
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Distortion-driven spin switching in electron-doped metal porphyrins†
Iulia
Cojocariu
 *ab,
Silvia
Carlotto
*cd,
Matteo
Jugovac
ab,
Luca
Floreano
e,
Maurizio
Casarin
c,
Vitaliy
Feyer
*af and
Claus Michael
Schneider
af
*ab,
Silvia
Carlotto
*cd,
Matteo
Jugovac
ab,
Luca
Floreano
e,
Maurizio
Casarin
c,
Vitaliy
Feyer
*af and
Claus Michael
Schneider
af
aPeter Grunberg Institute (PGI-6), Forschungszentrum Julich GmbH, Leo-Brandt-Strase, Julich, 52428, Germany. E-mail: i.cojocariu@fz-juelich.de; v.feyer@fz-juelich.de
bElettra-Sincrotrone, S.C.p.A, S.S. 14 - km 163.5 in AREA Science Park, Trieste, 34149, Italy
cDipartimento di Scienze Chimiche, Universita degli Studi di Padova, via F. Marzolo 1, Padova, 35131, Italy. E-mail: silvia.carlotto@unipd.it
dInstitute of Condensed Matter Chemistry and Technologies for Energy (ICMATE), National Research Council (CNR), c/o Department of Chemistry, University of Padova, via F. Marzolo 1, Padova, 35131, Italy
eCNR-IOM, Lab. TASC, S.S. 14 - km 163.5 in AREA Science Park, Trieste, 34149, Italy
fFakultat f. Physik and Center for Nanointegration Duisburg-Essen (CENIDE), Universitat Duisburg-Essen, Lotharstrase 1, Duisburg, 47048, Germany
First published on 6th June 2022
Abstract
Electron injection into electrode-supported metal complexes allows for charge redistribution within the molecule to be controlled. Here we show, for the first time, how the structural flexibility in electron-doped porphyrins is critical in defining charge localization by following the evolution of the spin state and charge distribution in the thermodynamically favored structure as a function of dopant dose and relaxation time. Two flexible transition metal-containing molecules are used as model systems, nickel and cobalt tetraphenylporphyrin, studied by combining a wide range of spectroscopic techniques with detailed DFT calculations.
Organic semiconductors, and in particular transition metal porphyrins (TMPs) and TM phthalocyanines (TMPcs), may be considered as a class of materials that can be used to create a wide range of adaptable and low-cost molecular-based electronic devices.1–4 To fully exploit the potential of these interfaces, the understanding and capability to tune the physical, chemical, and transport properties of organic semiconductor components is critical.5,6 Within this framework, the capacity of controlling the metal charge and spin states in the organic array is a step toward molecular spintronics realization, and it has been shown that single electron injection in the molecule can drastically change its properties. Along these lines, scanning tunneling microscopy (STM) investigations on a single-molecule junction have shown that electrons travel through the FePc molecule via two different 3d atomic orbitals (AOs), and the electron pathway within the tip-FePc-Au junction can be selected just by changing the magnetic field. Tunable giant magnetoresistance in this molecular device originates from the re-orientation of the iron magnetic moment, which alters the electron distribution in the Fe 3d AOs.7 As such, it has been recently reported that, under the force field of an STM tip, it is also possible to reversibly modify both the Fe oxidation and spin states of the Fe(III) octaethylporphyrin-chloride anchored on a Pb(111) substrate.8,9 In addition, both TMPs and TMPcs can coordinate electron acceptor/donor ligands to the metal center, thus resulting in a change of the valence electrons number in the chelated metal, which induces a variation in its oxidation and spin state.10,11 For instance, the spin state could be controlled even by coordinating single H atoms to the magnetic core of the adsorbed molecule.12–14
It is also noteworthy that X-ray magnetic circular dichroism (XMCD) measurements have recently brought to light that the molecular properties of surface-supported TMPc can also be modified by the donation of a single electron provided by an alkali metal (AM), with final oxidation and spin states being strongly TM dependent. Specifically, the TM(II) species in CuPc, NiPc, and FePc molecular layers decrease their oxidation state upon alkali metal deposition, whereas MnPc preserves its formal oxidation state Mn(II), but the magnetic configuration switches from an intermediate (IS) to a high-spin (HS) state.15 Besides the TM and AM nature, the electron donation is also influenced by the adsorption site and TM reduction is observed only when the AM adsorbs in TM proximity.16
Changes in the molecular properties of TMPcs and TMPs can also be induced by the coordination of polyatomic molecules to the TM. In this respect, it has been shown that axial coordination may be exploited to modulate the TM oxidation and spin states.17–19 In fact, the spin state of the Mn ion in the tetra-pyrrolic pocket may sweep across different configurations by subtle modification of the N ligand field, e.g., by changes of the Mn–ligand distance (HS in Mn–porphyrins,20 IS in Mn–Pc15) or of the ligand peripheral coordination (HS in Mn-TCNQ4 metal–organic covalent networks).21 Interestingly, it has been recently demonstrated that ruthenium tetraphenylporphyrin (RuTPP) on Ag(111) can be stabilized in two different conformations, saddle-shaped and planar, and only the former may axially interact with CO as a consequence of its greater molecular flexibility.22 As such, the role played by the molecular flexibility in prompting the electronic and magnetic properties of TMTPP after electron doping is an essential aspect whose potential applications are not yet sufficiently investigated.
In this work, the influence of the molecular conformation in the metal reduction of AM-doped TMTPP is tackled by combining X-ray photoemission and absorption spectroscopies with photoemission tomography and density functional theory (DFT) calculations. The charge donation induced by the bonding of a single K atom to NiTPP, and determining the Ni reduction, is almost unaffected by the K adsorption site. However, for higher K doses and then larger electron donation, the charge transfer is not limited to the Ni-based 3d AOs, but also involves the TPP-based lowermost unoccupied MOs. Two different adsorption configurations involving two K atoms per molecule are found to be energetically stable. In the flattest macrocycle, the Ni(II) + K → Ni(I) + K+ redox reaction takes place, while the relaxation to the energetically-favored distorted form leads to the concomitant nickel re-oxidation and the macrocycle doping increase. Such a result demonstrates the pivotal role played by the macrocycle flatness of the K-doped NiTPP in stabilizing the Ni(I) species. In addition to that, the TM role has been studied by investigating the K doping of CoTPP. The susceptibility to molecular conformation decreases for CoTPP and, unlike NiTPP, the Co(II) → Co(I) reduction takes place in both energetically stable configurations involving two K atoms per molecule.
