X.-G. Zhao, L. Kazmerski and A. Zunger, Formation and composition-dependent properties of alloys of cubic halide perovskites, Chem. Mater., 2019, 31, 2497–2506 CrossRef CAS.
- H. Kaslasi, Y. Feldman, Y. Rakita, D. Cahen and G. Hodes, Single-Crystal Growth and Thermal Stability of (CH3NH3) 1-x Cs x PbBr3, Cryst. Growth Des., 2020, 20, 4366–4374 CrossRef CAS.
- R. K. Singh, K. B. Masood, N. Jain, R. Tamrakar, J. Singh and R. Kumar, Exploration of structural, thermal stability and band-gap tunability of organic and inorganic mixed cation (MA) 1- x Cs x PbBr3 perovskite harvester via ultrasonication synthesis route, J. Phys.: Condens. Matter, 2021, 33, 245705 CrossRef CAS PubMed.
- C. F. Craig, W. R. Duncan and O. V. Prezhdo, Trajectory surface hopping in the time-dependent Kohn-Sham approach for electron-nuclear dynamics, Phys. Rev. Lett., 2005, 95, 163001 CrossRef PubMed.
- G. J. Hedley, C. Quarti, J. Harwell, O. V. Prezhdo, D. Beljonne and I. D. Samuel, Hot-hole cooling controls the initial ultrafast relaxation in methylammonium lead iodide perovskite, Sci. Rep., 2018, 8, 8115 CrossRef PubMed.
- W. Li, L. Zhou, O. V. Prezhdo and A. V. Akimov, Spin-orbit interactions greatly accelerate nonradiative dynamics in lead halide perovskites, ACS Energy Lett., 2018, 3, 2159–2166 CrossRef CAS.
- V. A. Hintermayr, L. Polavarapu, A. S. Urban and J. Feldmann, Accelerated carrier relaxation through reduced coulomb screening in two-dimensional halide perovskite nanoplatelets, ACS Nano, 2018, 12, 10151–10158 CrossRef CAS PubMed.
- S. A. Bretschneider, I. Ivanov, H. I. Wang, K. Miyata, X. Zhu and M. Bonn, Quantifying polaron formation and charge carrier cooling in lead-iodide perovskites, Adv. Mater., 2018, 30, 1707312 CrossRef PubMed.
- S. Sarkar, V. K. Ravi, S. Banerjee, G. R. Yettapu, G. B. Markad, A. Nag and P. Mandal, Terahertz spectroscopic probe of hot electron and hole transfer from colloidal CsPbBr3 perovskite nanocrystals, Nano Lett., 2017, 17, 5402–5407 CrossRef CAS PubMed.
- S. C. Boehme and S. T. Brinck, et al., Phonon-Mediated and Weakly Size-Dependent Electron and Hole Cooling in CsPbBr3 Nanocrystals Revealed by Atomistic Simulations and Ultrafast Spectroscopy, Nano Lett., 2020, 20, 1819–1829 CrossRef CAS PubMed.
- D. Niesner, H. Zhu, K. Miyata, P. P. Joshi, T. J. Evans, B. J. Kudisch, M. T. Trinh, M. Marks and X.-Y. Zhu, Persistent energetic electrons in methylammonium lead iodide perovskite thin films, J. Am. Chem. Soc., 2016, 138, 15717–15726 CrossRef CAS PubMed.
- P. P. Joshi, S. F. Maehrlein and X. Zhu, Dynamic screening and slow cooling of hot carriers in lead halide perovskites, Adv. Mater., 2019, 31, 1803054 CrossRef CAS PubMed.
- M. Li, S. Bhaumik, T. W. Goh, M. S. Kumar, N. Yantara, M. Grätzel, S. Mhaisalkar, N. Mathews and T. C. Sum, Slow cooling and highly efficient extraction of hot carriers in colloidal perovskite nanocrystals, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- M. R. Filip, G. E. Eperon, H. J. Snaith and F. Giustino, Steric Engineering of Metal-halide Perovskites with Tunable Optical Band Gaps, Nat. Commun., 2014, 5, 5757 CrossRef CAS PubMed.
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. Van Schilfgaarde and A. Walsh, Atomistic Origins of High-performance in Hybrid Halide Perovskite Solar Cells, Nano Lett., 2014, 14, 2584–2590 CrossRef CAS PubMed.
