
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of chromophore spacing on the stability and efficiency of host-free sky-blue dendrimer organic light emitting diodes†
Lorenz
Graf von Reventlow
ab,
Manikandan
Koodalingam
 c,
Christian
Siebert
a,
Philipp
Marlow
ab,
Emma V.
Puttock
c,
Paul L.
Burn
*c and
Alexander
Colsmann
*ab
c,
Christian
Siebert
a,
Philipp
Marlow
ab,
Emma V.
Puttock
c,
Paul L.
Burn
*c and
Alexander
Colsmann
*ab
aKarlsruhe Institute of Technology (KIT), Light Technology Institute, Engesserstrasse 13, 76131 Karlsruhe, Germany. E-mail: alexander.colsmann@kit.edu
bKarlsruhe Institute of Technology (KIT), Material Research Center for Energy Systems, Strasse am Forum 7, 76131 Karlsruhe, Germany
cCentre for Organic Photonics & Electronics (COPE), School of Chemistry & Molecular Biosciences, University of Queensland, St. Lucia, QLD 4072, Australia. E-mail: paul.burn@uq.edu.au
First published on 6th May 2022
Abstract
Combining light emission and charge carrier transport into one molecule simplifies the manufacturing of organic light-emitting diodes (OLEDs). In this work, phosphorescent iridium(III) complex-emitters are protected by conjugated dendrons to reduce intermolecular interchromophore interactions that lead to concentration quenching of the luminescence in host-free emission layers. When two dendrons are added to each ligand (doubly dendronized) of an iridium(III) complex at the core of the dendrimers (versus one), the core is found to have improved protection, leading to an enhancement of the external quantum efficiency of the OLEDs from 4% to more than 10%. Likewise, OLEDs comprising the doubly dendronized emitters exhibit improved device lifetimes. A detailed study of the degradation mechanisms reveals that exciton-induced ligand and/or dendron detachment promotes interchromophore interactions (aggregation) that lead to quenching sites and hence red electromer emission inside the OLED emissive layer, with the effects being less pronounced for the doubly dendronized emitter.
1. Introduction
Organic light-emitting diodes (OLEDs) are the prevalent technology in high-end displays such as those used in mobile phones and televisions. While red and green phosphorescent emitters are widely deployed in OLEDs, the implementation of highly efficient blue phosphorescent emitters is hampered by their lack of stability. To date, most OLED displays are fabricated using evaporation techniques, but the first products fabricated by ink-jet printing have now been introduced to the market.1 Solution processing, arguably, can enhance the resolution of large displays, reduce the material waste and hence lower the production costs.2 An important challenge in solution processing of OLEDs is the homogenous distribution of emitter molecules in the host. During the evaporation of the solvent (i.e., thin-film drying), emitter molecules can diffuse within the host and form aggregates, provoking concentration quenching of the luminescence and reducing the overall efficiency of the OLEDs.3 One of the most promising mitigation routes to avoid concentration quenching is to use a (macro)molecular species in the emission layer that enables charge carrier transport without the host, whilst ensuring spacing of the light-emitting moiety to avoid concentration quenching. Evaporated OLEDs featuring “host-free” emission layers have been reported in the literature, showing an encouraging EQE of 19.5% and an operational stability that even outperformed devices with mixed host/guest emission layers.4 In the case of solution processed materials, reduced concentration quenching can be realized by encapsulation of the emissive chromophore, featuring either thermally activated delayed fluorescence (TADF)5–8 or phosphorescence.9–12 For example, sky-blue solution processed host-free phosphorescent dendrimer OLEDs with first generation biphenyl-based dendrons have exhibited a maximum EQE of 7.9%.9 Furthermore, steric hindrance leading to twisting of chromophores can lead to enhanced properties of blue emissive materials.9,10 An example of this strategy is the enhanced EQE of 19.1% achieved by implementing highly twisted TADF molecules for blue emission.13 While much effort has gone into reducing the intermolecular interactions of the emissive chromophore, an alternative strategy is to enhance such interactions to achieve aggregation-induced emission. One example of this in the context of blue emission are films composed of TADF molecules with aggregation-induced emission properties which have produced an EQE of 17.2%.14 However, despite this promising performance of host-free OLEDs processed from solution, their stability has rarely been described in the literature.In this work, we investigate the effect of emissive core encapsulation by dendrons on the optoelectronic properties and the operational stability of host-free OLEDs composed of phosphorescent sky-blue emitting dendrimers. The dendrimers contain the same core and dendron type but differ in the number of dendrons attached to each ligand of the core iridium(III) complex – one or two.