1 Results and discussion
TM oxidation and spin state variations in TMPc and TMPs-based systems can be triggered by coordinating them to metal surfaces, and the charge transfer eventually taking place is strongly TM and substrate-dependent. Indeed, the adsorption on the highly reactive copper surface implies the filling of the adsorbate lowermost unoccupied MOs (up to the LUMO+3) and the concomitant TM(II) → TM(I) reduction.18,19,23,24 Charge injection into the metal complex may also be induced by exploiting alternative pathways such as AM doping. Aimed to look into the TM reduction process, a NiTPP layer deposited on the Au(111) surface has been stepwise K doped. The chemical inertness of the substrate assures that any variation of the NiTPP molecular properties eventually revealed can be traced back to the K doping. Valence band (VB) and near-edge X-ray absorption fine structure (NEXAFS) spectra recorded at the N K-edge were used to track the progressive filling of TPP-based MOs as the K doping is increased. Moreover, the K doping effect on the Ni 3d AOs was studied by using Ni 2p photoemission spectroscopy and Ni L2,3-edges NEXAFS measurements.1.1 Charge and spin donation to macrocycle molecular orbitals
Work function changes, Δϕ (a), the angle-integrated VB spectra (b) and the N K-edge spectra (d) of a single layer of NiTPP deposited on the Au(111) substrate as a function of the K doping dose are displayed in Fig. 1. Dose-dependent K 2p core-level spectra are reported in Fig. S1 in ESI,† together with a semi-quantitative analysis of the K coverage with respect to the molecular layer. Before going on, it may be useful to remind the reader that VB probes high-lying occupied levels, while NEXAFS provides information about electronic transitions from core levels to low-lying empty MOs. Spectra of the pristine NiTPP layer (0′) have also been recorded and added to the figure for comparison.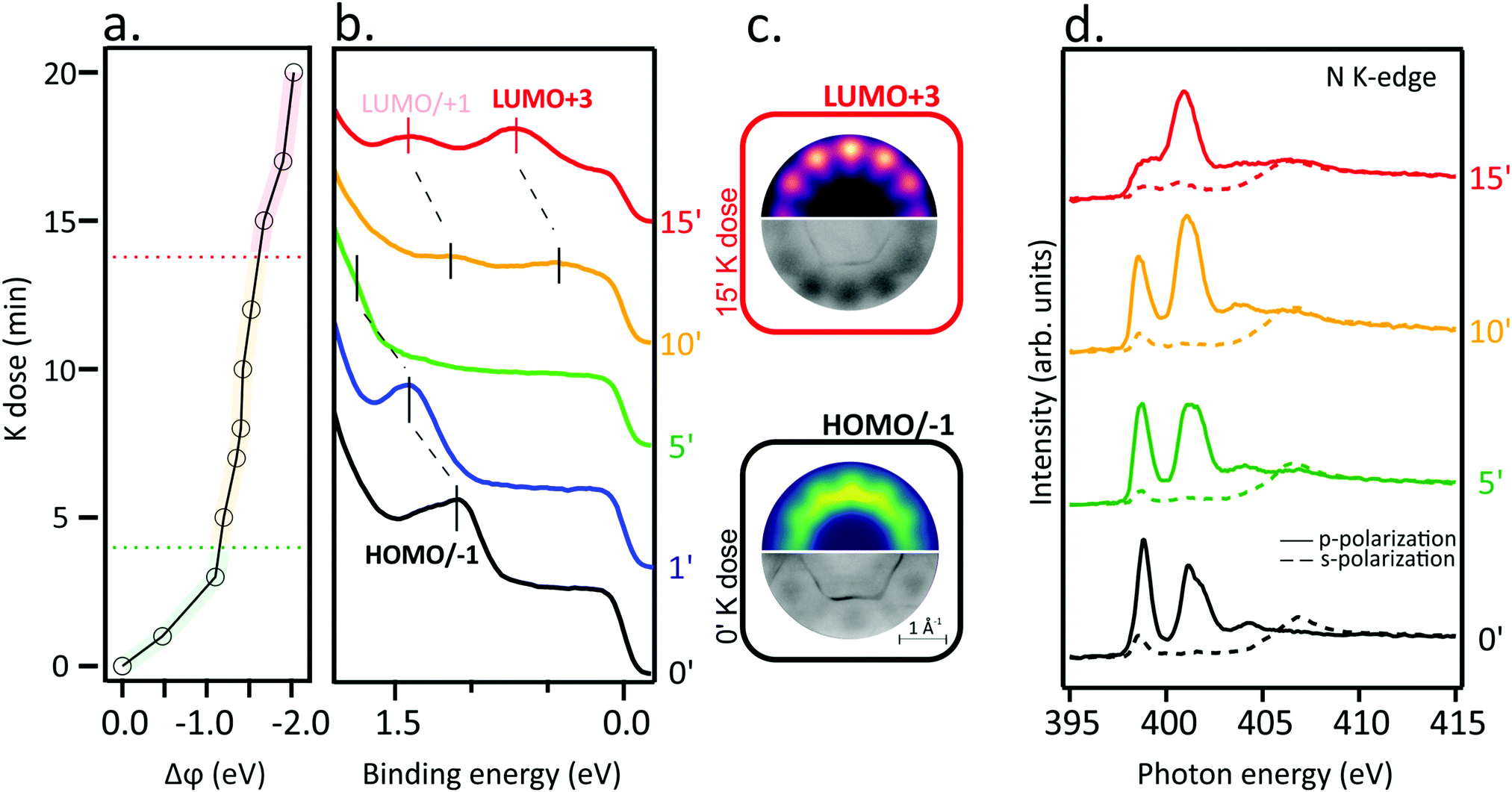 | ||
| Fig. 1 (a) Work function changes (Δϕ), (b) valence band spectra of NiTPP on Au(111) exposed to increasing K doses, (c) Simulated (top) and experimental (bottom) momentum maps identifying the last occupied molecular orbital before and after 15 min K dose, (d) N K-edge spectra of NiTPP on Au(111) exposed to increasing K doses (5, 10 and 15 minutes). | ||
The VB spectrum of the pristine NiTPP layer deposited on Au(111) is consistent with a weak adsorbate–substrate interaction: no peak due to charge transfer phenomena, frequently present at strongly interacting adsorbate–substrate interfaces,23,25,26 is observed and only features determined by the ionization from the topmost occupied MOs (HOMO/HOMO−1) are detected (see Fig. 1(b) and (c)) and assigned using the photoemission tomography (PT) method (see Methods section for further details). The assignment is in agreement with Scudiero et al.,27 according to whom the topmost occupied MOs correspond to the quasi-degenerate TPP-based a2u and a1u π MOs, as corroborated by exploiting the Slater transition state (STS) approximation.28
The weak interaction of NiTPP on Au(111) is confirmed by the shape of the N K-edge NEXAFS spectra (Fig. 1(d)). The major characteristics of the N K-edge NEXAFS spectra pre-edge region, both in terms of energy position and line-shape, are comparable to those recorded for NiTPP deposited on an inert substrate24,29 either as multilayer or as a self-assembled monolayer. The sharp peak at 398.9 eV is associated with excitations from N 1s into the doubly degenerate LUMO/LUMO+1 (eg) and LUMO+3 (b2u) levels having π* character.30 Excitations to higher-lying unoccupied π* MOs lead to the spectral feature at 401.1 eV.31 A σ*-symmetry resonance observed in the spectra obtained with s-polarized light at a photon energy of 398.6 eV results from the N 1s level to the nickel-based σ-type LUMO+2 (b1g) transition.29,30 The strong linear dichroism in the N K-edge spectra in the range of the π*-symmetry resonances indicates that the porphyrin macrocycle adsorbs parallel to the surface plane. On metal substrates, extended π-conjugated molecules frequently adopt such a comparable flat-lying geometry.11,32,33
The electron injection into low-lying empty MOs induced by the K doping should determine complementary changes in the NiTPP VB and NEXAFS spectra; specifically, the partial filling of TPP-based π* MOs should be accompanied by the emergence of new features in the VB spectra and intensity decrease of the low-lying NEXAFS resonances. Indeed, there is a dramatic change in the N K-edge NEXAFS spectra with increasing K doping, where the lowest energy resonance significantly alters the intensity weight after the K dosage, as a result of the partial filling of the lowermost unoccupied MOs. The inspection of Fig. 1(b) reveals that, at high K doses, new features enrich the VB spectrum around 0.7 and 1.4 eV. Theoretical and experimental momentum maps at the corresponding peak maximum (Fig. 1(c) top and bottom, respectively) provide information about the former empty MOs, now partially occupied by electron injection. In particular, simulated patterns are assigned to the LUMO/LUMO+1 and LUMO+3 levels, which is consistent with the NEXAFS results. Such a remarkable charge transfer (up to the LUMO+3) has already been revealed for the aforementioned