- D. Ghosh, A. Aziz, J. A. Dawson, A. B. Walker and M. S. Islam, Putting the Squeeze on Lead Iodide Perovskites: Pressure-Induced Effects to Tune their Structural and Optoelectronic Behaviour, Chem. Mater., 2019, 31, 4063 CrossRef CAS PubMed.
- Z. Zhang, W.-H. Fang, M. V. Tokina, R. Long and O. V. Prezhdo, Rapid decoherence suppresses charge recombination in multi-layer 2D halide perovskites: Time-domain ab initio analysis, Nano Lett., 2018, 18, 2459–2466 CrossRef CAS PubMed.
- A. V. Akimov and O. V. Prezhdo, The PYXAID program for non-adiabatic molecular dynamics in condensed matter systems, J. Chem. Theory Comput., 2013, 9, 4959–4972 CrossRef CAS PubMed.
- R. Long, J. Liu and O. V. Prezhdo, Unravelling the effects of grain boundary and chemical doping on electron-hole recombination in CH3NH3PbI3 perovskite by time-domain atomistic simulation, J. Am. Chem. Soc., 2016, 138, 3884–3890 CrossRef CAS PubMed.
- J. He, A. S. Vasenko, R. Long and O. V. Prezhdo, Halide composition controls electron-hole recombination in cesium-lead halide perovskite quantum dots: a time domain ab initio study, J. Phys. Chem. Lett., 2018, 9, 1872–1879 CrossRef CAS PubMed.
- J. He, W.-H. Fang, R. Long and O. V. Prezhdo, Increased Lattice Stiffness Suppresses Nonradiative Charge Recombination in MAPbI3 Doped with Larger Cations: Time-Domain Ab Initio Analysis, ACS Energy Lett., 2018, 3, 2070–2076 CrossRef CAS.
- G. Kieslich, S. Sun and A. K. Cheetham, Solid-state principles applied to organic-inorganic perovskites: new tricks for an old dog, Chem. Sci., 2014, 5, 4712–4715 RSC.
- E. M. Mozur, A. E. Maughan, Y. Cheng, A. Huq, N. Jalarvo, L. L. Daemen and J. R. Neilson, Orientational Glass Formation in Substituted Hybrid Perovskites, Chem. Mater., 2017, 29, 10168–10177 CrossRef CAS.
- D. Ferdani, S. Pering, D. Ghosh, P. Kubiak, A. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, S. Islam and P. J. Cameron, Partial Cation Substitution Reduces Iodide Ion Transport in Lead Iodide Perovskite Solar Cells, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- A. D. Jodlowski, C. Roldán-Carmona, G. Grancini, M. Salado, M. Ralaiarisoa, S. Ahmad, N. Koch, L. Camacho, G. De Miguel and M. K. Nazeeruddin, Large guanidinium cation mixed with methylammonium in lead iodide perovskites for 19% efficient solar cells, Nat. Energy, 2017, 2, 972 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. M. Jaeger, S. Fischer and O. V. Prezhdo, Decoherence-induced surface hopping, J. Chem. Phys., 2012, 137, 22A545 CrossRef PubMed.
- P. Hamm, Principles of Nonlinear Optical Spectroscopy: A Practical Approach, University ofZurich, 2005, vol. 41, p. 77 Search PubMed.
- A. V. Akimov and O. V. Prezhdo, Advanced capabilities of the PYXAID program: integration schemes, decoherence effects, multiexcitonic states, and field-matter interaction, J. Chem. Theory Comput., 2014, 10, 789–804 CrossRef CAS PubMed.
- J. Yin, P. Maity, R. Naphade, B. Cheng, J.-H. He, O. M. Bakr, J.-L. Bredas and O. F. Mohammed, Tuning hot carrier cooling dynamics by dielectric confinement in two-dimensional hybrid perovskite crystals, ACS Nano, 2019, 13, 12621–12629 CrossRef CAS PubMed.
- S. V. Kilina, A. J. Neukirch, B. F. Habenicht, D. S. Kilin and O. V. Prezhdo, Quantum zeno effect rationalizes the phonon bottleneck in semiconductor quantum dots, Phys. Rev. Lett., 2013, 110, 180404 CrossRef PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Volume in the FA1−xCsxPbI3 lattice, adiabatic time-dependent Kohn–Sham state energies for these systems, population of excited electron and hole states over time, and time-averaged magnitude of the carrier-phonon NACs in the valence and conduction bands for FA1−xCsx systems. See DOI: https://doi.org/10.1039/d2tc01413k |
| This journal is © The Royal Society of Chemistry 2022 |