2. Results and discussion
2.1 Synthesis
The synthetic procedure for the singly dendronized homoleptic iridium(III) complex Ir(SDTri)3 (structure shown in Scheme 1) was reported earlier.15 The synthetic routes to the doubly dendronized triazole-based ligand, DDTri, and its corresponding homoleptic iridium(III) complex, Ir(DDTri)3, are shown in Scheme 1. A convergent approach was taken for the synthesis, which began with preparing the dendron to be attached to the triazole moiety of the ligand. In the first step, methyl 3,5-dibromo-4-methylbenzoate was coupled to 4-tert-butylphenylboronic acid under Suzuki conditions to give the first generation dendron 3 with a methyl ester at its foci in a 90% yield. The ester was then reduced to the corresponding benzyl alcohol 4 with lithium aluminum hydride in a 93% yield. Treatment of 4 with phosphorous tribromide then gave benzyl bromide 5 in a 90% yield. The first generation dendron 5 was then converted into the singly dendronized triazole ligand using a three-step procedure in an overall yield of 44%. It was first reacted with tert-butyl 2-[propan-2-ylidene]hydrazine-1-carboxylate 6 to form the corresponding tert-butyloxycarbonyl-protected hydrazine 7, which was subsequently deprotected with hydrochloric acid to form the salt 8. 8 was then cyclized with ethyl-trans-N-[3-bromobenzoyl]butyrimidate to give 10. To form the doubly dendronized ligand DDTri for comparative purposes, the singly dendronized triazole ligand 10 was coupled to the first generation boronate ester 1116 under Suzuki conditions to give DDTri in a yield of 62%. We initially attempted to form the doubly dendronized dendrimer Ir(DDTri)3 from DDTri using the normal two step procedure – reaction with the iridium(III) chloride hydrate to form the bis-iridium(III) bis-chloro-bridged dimer followed by cracking with excess ligand at high temperature.9 However, although we were able to isolate the desired Ir(DDTri)3, the steric bulk of the ligand hindered the formation of the complex, which resulted in a poor yield of 6%. We therefore took an alternative approach in which the singly dendronized ligand 10 was used to form the mono-dendronized complex 13 following the standard two-step procedure.910 and iridium(III) chloride hydrate were heated in a 2-ethoxyethanol/water mixture to give the bis-iridium(III) bis-chloro-bridged dimer 12, which was then reacted with excess 10 in the presence of silver triflate in the melt at 166 °C to give 13 in a yield of 45%. The mono-dendronized 13 was then reacted with the first generation boronate ester under Suzuki conditions to give the target dendrimer Ir(DDTri)3 in a yield of 49%. The 1H NMR of Ir(DDTri)3 was consistent with the formation of the fac-isomer and gel permeation chromatography (GPC) showed that the dendrimer was monodisperse (Fig. S8, ESI†). Given that the dendrimer was monodisperse![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) v/n = 117 and using the n from the GPC measurement and the Hester-Mitchell equation in combination with the Mark–Houwink relationship18 enabled determination of the hydrodynamic radius, which was 10.2 Å. The hydrodynamic radius was larger than of the singly dendronized material [Ir(SDTri)3 = 9.0 Å], which is consistent with the extra dendron being attached to each ligand. Thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) showed Ir(DDTri)3 had excellent thermal stability with a decomposition temperature corresponding to a 5% weight loss at 382 °C and no glass transition was observed between −50 °C and 250 °C. Also, no glass transition was observed for Ir(SDTri)3 in the same temperature range.
v/n = 117 and using the n from the GPC measurement and the Hester-Mitchell equation in combination with the Mark–Houwink relationship18 enabled determination of the hydrodynamic radius, which was 10.2 Å. The hydrodynamic radius was larger than of the singly dendronized material [Ir(SDTri)3 = 9.0 Å], which is consistent with the extra dendron being attached to each ligand. Thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) showed Ir(DDTri)3 had excellent thermal stability with a decomposition temperature corresponding to a 5% weight loss at 382 °C and no glass transition was observed between −50 °C and 250 °C. Also, no glass transition was observed for Ir(SDTri)3 in the same temperature range.