NiTPP/Cu(100) and CoTPP/Cu(100) interfaces.18,19,23 As a final remark, the peculiar work function (WF) trend as a function of the K dose (see Fig. 1(a)) deserves to be highlighted: (i) a significant WF drop, indicative of the formation of a new surface dipole induced by the adsorption of the K atoms at the interface, is present at the lower K doping level; such WF change is accompanied by a HOMO blue shift with respect to the Fermi level; (ii) a plateau region leading to the complete filling of the gas-phase unoccupied MOs up to LUMO+3 characterizes the intermediate K coverage; this range corresponds to the region of interest for the electron doping of the molecular layer; (iii) a further, less pronounced, WF drop determining a blue-shift of low-lying empty MO (LUMO/+1 and LUMO+3) takes place at higher K doses, which corresponds to the appearance of neutral K species in the K 2p XPS spectrum (see Fig. S1 in ESI†). Similar effects on the VB spectra were previously reported for AM-doped TMPc layers.34–36
1.2 Spin and oxidation states of the Ni ion in electron-doped NiTPP
So far, the effects associated with the filling of the low-lying empty TPP-based MOs have been discussed just by referring to the VB and N K-edge NEXAFS outcomes. Next, we look at the K doping effect on the nickel 3d levels. To that end, X-ray photoemission spectroscopy (XPS) spectra at the Ni 2p core level and Ni L3-edge NEXAFS measurements on the pristine NiTPP deposited on Au(111) have been recorded; their evolution as a function of the K doping has been then tracked (see Fig. 2). The Ni 2p photoemission spectrum of the undoped NiTPP layer on Au(111) (see Fig. 2(a)) shows a single and well-resolved peak at 855.1 eV accompanied by a satellite feature located at higher BE. Such evidence would indicate a Ni(II) oxidation state, which has been confirmed by the corresponding Ni L3-edge spectrum (see Fig. S2 in ESI†). The L3-spectra is characterized by a primary peak at 854.3 eV and the strong satellite A at higher photon energy (856.2 eV). The observed spectral shape is a fingerprint of a Ni(II) in a tetra-coordinated square-planar geometry.15,24,37–39 Moreover, the presence of the extra satellite peak A in the spectra of NiTPP/Au(111) is indicative of a nickel in a low spin (LS) 3d8 electronic configuration.15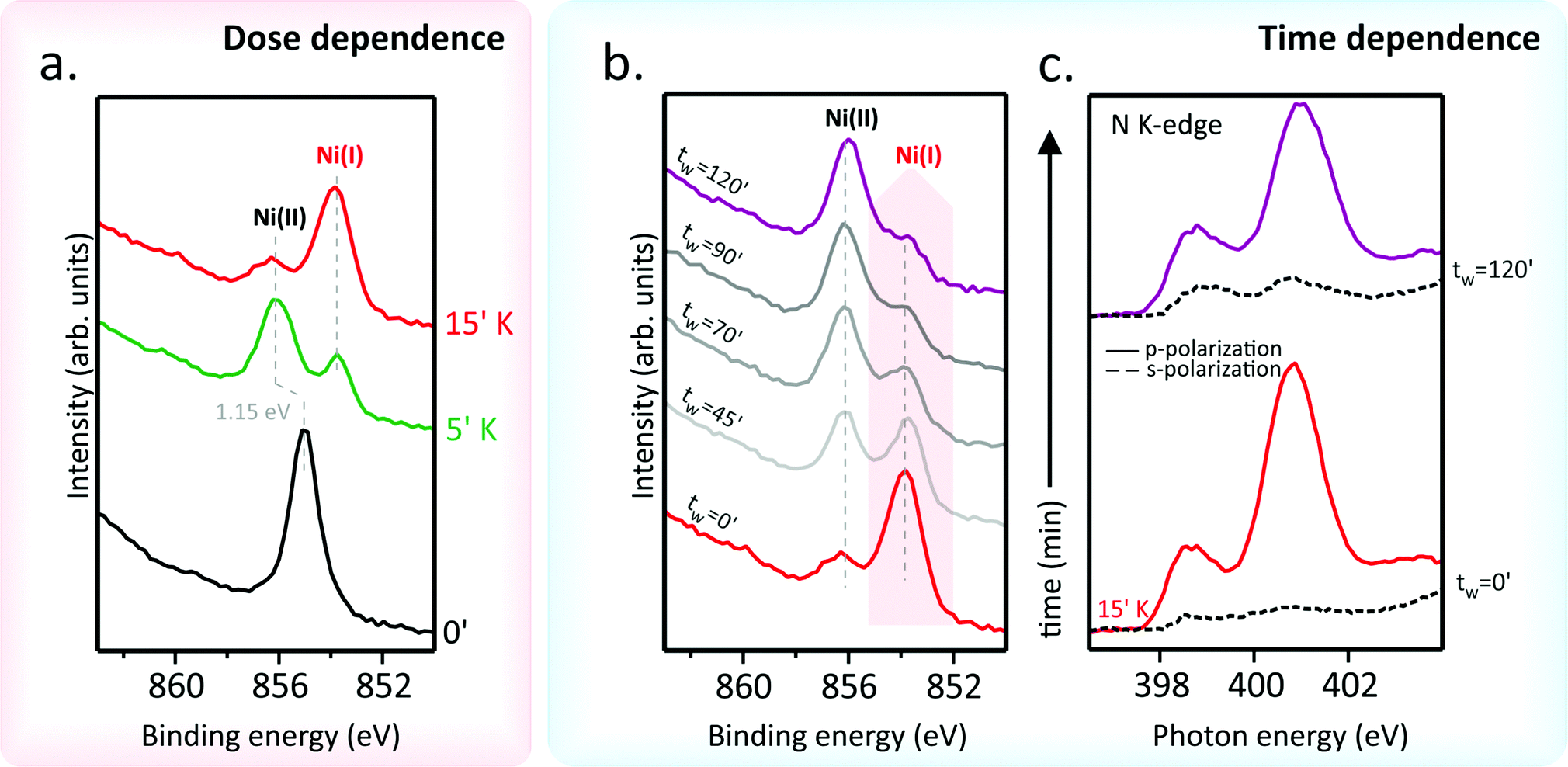 | ||
| Fig. 2 (a) Ni 2p3/2 XPS spectra measured on the pristine NiTPP/Au(111) layer and after exposure to increasing K doses (5 and 15 minutes). Nickel oxidation states are evidenced in the spectra. Time dependent (b) Ni 2p3/2 XPS and (c) N K-edge NEXAFS spectra. The waiting time (tw) after the dose is evidenced for every spectrum. The reference spectrum (tw = 0′) is recorded immediately after the 15 minutes K dose on freshly prepared NiTPP/Au(111). | ||
The K doping of the NiTPP monolayer significantly affects both XPS and NEXAFS spectra. In particular, the gradual Ni(II) → Ni(I) reduction induced by the electron injection due to the K adsorption on the NiTPP layer is well evident in the Ni 2p3/2 core-level spectra (Fig. 2(a)) where a new, red-shifted feature with BE = 852.8 eV progressively increases its intensity with K doping. Likewise, in the Ni L3-edge NEXAFS spectra (see Fig. S2 in ESI†), as the K concentration increases, a new peak emerges on the low-energy side of the original L3 main peak. The shift to lower photon energies, as well as the energy position of the new resonance, is consistent with the Ni ion reduction24 induced by the electron doping via AM. As such, it has to be noted that the intensity of the new Ni(I) peak increases at the expense of the Ni(II) resonance and its satellite A, whose drop in intensity implies a partial filling of the Ni 3dx2−y2-based b1g MO. Filling one of the spin-paired holes in the dx2−y2 orbitals results in a free spin 1/2, thus inducing a magnetic moment in the Ni ion. Current data reveal that NiTPP behaves similarly to NiPc, where magnetism is induced by the adsorption of Li atoms on top of the NiPc layer, as confirmed by XMCD measurements.15 To investigate the stability of the Ni(I)TPP species, Ni 2p XPS spectra have been collected as a function of the time (tw) after the 15 minutes K dose (see Fig. 2(b)). To this end, the NiTPP/Au(111) interface has been freshly reprepared before the 15 minutes K doping. The main feature of the pristine Ni 2p3/2 core level spectrum has been assigned to Ni(I) which, after tens of minutes, re-oxidizes to Ni(II). Such a process takes a 90 minute tw interval to complete at room temperature. This reconversion process is also observed on the same time scale for smaller doses of K, namely 5 and 10 minutes, proving to be dose-independent. In addition, time-dependent N K-edge NEXAFS spectra (Fig. 2(c)) show distinct changes when the Ni(I) to Ni(II) transition takes place. As such, it is noteworthy that the spectra linear dichroism at tw = 0′ is perfectly consistent with a flat adsorption geometry of the porphyrin macrocycle, while the dichroic behavior of π* resonances in s-polarization at tw = 120′ suggests its distortion. Moreover, the different relative intensity of the two π* resonances in p-polarization at tw = 0′ and 120′ indicates an increase of the overall charge transfer on the macrocycle MOs when observing the Ni(I) → Ni(II) conversion.
1.3 Structural determination of the Ni(I) and Ni(II) molecular species
At this point, the question that spontaneously arises concerns the structure of the K doped NiTPP species, not only because it is probably related to the Ni(I) → Ni(II) conversion, but it also involves the bonding site of K atoms on NiTPP. As such, DFT-based numerical experiments may provide insights into the electronic and geometrical structure of adsorbed species upon increasing the K dosing. As previously remarked, the NEXAFS measurements of the bare molecular adlayer reveal that its electronic structure is unaffected by the Au substrate, hence it may be regarded as a free-standing layer. In this case, a molecular cluster approach provides a quantitative analysis of the atomic and molecular orbitals modification and corresponding charge exchange.15,21 We tested different adsorption geometries by increasing the number of K atoms directly bonded to NiTPP.Low K doses have been modeled by bonding a single K atom to NiTPP. Different stable K adsorption sites are found: (i) K directly bonded to Ni in the top position, (ii) K adsorbed in the hollow position, and (iii) K adsorbed in the bridge position (see Fig. 3). While the K atom has a single unpaired electron in its isolated electronic structure, the chelated TM ion in the NiTPP on the gold surface is stabilized in the Ni(II) LS 3d8 closed-shell configuration with no unpaired electron. A single unpaired electron is then assumed for the different K-NiTPP clusters. The structure analysis (see Table 1) reveals that the hollow adsorption configuration is more stable than the top and bridge ones by 0.12 eV and 0.07 eV, respectively. The higher stability of the hollow configuration may be justified by the formation of a higher number of interactions between K and the TPP moiety, as can be appreciated in the structural models reported in Fig. 3. Such small energy differences among the structures suggest that all absorption sites are accessible at room temperature. Data reported in Table 1 also point out that, independently of the assumed geometry, the Ni(II) + K → Ni(I) + K+ redox reaction systematically takes place. It is of some interest to underline that, at large K⋯NiTPP distances (see Fig. 3 and Table 1), the Ni(II) reduction is inhibited and the K⋯NiTPP system is destabilized by 1.20 eV with respect to the hollow geometry. In tune with the Ni–N antibonding character of the Ni 3dx2−y2-based MO, its partial filling induced by electron injection implies that the Ni–N Nalewajski–Mrozek index (NMINi–N in Table 1) decreases upon K binding; the opposite is true for NMINi–K.
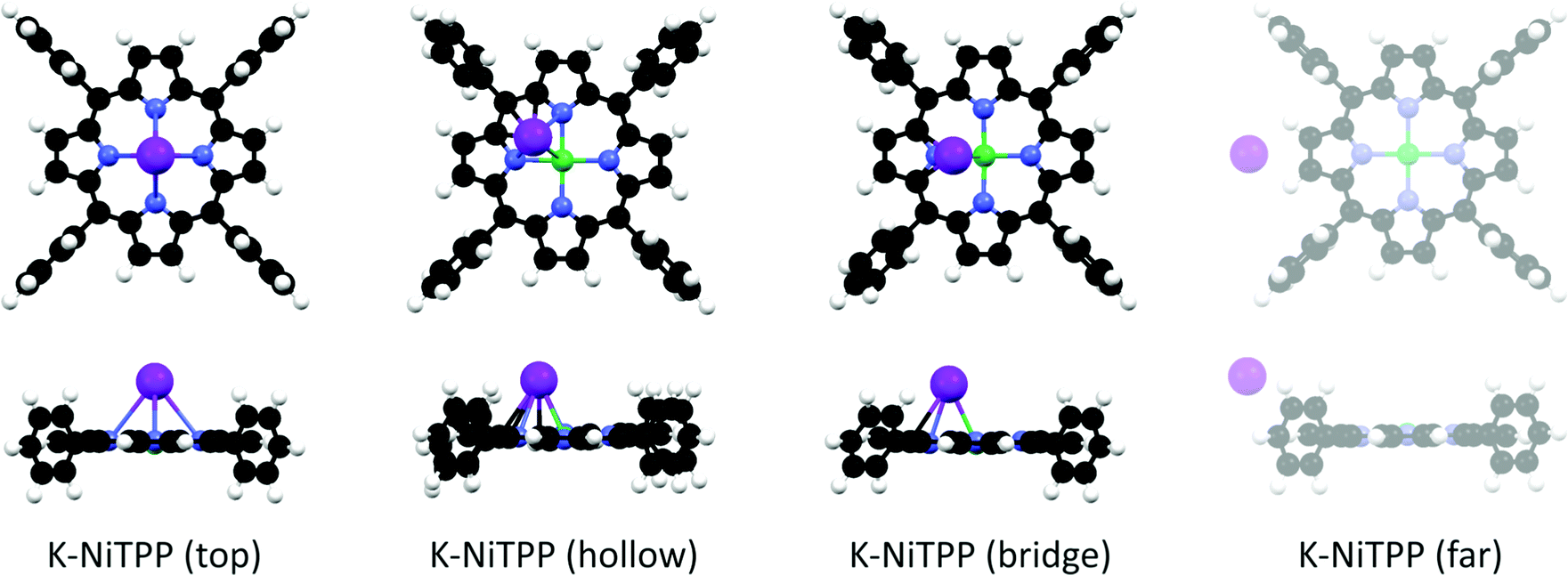 | ||
| Fig. 3 A schematic representation of the different K adsorption geometries calculated for the K-NiTPP complex. Black, white, blue, green, and violet spheres represent C, H, N, Ni, and K, respectively. | ||
| ΔE eV | E ads eV | Ni unpaired el. | Ni ox state | NMINi–N avg | NMINi–K | |
|---|---|---|---|---|---|---|
| *In the K-NiTPP (far) complex, there is not a direct Ni–K bond. See methods for details. | ||||||
| K-NiTPP (top) | 0.12 | −1.24 | 0.88 | Ni(I) d9 | 0.380 | 0.136 |
| K-NiTPP (hollow) | 0.00 | −1.37 | 0.75 | Ni(I) d9 | 0.387 | 0.095 |
| K-NiTPP (bridge) | 0.07 | −1.30 | 0.86 | Ni(I) d9 | 0.383 | 0.115 |
| K-NiTPP (far) | 1.20 | −0.16 | 0.32 | Ni(II) d8 | 0.416 | —* |
Higher K doses on the NiTPP/Au(111) interface, which retrace experimental data, have been modeled by bonding two K atoms to NiTPP. Numerical experiments outcomes indicate that two stable structures are present, labeled as A and B in Fig. 4.