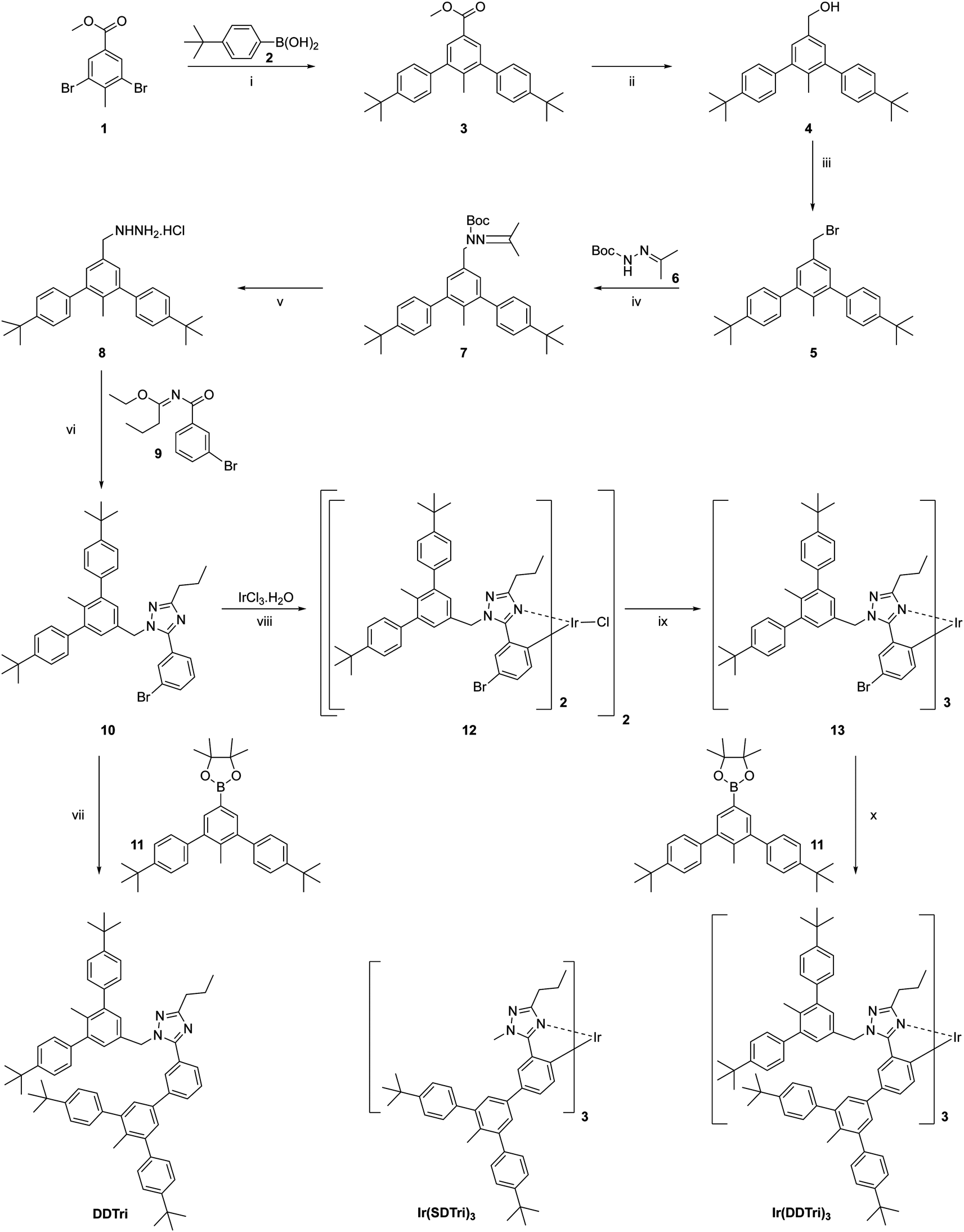 | ||
| Scheme 1 Reagents and conditions: (i) 2, Na2CO3, Pd(PPh3)4, ethanol, toluene, H2O, 95 °C, Ar, 18 h; (ii) LiAlH4, tetrahydrofuran, 0 °C to rt, Ar, 3 h, (iii) PBr3, 0 °C to 90 °C, Ar, 18 h; (iv) 6, KOH, tetra-n-butylammonium hydrogen sulfate, toluene, 50 °C to 80 °C, Ar, 3 h; (v) tetrahydrofuran, 3 M HCl, 95 °C, Ar, 18 h; (vi) 9, Et3N, dichloromethane, rt, 18 h; (vii) 11, Na2CO3, Pd(PPh3)4, ethanol, toluene, H2O, 95 °C, Ar, 18 h; (viii) iridium(III) trichloride hydrate, 2-ethoxyethanol, H2O, 126 °C, Ar, 18 h; (ix) 10, silver triflate, 166 °C, Ar, 18 h, (x) 11, Na2CO3, Pd(PPh3)4, ethanol, toluene, H2O, 95 °C, Ar, 36 h. | ||
In common with the singly dendronized complex,12 only a single chemically reversible oxidation (E1/2 = 0.3 V versus the ferrocene/ferrocenium couple) was observed for Ir(DDTri)3 within the solvent window (ESI,† Fig. S9). The similarity of the oxidation potentials for the singly (0.2 V) and doubly dendronized complexes indicates that the electroactive component of the dendrimer remains the emissive core.12
2.2 Photophysical characterization
To assess the principal applicability of the dendrimers for light-emitting applications, we investigated their photophysical properties. The solution absorbance and photoluminescence (PL) spectra of both dendrimers in degassed tetrahydrofuran (THF) as well as the PL of neat films of the dendrimers are depicted in Fig. 1. In common with other iridium(III) complex-cored dendrimers, the solution UV-Vis spectra show two regimes: the short-wavelength absorption (<350 nm) can be attributed to π–π* transitions on the ligands and the dendrons, whereas the longer wavelength absorption (>350 nm) can be assigned to a mixture of singlet and triplet metal-to-ligand charge transfer (MLCT) transitions. Ir(SDTri)3 and Ir(DDTri)3 exhibit very similar sky-blue emission in THF with emission maxima at 473 nm and 470 nm, respectively. We observed only a minor difference in the contribution from the lower-energy triplet sublevels visible as a weak shoulder at around 550 nm in the PL spectrum.19 That is, the addition of the dendron onto the triazolyl unit of the ligands does not perturb the emission color. Our results are consistent with previous reports that show that providing the triplet energy of the dendrons is higher than the emissive core the emission occurs from the core.9,10,20 The photoluminescence quantum yields (PLQYs) of Ir(SDTri)3 (70 ± 7%) and Ir(DDTri)3 (79 ± 8%) are also comparable. These matching photophysical and electrochemical properties of the macromolecules in solution render the two emitters very good candidates to study the effects of molecular encapsulation on the optoelectronic properties in the solid state.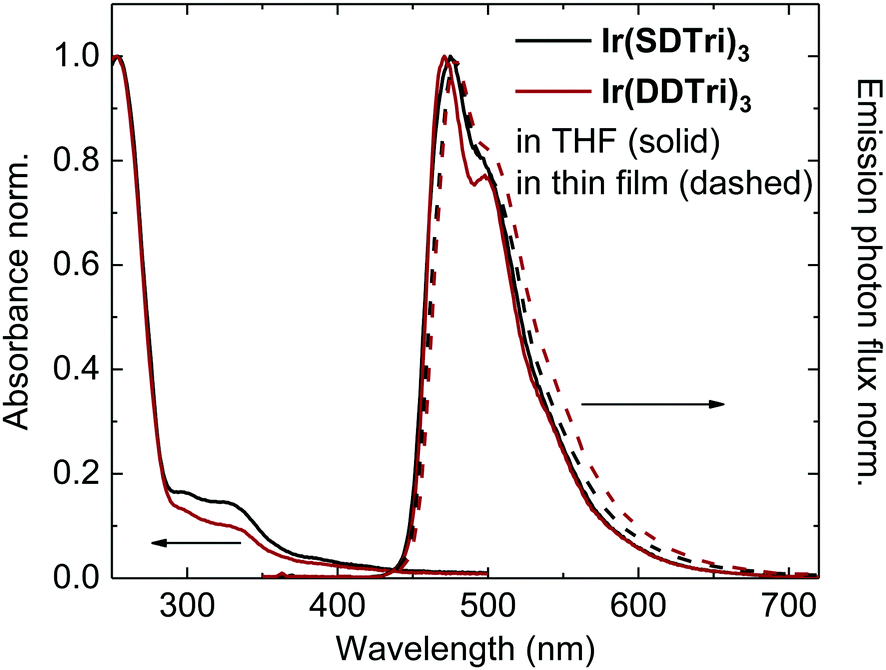 | ||
| Fig. 1 Absorbance and photoluminescence spectra of Ir(SDTri)3 (black) and Ir(DDTri)3 (red) in degassed tetrahydrofuran (solid line) and in thin films (dashed line), normalized to the maxima in each case. | ||
While both emitters exhibit the same photophysical properties in solution, their PLQYs differ significantly in thin films. Upon transformation to the solid state, the PLQYs of Ir(SDTri)3 and Ir(DDTri)3 were reduced to 15% and 45%, respectively. At the same time, the phosphorescence lifetimes drop and transform from mono- to biexponential decays. The reduced PLQY paired with the biexponential lifetime decay indicates luminescence concentration quenching and more than one emitting species in the thin films, which is often observed in iridium(III)-complexes.16 The higher PLQY of Ir(DDTri)3 is a first indication of the superior properties of the doubly dendronized Ir(DDTri)3 in the solid state. Both emitters furthermore show only a small red-shift of the PL spectrum (vs. solution) with a PL maximum at 478 nm.
2.3 Organic light emitting diodes: device performance
Both emitters were implemented in the host-free OLED architecture that is depicted in Fig. 2(a). In addition to controlling the intermolecular interactions of the chromophores at their core, the dendrons facilitated solution processing of the emitter chromophores to form films of sufficient thickness and quality for use in devices. Holes were injected from an indium tin oxide (ITO) electrode through a poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) hole transport layer (HTL) into the emission layer (EML). Electrons were injected from an aluminum electrode through (8-hydroxyquinolinato)lithium (Liq) and a 1,3,5-tri(m-pyridin-3-ylphenyl)benzene (TmPyPB) electron transport layer (ETL). PEDOT:PSS and the emission layer were spin-coated from solution, whereas TmPyPB, Liq and Al were thermally evaporated in vacuum for best device reproducibility.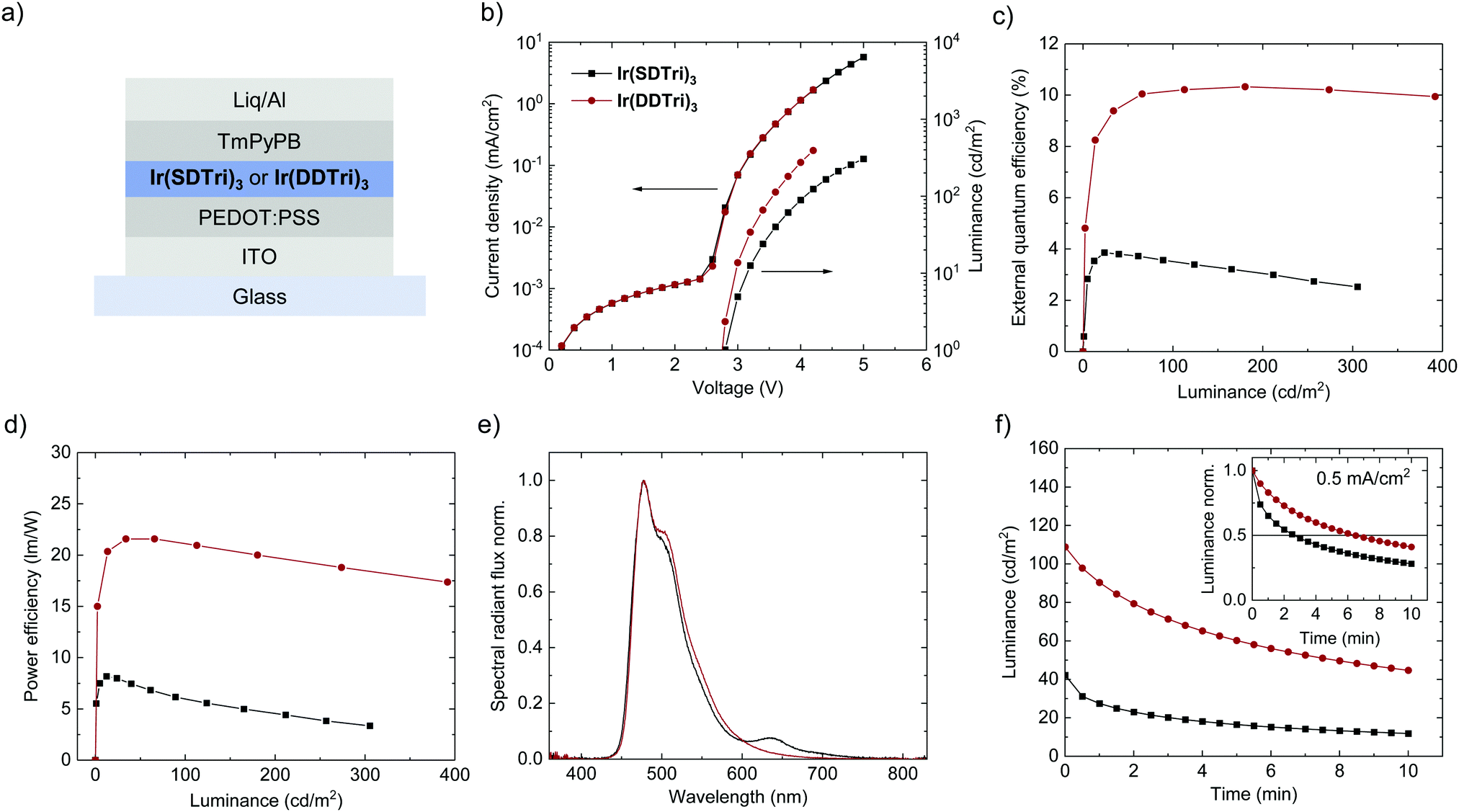 | ||
| Fig. 2 (a) Host-free OLED architecture. (b) J–V–L characteristics of OLEDs comprising Ir(SDTri)3 or Ir(DDTri)3 (initial measurement). (c) External quantum efficiency (EQE) vs. luminance. (d) Power efficiency vs. luminance. (e) Electroluminescence spectra of the OLEDs: [Ir(SDTri)3 at 305 cd m−2; Ir(SDTri)3 at 391 cd m−2]. (f) Device degradation: luminance vs. time (L–t) at J = 0.5 mA cm−2. Inset: L–t normalized to the initial luminance. LT50 is marked with a black line. | ||
OLEDs comprising either Ir(SDTri)3 or Ir(DDTri)3 exhibited essentially identical electrical properties. Fig. 2(b) shows nearly matching current density–voltage (J–V) curves, which allows us to conclude that the second dendron on Ir(DDTri)3 has a negligible effect on the net transport of charge carriers. Likewise, both devices show matching turn-on voltages of 2.75 V, indicating equal barrier-free charge carrier injection into the EML. Yet, we observed differences in the luminance–voltage (L–V) curves of the OLEDs, their external quantum efficiency (EQE) and their power efficiency as depicted in Fig. 2(b)–(d), altogether indicating different charge-carrier-to-photon conversion efficiencies. In particular, the Ir(DDTri)3-based OLEDs exhibit an EQE of more than 10% (10.5 ± 0.6%) which is more than twice the EQE of the Ir(SDTri)3-based OLEDs (3.9 ± 0.2%), and which reflects the different solid-state PLQYs of the two compounds. It is interesting to note that the maximum EQE of both devices is the best that is expected based on the film PLQYs and the out-coupling of light from a bottom emitting device. The electroluminescence (EL) spectra of both OLEDs in Fig. 2(e) show very similar sky-blue emission with CIE1931-coordinates of (x;y) = (0.18;0.37) and (x;y) = (0.18;0.40) for Ir(SDTri)3 and Ir(DDTri)3, respectively. Towards higher voltages, the Ir(SDTri)3-OLEDs exhibited an additional red emission at 640 nm, which will be discussed later. This red emission was not visible in Ir(DDTri)3-OLEDs in the initial scan.