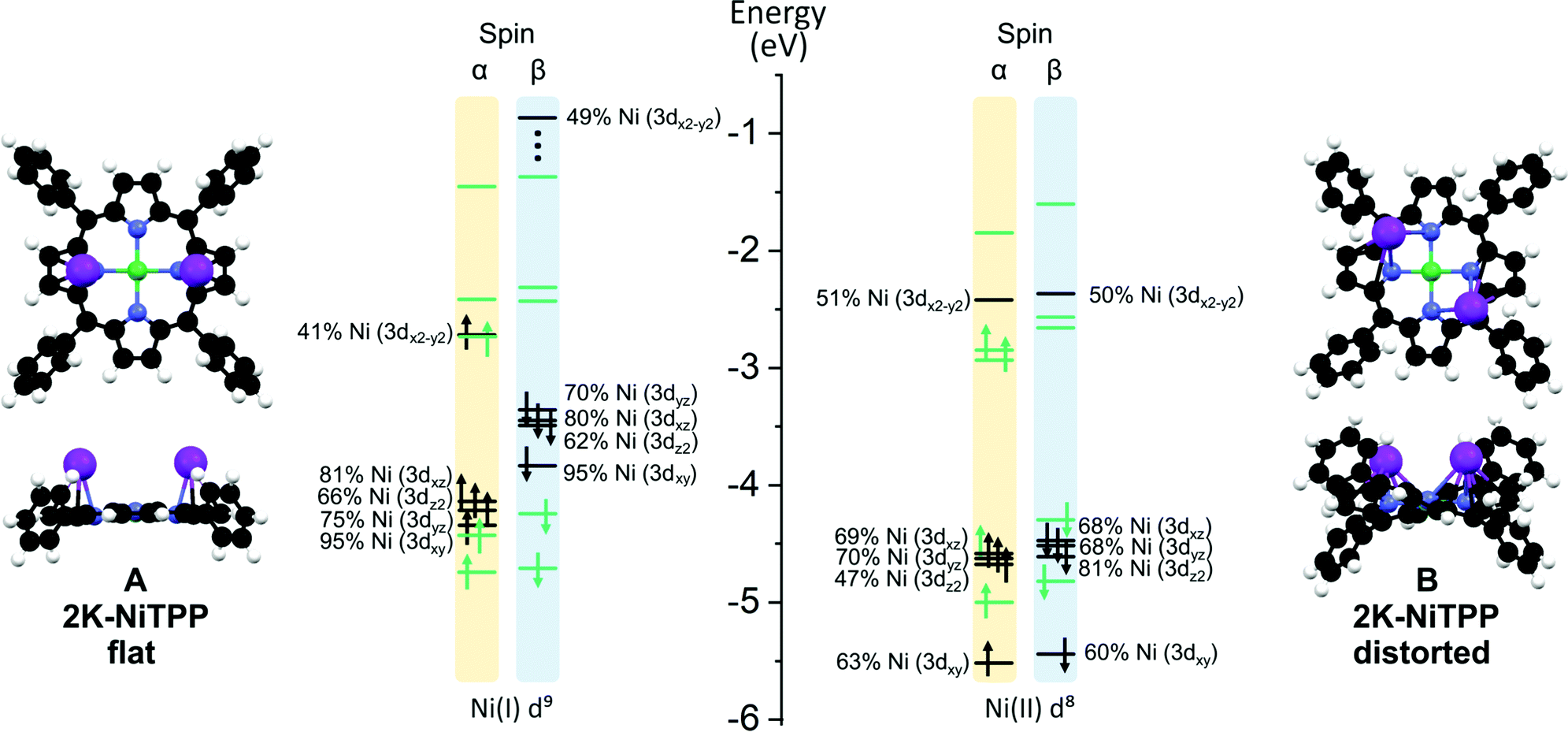 | ||
| Fig. 4 A schematic representation of the different conformations (A) and (B) calculated for the 2K-NiTPP complex. Color codes are the same adopted in Fig. 3. On the right side of each configuration is reported the energy level diagram for selected MOs, Ni-based and TPP-based levels are highlighted in black and green, respectively. Both α and β spin are reported. | ||
The inspection of the results reported in Table 2 indicates that, both in A and B, the dopant species is present in its oxidized form. Interestingly, the porphyrin macrocycle in A is very flat, while it is significantly distorted in B (see Fig. 4); in addition, the former conformation is less stable than the latter one by 0.23 eV (see Table 2). These conformational changes induce a different charge donation from K atoms to NiTPP. More specifically, the electron injection determined by the high K dosing involves both the Ni(II) 3dx2−y2-based MO and a TPP2−-based MO in A, while in B, the charge transfer does not involve the metal center, which maintains the same LS 3d8 configuration of the pristine NiTPP on Au(111). Similarly to K-NiTPP, the Ni(II)–N bond is stronger than the Ni(I)–N one (see the NMINi–N in Table 2). As a whole, the DFT outcomes pertaining to the models adopted to mimic low and high K doping of the NiTPP/Au(111) interface agree well with experimental evidence. In particular, they provide a rationale for the experimental temporal evolution from the flat conformation A, characterized by the presence of the paramagnetic Ni(I) species, to the thermodynamically most stable distorted geometry B typified by the occurrence of the diamagnetic Ni(II) species (see Fig. 2(b) and (c)).
| ΔE eV | E ads eV | Ni unpaired el. | Ni ox. state | NMINi–N avg | NMINi–K avg | |
|---|---|---|---|---|---|---|
| A – 2K-NiTPP flat | 0.23 | −2.19 | 1.00 | Ni(I) d9 | 0.384 | 0.075 |
| B – 2K-NiTPP distorted | 0.00 | −2.41 | 0.04 | Ni(II) d8 | 0.492 | 0.038 |
The interaction of NiTPP with three K atoms has also been considered and, in agreement with the experiment and 2K-NiTPP theoretical results, the 3K-NiTPP outcomes confirm that: (i) once again, the most stable species is associated with the Ni(II) presence (see Table S1 in ESI†); (ii) Ni(II) is always associated to the most distorted geometries (see Fig. S3 in ESI†), thus evidencing the relevant role played by the molecular distortion in determining the Ni oxidation state even in the presence of a large number of the electron donor K atoms.
2 From NiTPP to CoTPP
TMPcs with diverse TM act differently upon AM doping;15 in the following, we focused on CoTPP deposited on Au(111) to look into its eventually different behavior from NiTPP upon the AM dosing.The Co 2p3/2 core level spectrum of CoTPP self-assembled on the Au(111) surface (see Fig. 5(a)) is similar to the spectra reported in ref. 40, and analogously to the free molecule, CoTPP deposited on Au(111) has an LS 3d7 configuration with its single unpaired electron occupying the Co(II) 3dz2-based MO.17,33,41,42 The K binding to CoTPP red-shifts the spectrum main feature by 0.85 eV, a behavior tightly linked to the Co(II) → Co(I) reduction.19 Moreover, the inspection of Fig. 5(b), where the N K-edge spectra of the pristine CoTPP/Au(111) and after 15 minutes K dosing in s- and p-polarization are reported, clearly shows that the macrocycle distortion takes place right after the K doping.
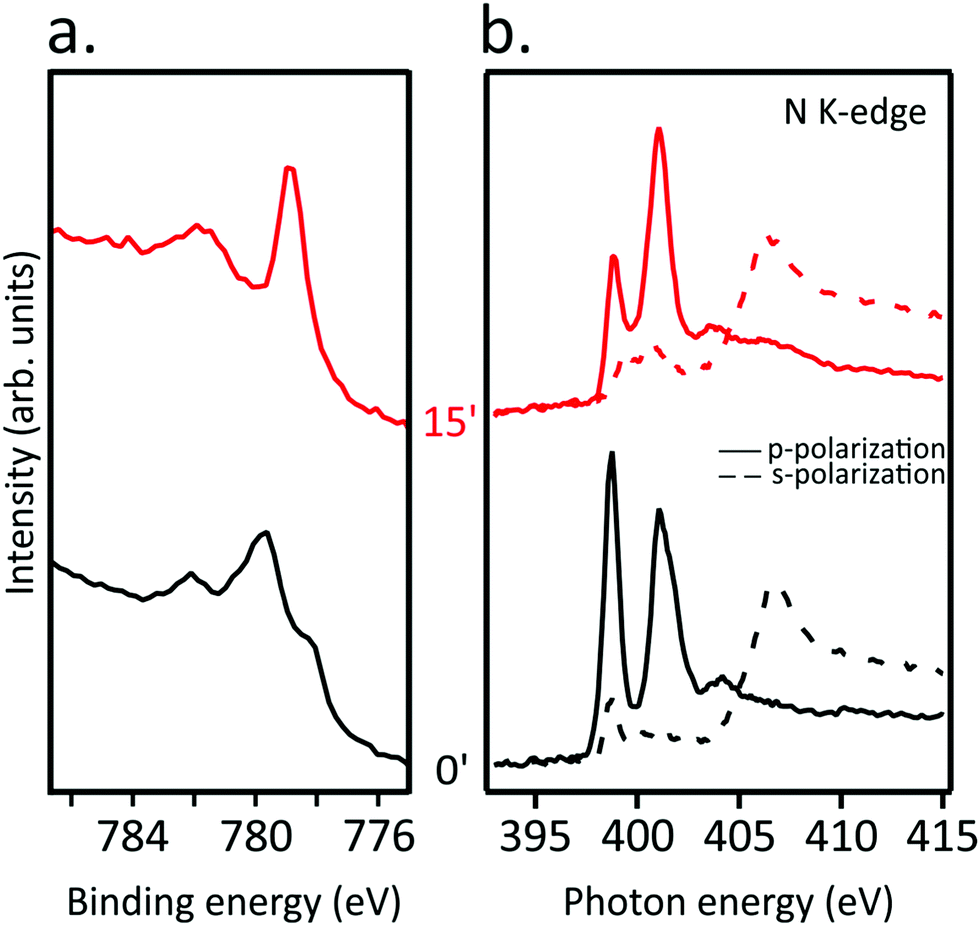 | ||
| Fig. 5 (a) Co 2p3/2 core level and (b) N K-edge NEXAFS spectra for pristine CoTPP/Au(111) (black curves) and after exposure to K for 15 minutes (red curves). | ||
Similarly to NiTPP, different K doses have been modeled by directly bonding either one or two K atoms to CoTPP. K-NiTPP and K-CoTPP DFT results are very similar; specifically: (i) three distinct adsorption sites are possible when a single K atom is considered (top, bridge, hollow position) (see Fig. 6(a)), (ii) the most stable configuration is the one with the K atom in the hollow site, (iii) followed by the bridge (+0.05 eV) and the top (+0.11 eV) adsorption structures (see Table 3), closely spaced in energy. In addition, the bonding energy of the K-CoTPP cluster is tightly and directly related to the K-CoTPP bonding interaction, which determines the distortion of the macrocycle fragment. Indeed, the largest distortion is predicted for the most stable hollow configuration (see Fig. 6), while an almost CoTPP flat geometry is foreseen for the top position. K-CoTPP adsorption energies are −1.76, −1.82 and −1.87 eV for top, bridge and hollow sites, respectively. Interestingly, the comparison of theoretical outcomes pertaining to K-CoTPP and K-NiTPP clearly indicates that the K-CoTPP interaction is stronger than the Ni-TPP one (see Tables 1 and 3).