2.4 Organic light emitting diodes: operational lifetime
Another important difference of both emitters became visible in the operational lifetime of the OLEDs (LT50, i.e., the time to 50% of the initial luminance), which is depicted in Fig. 2(f). At a constant current density of 0.5 mA cm−2, the LT50 = 6.5 min of the OLEDs comprising the doubly dendronized Ir(DDTri)3 exceeds the LT50 = 2.5 min of devices composed of the singly dendronized Ir(SDTri)3 – an increase of more than a factor of two. Notably, the Ir(SDTri)3-based OLED exhibited a much lower luminance during the entire lifetime measurement, due to the lower EQE. The difference in the OLED operational lifetime was similar to the difference in the EQE and PLQY, which may be a first indication that increased non-radiative exciton decay pathways in the Ir(SDTri)3-based OLEDs result in a faster degradation.The degradation of the OLED does not only show in the overall device performance, but also becomes visible in the EL spectra that are depicted in Fig. 3(a) and (b). The initial emission spectrum (i.e., when the device turns on) of the Ir(SDTri)3-based OLED in Fig. 3(a) exhibits a secondary emission peak at 640 nm in addition to the sky-blue emission. This secondary peak was found to increase in intensity over time. We also observed the appearance and growth of an emission peak at the same wavelength for the Ir(DDTri)3-based OLEDs (see Fig. 3(b)), but the absolute intensity was much smaller. To determine whether the long wavelength emission was similar in nature for the two devices, we subtracted the normalized initial spectra from the spectra after 10 min of electrical stress at 0.5 mA cm−2. Fig. 3(c) clearly shows that the EL spectral component at around 640 nm was similar for both the singly and the doubly dendronized emitters, which indicates a similar degradation mechanism for each device type. The major difference is the faster degradation of the Ir(SDTri)3-based OLEDs. The red emission could arise from a number of sources including degradation of the emitter molecule, formation of charge-transfer states at interfaces, or from electromers.21 In the following part of the study we used the red emission as a probe to locate and understand the origin of the reduction in device performance over time.
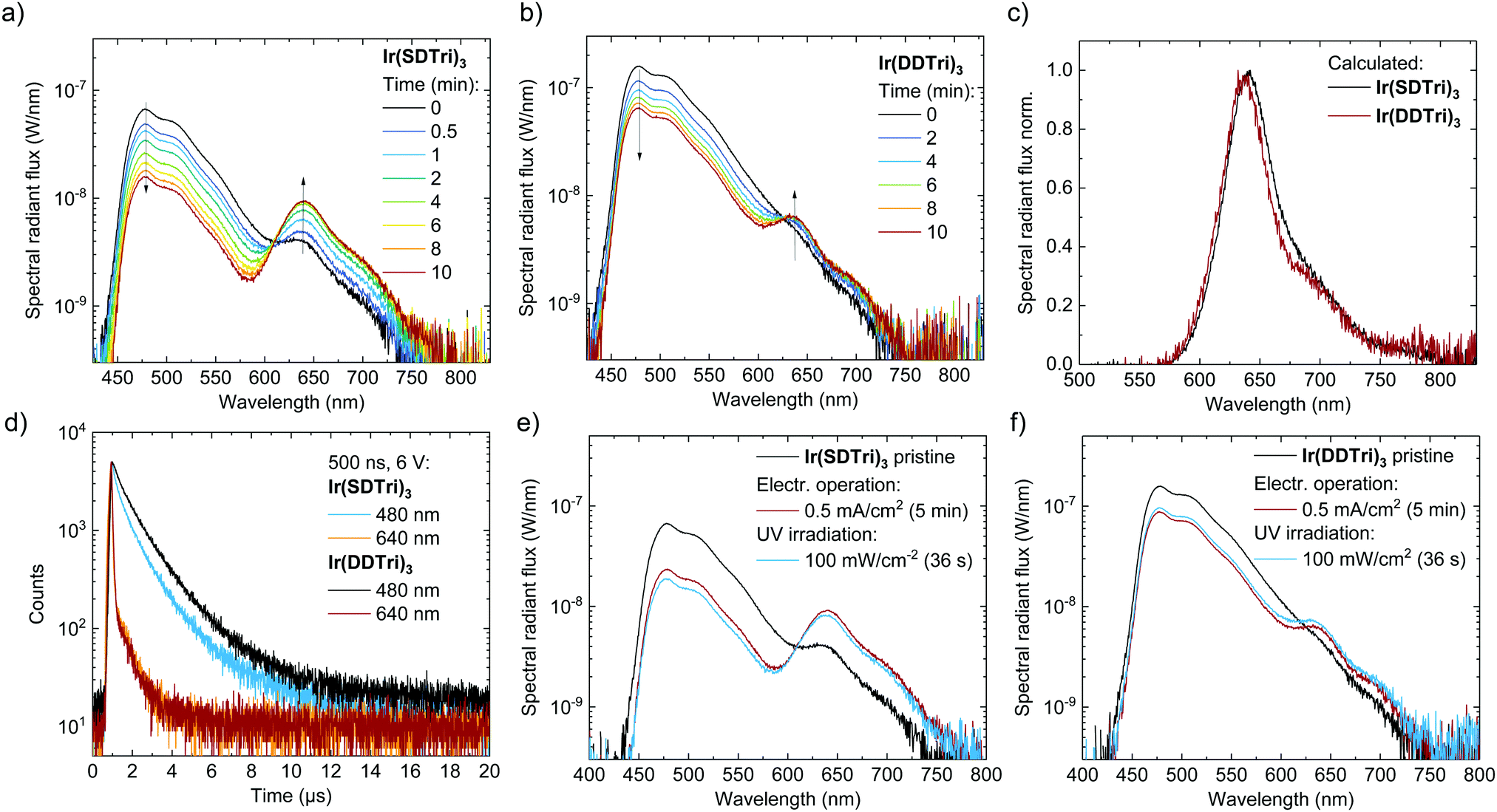 | ||
| Fig. 3 Electroluminescence spectra of OLEDs comprising (a) Ir(SDTri)3 and (b) Ir(DDTri)3 under continuous operation at J = 0.5 mA cm−2, measured for up to 10 min after initial characterization. (c) Red emission that evolves during OLED operation extracted from the initial emission spectrum and the emission spectrum after 10 min of operation. (d) Transient electroluminescence of aged OLEDs (after 10 min) at different emission wavelengths under pulsed operation (amplitude: 6 V, pulse width: 500 ns, repetition rate: 25 kHz). (e) Electroluminescence spectra of OLEDs comprising Ir(SDTri)3 and (f) Ir(DDTri)3 at J = 0.5 mA cm−2 after turn-on (black line) and after 5 min of operation in comparison to another pristine OLED that was irradiated with UV (400 nm, 100 mW cm−2, 36 s). The EL spectra of the UV-irradiated OLEDs was collected immediately after turn-on. | ||
2.5 Locating the layer in which degradation occurs