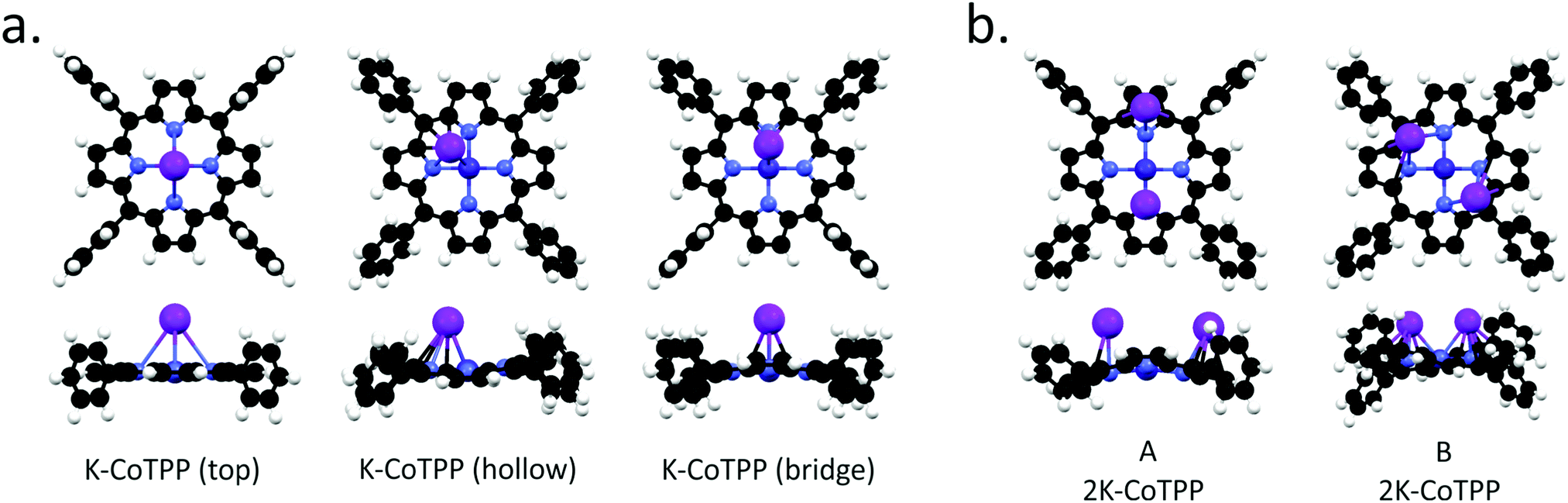 | ||
| Fig. 6 A schematic representation of the different K adsorption geometries calculated for the K-CoTPP (a) and 2K-CoTPP (b) complexes. Black, white, blue, light blue, and violet spheres represent C, H, N, Co, and K, respectively. | ||
| ΔE eV | E ads eV | Co unpaired el. | Co ox. state | NMICo–N avg | NMICo–K | |
|---|---|---|---|---|---|---|
| K-CoTPP (top) | 0.11 | −1.76 | 0.36 | Co(I) d8 | 0.659 | 0.253 |
| K-CoTPP (hollow) | 0.00 | −1.87 | 0.42 | Co(I) d8 | 0.662 | 0.191 |
| K-CoTPP (bridge) | 0.05 | −1.82 | 0.40 | Co(I) d8 | 0.662 | 0.210 |
| A – 2K-CoTPP | 0.30 | −2.59 | 0.39 | Co(I) d8 | 0.644 | 0.142 |
| B – 2K-CoTPP | 0.00 | −2.88 | 0.37 | Co(I) d8 | 0.636 | 0.158 |
Both the free CoTPP molecule and the K atom hold a single unpaired electron in their electronic structure hence two spin states are possible in the K-CoTPP complexes: zero or two unpaired electrons. The zero unpaired electron structure is more stable than the two unpaired electrons one by more than 1 eV. The detailed electronic analysis reveals that in all structures reported in Fig. 6, the chelated Co ion gains an electron from K (see Table 3) and is stabilized in the closed-shell configuration with the empty 3dx2−y2 MO. This scenario is consistent with an electron transfer from K to Co and the stabilization of the Co(I) and K+ ions in K-CoTPP complexes.
Analogously to NiTPP, higher K doses have been modelled by bonding to CoTPP two K atoms. Even though once again, LS (one unpaired electron) and HS (three unpaired electrons) states are possible, the LS one is always more stable than HS by 0.7 eV. Similarly to 2K-NiTPP, two different stable structures (see Fig. 6) are obtained for LS; moreover, the B arrangement is more stable than the A one by 0.30 eV (see Table 3). Incidentally, the presence of two K atoms induces significant structural perturbations both in A and B arrangements but does not affect the Co(I) oxidation state. As such, the A and B spin population analysis indicate that, consistently with a Co(I) 3d8 configuration, Co has 0.39 unpaired electron in A and 0.37 in B. Such a result clearly states that the two electrons transferred from K to CoTPP in 2K-CoTPP are used for inducing the Co(II) → Co(I) reduction and the partial occupation of a low-lying TPP-based empty MO. Differently from NiTPP, no stable structure with two K atoms and Co(II) is found.
DFT outcomes agree well with the experimental evidence. In particular, time-dependent Co 2p3/2 spectra on a freshly prepared K-doped CoTPP layer demonstrate, differently from NiTPP, the Co(I) high stability not only at room temperature but also when annealed up to 500 K (see Fig. S4 in ESI†). The charge donation from K atoms in both the CoTPP conformations stabilizes the chelated ion in the Co(I) oxidation state. Moreover, the time-dependent NEXAFS spectra measured at N K-edge, in particular the linear dichroic spectral behavior, show strong distortion of TPP macrocycle already in the as-deposited configuration and no significant time-dependent change is observed.