The first step in the analysis was to determine which of the layers was primarily responsible for the red EL peak. To investigate whether interface effects or the charge transport materials promoted the formation of the red emission component, we fabricated OLEDs without the hole transport layer or electron transport layer. In all cases the red emission appeared over time (ESI,† Fig. S1), which indicates that it originates from degradation within the emissive layer.The question that therefore arises is whether the red emission occurs through energy transfer from the excitons responsible for the sky-blue emission, leading to the quenching of the luminescence, or direct electrical excitation of the new emissive species. Exciton transfer can be studied using transient electroluminescence experiments. To investigate the lifetime of the excited species, we powered the degraded OLEDs with short electrical pulses (500 ns, 6 V) and measured the EL transients of the blue phosphorescence (480 nm) and red emission (640 nm), which are depicted in Fig. 3(d). The observed lifetime on a quenching species (exciton acceptor) will always be equal to or longer than the lifetime of the exciton donor after donor excitation. The exciton lifetimes of the Ir(SDTri)3- and Ir(DDTri)3-based OLEDs in the blue regime are in the microsecond range typical for phosphorescent emitters. The transients of the red emission are similar for both emitters and can be fitted with two exponents. The first component has a lifetime of 40 ns, corresponding to the RC-components of the OLEDs and the pulse generator. Thus, under the experimental conditions an exciton lifetime shorter than 40 ns cannot be identified or excluded. The second component has a lifetime of 700 ns. Interestingly, the long lifetime component can be reduced by applying a negative bias directly after the forward bias (ESI,† Fig. S2). Such behavior has been previously attributed to delayed emission from trapped charge carriers.15,22 Most importantly, the lifetime of the red emission is shorter than the lifetime of the blue emission, which lets us conclude that this red-emitting species is formed by direct electrical excitation and not by energy transfer. That is, the red-emitting species forms a competing exciton generation pathway, reducing the EQE in the blue region of the spectrum. Also, the exciton lifetime of 40 ns of the red emitting species implies that it is fluorescent rather than phosphorescent in nature.
To summarize thus far, the doubly-dendronized Ir(DDTri)3 surpasses the singly-dendronized Ir(SDTri)3 not only in efficiency but also in device lifetime. The degradation process that occurs for devices composed of either emitter appears to be similar, with the former slower than the latter. The degradation is associated with the emissive layer, with the red emission arising from direct electrical excitation and not from energy transfer.
2.6 Degradation mechanisms of the dendrimers
The appearance of the red emission peak during device operation being associated with the emissive layer suggests that the dendrimers are undergoing chemical decomposition. Chemical decomposition can have various origins: (i) Joule heating leading to thermal degradation; (ii) the oxidized or reduced species during charge carrier transport are highly reactive and could undergo further reactions; (iii) the excitons on the emitter molecules could undergo non-radiative decay with energy released leading to bond scission; and/or (iv) triplet-polaron-annihilation (TPA) or triplet–triplet-annihilation (TTA) could lead to excited states with even higher energy and a greater probability of irreversible bond scission.23Molecular decomposition arising simply from Joule heating during device operation can be discounted as thermal gravimetric analysis showed that the thermal decomposition of Ir(DDTri)3 was much higher than 300 °C, well above the thermal annealing temperature used during the device fabrication process. Furthermore, thermal annealing of the OLEDs (80 °C, 10 min, no electrical bias) did not reduce the EQE during subsequent testing (ESI,† Fig. S3).
To investigate the stability of the excited emitters we irradiated the OLEDs with UV (400 nm, 100 mW cm−2, 36 s). At an excitation wavelength of 400 nm, only the emitters exhibit significant absorption. As depicted in Fig. 3(e) and (f), exposing the OLEDs to UV irradiation caused a drop of the blue EL and the emergence of the same red feature at 640 nm which was observed during electrical-only stressing of the samples. We monitored the temperature of the OLEDs during UV irradiation, which was found not to exceed 50 °C, well below the thermal degradation temperature of the emissive materials. After UV irradiation, the blue EL photon flux was found to decrease by 39%, while the contribution of the red emission to the total photon flux of the Ir(DDTri)3-based OLED increased to 5%. Interestingly, the decrease in blue photon flux corresponded well to the reduction in the PLQY of an UV irradiated Ir(DDTri)3 layer of the same thickness on a quartz substrate (38% rel. loss). The effect of the UV irradiation on the PL properties of the films will be discussed in more detail later. That said, these results provide strong evidence that the red emissive species arises from degradation caused by excitons. The fact that the process that leads to the red emission occurs when no electrical bias is applied indicates that the degradation is not due to charge carrier injection or TPA. The drop in the blue EL photon flux for the singly dendronized Ir(SDTri)3-based OLEDs was higher than the drop in the corresponding thin film PLQY (72% vs. 45% rel.). This is a direct consequence of the large amount of red emission from the OLEDs, which is not visible in the solid-state PL of thin films. After UV irradiation, about one third of the electroluminescence flux in Fig. 3(e) originates from red emission, which decreases the exciton generation efficiency for blue emission dramatically.
To investigate if the degradation is caused by a bimolecular process like TTA or by a monomolecular process, we measured the operational lifetime of Ir(DDTri)3-based OLEDs at different current densities and hence different exciton densities. The dependence of LT50 on the current density in a double-logarithmic plot gives information on the number of excitons that are involved in the degradation process. For TTA, which is a bimolecular process, we would expect a slope of 2. Our experiments produced a slope of 1.3 indicating that the degradation is a monomolecular process (ESI,† Fig. S4). Therefore, degradation in the dendrimer OLEDs is mostly caused by monomolecular non-radiative exciton decay in the emission layer.
2.7 Molecular decomposition under photoexcitation
Since the observations strongly indicated that the changes giving rise to the red emission from the OLEDs are caused by changes to the structure of a proportion of the emitters, in the next part of the work we investigated the potential changes in molecular structure that occurred on excitation. An earlier computational study calculated that one of the Ir–N bonds in the parent molecule (without the dendrons) can dissociate in the excited state leading to ligand detachment and permanent degradation of the molecule.24A common approach to investigate decomposition products from aged OLEDs is laser desorption/ionization time-of-flight mass spectroscopy (LDI-ToF).25,26 An important prerequisite for conducting this measurement is the stability of the materials upon evaporation. As Ir(SDTri)3 and Ir(DDTri)3 cannot be thermally evaporated due to their high molecular weight, strong molecular fragmentation during laser desorption is very likely to occur. Therefore, we chose to use matrix assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-ToF) to analyze the degradation of the dendrimers. Thin films of the dendrimers on quartz substrates were exposed to UV-irradiation (400 nm, 100 mW cm−2, 2 min), collected and then measured using MALDI-ToF mass spectrometry (2-({2E}-3-{4-tert-butylphenyl}-2-methylprop-2-enylidene)malononitrile (DCTB) was used as the matrix). Pristine samples were also measured to ensure that materials did not fragment under the experiment conditions (ESI,† Fig. S10). The mass spectrum of the UV exposed Ir(SDTri)3 sample (Fig. 4(a)) exhibits mass peaks that correspond to the dendrimer that had a dendronized ligand missing (1300 Da), although the exact structure of the product could not be elucidated. For Ir(DDTri)3 we observed a small peak at 2010 Da that corresponded to a doubly dendronized ligand being detached (Fig. 4(b)). In addition, there was a stronger peak originating from the emitter molecule with one dendron missing (2550 Da). From the observed molecular weight of the fragment we believe that the dendron that has been lost from Ir(DDTri)3 was originally attached to the triazole ring. This is consistent with carbon–nitrogen bond being weaker than a carbon–carbon bond.
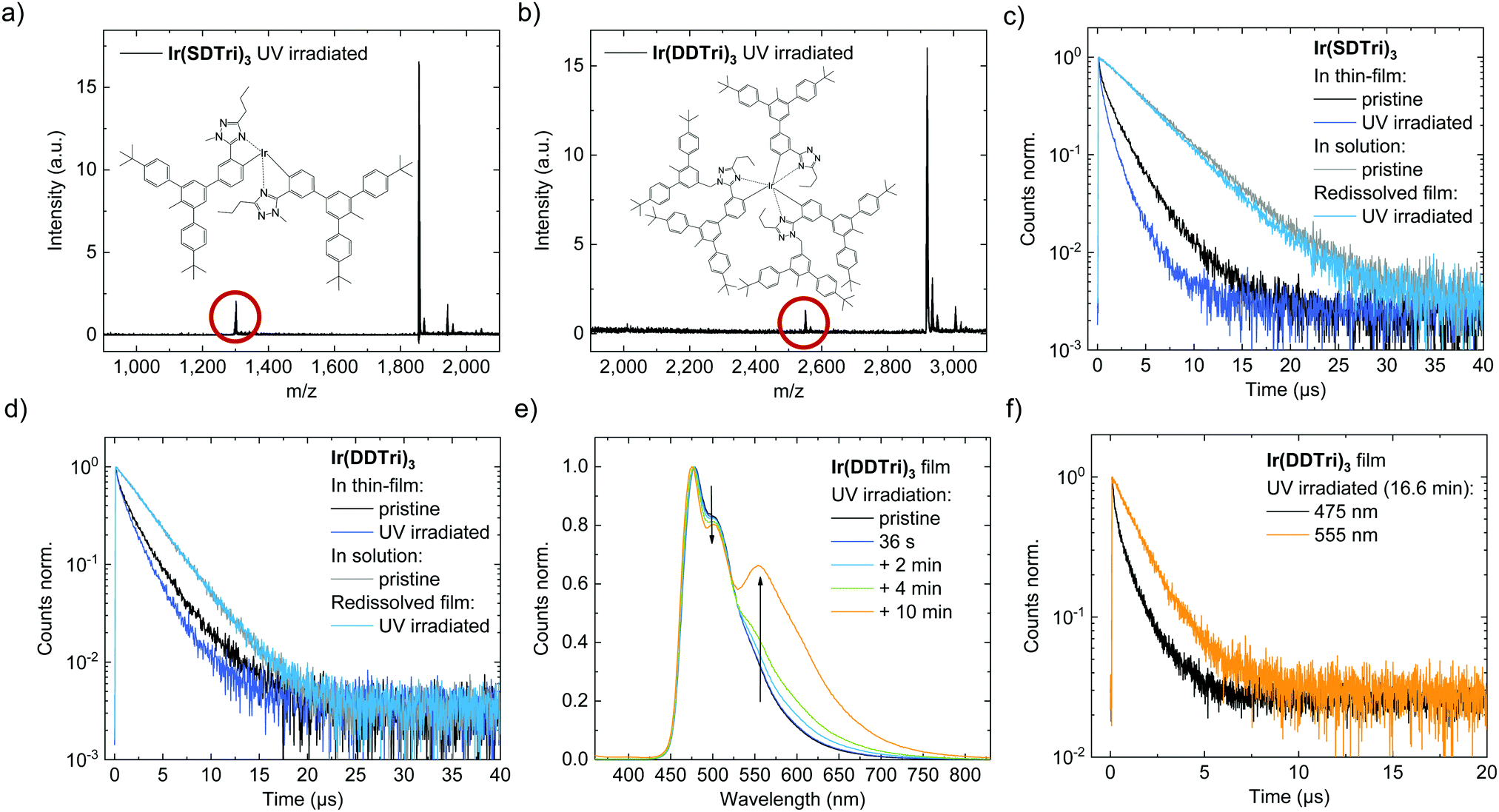 | ||