3 Conclusion
In this paper, we investigated the role of molecular conformation on the reduction of a chelated metal ion and, as a result, the stabilization of various oxidation and spin states. Two different MTPPs, characterized by a different electron affinity towards the K dopant, have been studied. We show that when the alkali metal is strongly bonded to the central ion, the doping is very little influenced by molecular distortion, while for the more labile 2K-NiTPP complex, the molecular distortion plays a crucial role. Moreover, the flattening of the macrocycle moiety of AM doped metal TPP is a key factor in the Ni(II) → Ni(I) reduction and this transition is not observed in distorted NiTPP molecules. Thus, multiple spin configurations can be achieved depending on the conformational shape, proving the importance of flexibility in determining the molecular properties.4 Methods
4.1 Sample preparation
The clean Au(111) surface was prepared by a standard procedure involving repeated cycles of Ar+ ion sputtering at 2.0 keV followed by annealing at 800 K. NiTPP and CoTPP molecules (Porphyrin system, ≥95% purity) were thermally sublimated at 520 K from a home-made Knudsen cell-type evaporator. Before the experiments, the molecules were carefully outgassed at 480 K for several days while the UHV system base pressure was monitored. The coverage has been calibrated with a quartz micro-balance. The resulting deposition rate was 15 min mL−1. The nominal coverage for all the experiments presented here is monolayer coverage. Potassium was sublimated from a properly outgassed SAES-getter dispenser, keeping the sample at room temperature and without any detectable pressure enhancement during deposition.4.2 X-ray photoemission and absorption spectroscopies
The valence band photoemission spectra were measured at the NanoESCA beamline of Elettra, the Italian synchrotron radiation facility in Trieste, using an electrostatic photoemission electron microscope (PEEM) set-up described in detail in ref. 43. Data were collected with a photon energy of 30 eV and a total energy resolution of 100 meV, using p-linearly polarized light, while keeping the sample at 80 K. The XPS and NEXAFS spectra were collected using the end-station at the ALOISA beamline situated at the Elettra synchrotron radiation facility in Trieste and analyzed following the procedure described elsewhere.44,45 The orientation of the surface in the NEXAFS experiment with respect to the linear polarization (s and p) of the synchrotron beam was changed by rotating the sample around the beam axis while keeping the incident angle (6°) of the synchrotron light fixed. The binding energy scale was referred to as the metallic Fermi level. XPS spectra at the Ni 2p and Co 2p levels were acquired using a photon energy of 1020 and 980 eV, respectively.4.3 Photoemission tomography and DFT calculations
The photoemission tomography (PT) method combines two-dimensional momentum mapping with density functional theory simulations. The momentum pattern originating from a certain initial molecular state can be represented as the Fourier transform (FT) of the initial-state wave function within certain assumptions about the final state of the photo-emitted electron.46,47 The momentum maps, or photoelectron momentum distribution patterns at a specific binding energy, allow each emission to be ascribed to a specific molecular orbital, allowing for a proper description of the energy level alignment.23,48–51 Within the PT approach, density functional calculations for gas-phase NiTPP (NiC44N4H28) have been performed by employing the NWCHEM code.52 The geometries of the molecules have been optimized using the Becke, 3-parameter, Lee–Yang–Parr (B3LYP) exchange-correlation functional53,54 and the 6-31G* basis set. The simulated momentum maps of the gas-phase molecules were obtained as the Fourier transforms of the respective Kohn–Sham orbitals as described previously.7,46 Calculations can be found online at the Organic Metal Interfaces database (https://143.50.77.12:5001). All optimized structures have been obtained without any constraint by exploiting the Amsterdam Density Functional (ADF) suite of programs55 by using the GGA functional BP86,56,57 by employing a triple-ζ with one polarization function Slater-type basis set58 for all the atoms and by freezing throughout the calculations the K, C, N and O 1s atomic orbital (AO), the Co and Ni 1s–2p AOs. 3D contour plots (CPs) have also been obtained to get information about the localization and the character of frontier MOs. To compare the different structures, their stabilities, the adsorption energies (Eads) and the Nalewajski–Mrozek bond multiplicity indexes59 are considered. The stabilities (ΔE) are calculated with respect to the most stable structure. Thus, they are relative stabilities, but allow to compare different systems with the same number of K dopant in different positions. Adsorption energies (Eads) of the K dopant to the TMTPP have been obtained as the difference between system after the interaction with the dopant and the isolated ones:| Eads = EK-TMTPP − (EK + ETMTPP) |
Conflicts of interest
There are no conflicts to declare.References
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- A. A. Khajetoorians, J. Wiebe, B. Chilian and R. Wiesendanger, Science, 2011, 332, 1062–1064 CrossRef CAS.
- F. D. Natterer, K. Yang, W. Paul, P. Willke, T. Choi, T. Greber, A. J. Heinrich and C. P. Lutz, Nature, 2017, 543, 226–228 CrossRef CAS.
- D. A. Reed, B. K. Keitz, J. Oktawiec, J. A. Mason, T. Runčevski, D. J. Xiao, L. E. Darago, V. Crocellà, S. Bordiga and J. R. Long, Nature, 2017, 550, 96–100 CrossRef CAS PubMed.
- V. A. Dediu, L. E. Hueso, I. Bergenti and C. Taliani, Nat. Mater., 2009, 8, 707–716 CrossRef CAS.
- W. Xu, G. J. Szulczewski, P. LeClair, I. Navarrete, R. Schad, G. Miao, H. Guo and A. Gupta, Appl. Phys. Lett., 2007, 90, 072506 CrossRef.
- K. Yang, H. Chen, T. Pope, Y. Hu, L. Liu, D. Wang, L. Tao, W. Xiao, X. Fei, Y.-Y. Zhang, H.-G. Luo, S. Du, T. Xiang, W. A. Hofer and H.-J. Gao, Nat. Commun., 2019, 10, 3599 CrossRef.
- B. W. Heinrich, L. Braun, J. I. Pascual and K. J. Franke, Nano Lett., 2015, 15, 4024–4028 CrossRef CAS PubMed.
- B. W. Heinrich, C. Ehlert, N. Hatter, L. Braun, C. Lotze, P. Saalfrank and K. J. Franke, ACS Nano, 2018, 12, 3172–3177 CrossRef CAS PubMed.
- W. Hieringer, K. Flechtner, A. Kretschmann, K. Seufert, W. Auwärter, J. V. Barth, A. Görling, H.-P. Steinrück and J. M. Gottfried, J. Am. Chem. Soc., 2011, 133, 6206–6222 CrossRef CAS PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- L. Liu, K. Yang, Y. Jiang, B. Song, W. Xiao, L. Li, H. Zhou, Y. Wang, S. Du, M. Ouyang, W. A. Hofer, A. H. Castro Neto and H.-J. Gao, Sci. Rep., 2013, 3, 1210 CrossRef PubMed.
- Y. Wang, X. Li, X. Zheng and J. Yang, J. Chem. Phys., 2017, 147, 134701 CrossRef.
- B. Liu, G. Miao, W. Zhong, X. Huang, N. Su, J. Guo and W. Wang, ACS Nano, 2022, 16, 2147–2153 CrossRef CAS PubMed.
- S. Stepanow, A. Lodi Rizzini, C. Krull, J. Kavich, J. C. Cezar, F. Yakhou-Harris, P. M. Sheverdyaeva, P. Moras, C. Carbone, G. Ceballos, A. Mugarza and P. Gambardella, J. Am. Chem. Soc., 2014, 136, 5451–5459 CrossRef CAS.
- C. Krull, R. Robles, A. Mugarza and P. Gambardella, Nat. Mater., 2013, 12, 337–343 CrossRef CAS.
- C. Wäckerlin, D. Chylarecka, A. Kleibert, K. Müller, C. Iacovita, F. Nolting, T. A. Jung and N. Ballav, Nat. Commun., 2010, 1, 61 CrossRef PubMed.
- H. M. Sturmeit, I. Cojocariu, A. Windischbacher, P. Puschnig, C. Piamonteze, M. Jugovac, A. Sala, C. Africh, G. Comelli, A. Cossaro, A. Verdini, L. Floreano, M. Stredansky, E. Vesselli, C. Hohner, M. Kettner, J. Libuda, C. M. Schneider, G. Zamborlini, M. Cinchetti and V. Feyer, Small, 2021, 17, 2104779 CrossRef CAS.
- I. Cojocariu, S. Carlotto, G. Zamborlini, M. Jugovac, L. Schio, L. Floreano, M. Casarin, V. Feyer and C. M. Schneider, J. Mater. Chem. C, 2021, 9, 12559–12565 RSC.
- M. E. Ali, B. Sanyal and P. M. Oppeneer, J. Phys. Chem. C, 2009, 113, 14381–14383 CrossRef CAS.
- S. Carlotto, J. D. Fuhr, A. Cossaro, A. Verdini, M. Casarin, M. Lingenfelder, J. E. Gayone, L. Floreano and H. Ascolani, Appl. Surf. Sci., 2021, 551, 149307 CrossRef CAS.
- P. Knecht, J. Reichert, P. S. Deimel, P. Feulner, F. Haag, F. Allegretti, M. Garnica, M. Schwarz, W. Auwärter, P. T. P. Ryan, T.-L. Lee, D. A. Duncan, A. P. Seitsonen, J. V. Barth and A. C. Papageorgiou, Angew. Chem., Int. Ed., 2021, 60, 16561–16567 CrossRef CAS.
- G. Zamborlini, Z. Lüftner, D. Feng, B. Kollmann, P. Puschnig, C. Dri, M. Panighel, G. Di Santo, A. Goldoni, G. Comelli, M. Jugovac, V. Feyer and C. M. Schneider, Nat. Commun., 2017, 8, 335 CrossRef PubMed.
- G. Zamborlini, M. Jugovac, A. Cossaro, A. Verdini, L. Floreano, D. Lüftner, P. Puschnig, V. Feyer and C. M. Schneider, Chem. Commun., 2018, 54, 13423–13426 RSC.
- M. Wießner, D. Hauschild, A. Schöll, F. Reinert, V. Feyer, K. Winkler and B. Krömker, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 045417 CrossRef.
- I. Cojocariu, S. Carlotto, H. M. Sturmeit, G. Zamborlini, M. Cinchetti, A. Cossaro, A. Verdini, L. Floreano, M. Jugovac, P. Puschnig, C. Piamonteze, M. Casarin, V. Feyer and C. M. Schneider, Chem. – Eur. J., 2021, 27, 3526–3535 CrossRef CAS PubMed.
- L. Scudiero, D. E. Barlow, U. Mazur and K. W. Hipps, J. Am. Chem. Soc., 2001, 123, 4073–4080 CrossRef CAS PubMed.
- J. C. Slater and J. C. Phillips, Phys. Today, 1974, 27, 49–50 CrossRef.
- I. Cojocariu, H. M. Sturmeit, G. Zamborlini, A. Cossaro, A. Verdini, L. Floreano, E. D'Incecco, M. Stredansky, E. Vesselli, M. Jugovac, M. Cinchetti, V. Feyer and C. M. Schneider, Appl. Surf. Sci., 2020, 504, 144343 CrossRef CAS.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- G. Mangione, M. Sambi, S. Carlotto, A. Vittadini, G. Ligorio, M. Timpel, L. Pasquali, A. Giglia, M. V. Nardi and M. Casarin, Phys. Chem. Chem. Phys., 2016, 18, 24890–24904 RSC.
- F. Petraki, H. Peisert, U. Aygül, F. Latteyer, J. Uihlein, A. Vollmer and T. Chassé, J. Phys. Chem. C, 2012, 116, 11110–11116 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef.
- M. G. Betti, F. Crispoldi, A. Ruocco and C. Mariani, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 125407 CrossRef.
- V. Aristov, O. Molodtsova, V. Maslyuk, D. Vyalikh, T. Bredow, I. Mertig, A. Preobrajenski and M. Knupfer, Org. Electron., 2010, 11, 1461–1468 CrossRef CAS.
- A. Calabrese, L. Floreano, A. Verdini, C. Mariani and M. G. Betti, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115446 CrossRef.
- H. Wang, P. Ge, C. G. Riordan, S. Brooker, C. G. Woomer, T. Collins, C. A. Melendres, O. Graudejus, N. Bartlett and S. P. Cramer, J. Phys. Chem. B, 1998, 102, 8343–8346 CrossRef CAS.
- H. Wang, C. Y. Ralston, D. S. Patil, R. M. Jones, W. Gu, M. Verhagen, M. Adams, P. Ge, C. Riordan, C. A. Marganian, P. Mascharak, J. Kovacs, C. G. Miller, T. J. Collins, S. Brooker, P. D. Croucher, K. Wang, E. I. Stiefel and S. P. Cramer, J. Am. Chem. Soc., 2000, 122, 10544–10552 CrossRef CAS.
- C. Y. Ralston, H. Wang, S. W. Ragsdale, M. Kumar, N. J. Spangler, P. W. Ludden, W. Gu, R. M. Jones, D. S. Patil and S. P. Cramer, J. Am. Chem. Soc., 2000, 122, 10553–10560 CrossRef CAS.
- Y. Bai, M. Sekita, M. Schmid, T. Bischof, H.-P. Steinrück and J. M. Gottfried, Phys. Chem. Chem. Phys., 2010, 12, 4336–4344 RSC.
- C. Wäckerlin, K. Tarafder, D. Siewert, J. Girovsky, T. Hählen, C. Iacovita, A. Kleibert, F. Nolting, T. A. Jung, P. M. Oppeneer and N. Ballav, Chem. Sci., 2012, 3, 3154–3160 RSC.
- S. Carlotto, I. Cojocariu, V. Feyer, L. Floreano and M. Casarin, Nanomaterials, 2022, 12, 218 CrossRef CAS.
- C. Schneider, C. Wiemann, M. Patt, V. Feyer, L. Plucinski, I. Krug, M. Escher, N. Weber, M. Merkel, O. Renault and N. Barrett, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 330–339 CrossRef CAS.
- L. Floreano, G. Naletto, D. Cvetko, R. Gotter, M. Malvezzi, L. Marassi, A. Morgante, A. Santaniello, A. Verdini, F. Tommasini and G. Tondello, Rev. Sci. Instrum., 1999, 70, 3855–3864 CrossRef CAS.
- L. Floreano, A. Cossaro, R. Gotter, A. Verdini, G. Bavdek, F. Evangelista, A. Ruocco, A. Morgante and D. Cvetko, J. Phys. Chem. C, 2008, 112, 10794–10802 CrossRef CAS.
- P. Puschnig, S. Berkebile, A. J. Fleming, G. Koller, K. Emtsev, T. Seyller, J. D. Riley, C. Ambrosch-Draxl, F. P. Netzer and M. G. Ramsey, Science, 2009, 326, 702–706 CrossRef CAS.
- D. Brandstetter, X. Yang, D. Lüftner, F. S. Tautz and P. Puschnig, Comput. Phys. Commun., 2021, 263, 107905 CrossRef CAS.
- N. Haag, D. Lüftner, F. Haag, J. Seidel, L. L. Kelly, G. Zamborlini, M. Jugovac, V. Feyer, M. Aeschlimann, P. Puschnig, M. Cinchetti and B. Stadtmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 2020, 101, 165422 CrossRef CAS.
- X. Yang, L. Egger, P. Hurdax, H. Kaser, D. Lüftner, F. C. Bocquet, G. Koller, A. Gottwald, P. Tegeder, M. Richter, M. G. Ramsey, P. Puschnig, S. Soubatch and F. S. Tautz, Nat. Commun., 2019, 10, 3189 CrossRef PubMed.
- M. Grimm, C. Metzger, M. Graus, M. Jugovac, G. Zamborlini, V. Feyer, A. Schöll and F. Reinert, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 98, 195412 CrossRef CAS.
- P. Kliuiev, G. Zamborlini, M. Jugovac, Y. Gurdal, K. v Arx, K. Waltar, S. Schnidrig, R. Alberto, M. Iannuzzi, V. Feyer, M. Hengsberger, J. Osterwalder and L. Castiglioni, Nat. Commun., 2019, 10, 5255 CrossRef.
- M. Valiev, E. Bylaska, N. Govind, K. Kowalski, T. Straatsma, H. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. Windus and W. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- R. F. Nalewajski and J. Mrozek, Int. J. Quantum Chem., 1994, 51, 187–200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc01253g |
| This journal is © The Royal Society of Chemistry 2022 |