| Fig. 4 (a) MALDI-ToF mass spectrum of an Ir(SDTri)3 thin film that had been UV irradiated (400 nm, 100 mW cm−2, 2 min). Inset: Chemical structure of the most prominent decomposition product. (b) MALDI-ToF mass spectrum of a UV irradiated Ir(DDTri)3 thin film. Inset: Chemical structure of the most prominent decomposition product. (c) PL lifetime of neat and UV irradiated (dark blue, 72 mW cm−2) Ir(SDTri)3 thin films. Upon redissolution of the UV irradiated thin film (light blue), the PL lifetime recovers and is similar to untreated Ir(SDTri)3 in solution (grey). (d) Corresponding PL lifetime measurements of Ir(DDTri)3. (e) Evolution of the photoluminescence spectrum of Ir(DDTri)3 upon UV irradiation. After several minutes, an emission band at 555 nm evolves. (f) The PL lifetime of an UV irradiated Ir(DDTri)3 thin film is longer at 555 nm than at 475 nm. | ||
UV irradiation of Ir(DDTri)3 in degassed toluene solution causes neither a decrease in the PLQY nor in the phosphorescence lifetime, demonstrating a certain stability of the emitter against photo-stressing. This indicates that the reduced freedom of movement in the solid state promotes the degradation. An effect of the degradation mechanism that is more likely to occur in the solid state than in solution is the formation of aggregates.
2.8 Effect of UV-excitation on the photophysical properties
The results thus far indicate that the degradation of the Ir(SDTri)3- and the Ir(DDTri)3-based OLEDs is mostly caused by excitons formed in the emissive layer. We therefore studied the effect of UV irradiation on neat emission layers deposited on quartz. As depicted in Fig. 4(c) and (d), UV irradiation causes a reduction of the PL lifetime and the PLQY. Interestingly, when the UV treated thin films were dissolved in toluene, the PL lifetime was the same as that of pristine emitters in solution. That is, there is not significant energy transfer between the different species in solution and the degradation products observed in the MALDI-ToF do not act as significant quenchers. These observations suggest that the chemical changes in the films lead to a change in interchromophore interactions (aggregation) in the thin film that leads to alternate exciton decay pathways. Dissolution of the thin films leads to dissociation of these interactions.Further evidence for the formation of interchromophore interactions in the film can be gained from the emission spectrum shown in Fig. 4(e). Interchromophore interactions can lead to a red-shift in the emission spectrum and the appearance of new emission bands.27 As depicted in Fig. 4(e), upon irradiation of an Ir(DDTri)3 thin film for 36 s, the PL spectrum does not change, but at longer timescales we observed the gradual appearance of a new lower-energy emission band at 555 nm. The emission at 555 nm exhibits a longer PL lifetime (τ = 1.5 μs) than the emission at 480 nm (τ1 = 0.2 μs, τ2 = 1.2 μs). The longer PL lifetime at 555 nm suggests a triplet state that could potentially act as a quenching state of the blue emission. Notably, this emission band differs from the red emission at 640 nm in the EL spectra of the degraded OLEDs. Also, the UV dose that led to the emergence of a pronounced peak at 555 nm was nearly 30 times higher than for that used the UV-degradation of the OLEDs in Fig. 3(e) and (f). The temperature of the thin films during the long UV irradiation treatment reached 100 °C. Using a heat sink to control the temperature to below 60 °C resulted in a slower appearance of the emission band at 555 nm, but the effect prevailed. Thermal annealing alone in the absence of UV irradiation did not change the photophysical properties of the thin film. The Ir(SDTri)3 thin films were found to have similar changes in the photophysical properties upon UV irradiation (ESI,† Fig. S5). A plausible explanation of these results is that the detached ligands act as a plasticizer for the film enabling the dendrimers to move, and doing so, increases the intermolecular interchromophore interactions that lead to aggregate emission. As no glass transition temperature was observed below 250 °C, movement of dendrimers at common operation temperatures (<80 °C) should otherwise be suppressed.28,29 Alternatively, and more likely, is that the new emission arises from the less encapsulated chromophores being responsible for the change in emission through interchromophore interactions. Aggregates are also not visible in atomic force microscope measurements (ESI,† Fig. S6). Taken altogether, the reduction of the phosphorescence lifetime and the appearance of another lower-energy emission peak, as well as the recovery of the PL lifetime by re-dissolving the thin films are a strong indication for chemical changes caused by the UV irradiation. These chemical changes promote interchromophore interactions of molecules (molecular aggregation) that create new non-radiative pathways that can lead to a reduction in efficiency of the blue phosphorescence emission.27,30–32
2.9 Red electromer emission in degraded OLEDs
The final piece in the study was to understand why the PL and EL of the emissive thin films containing degraded dendrimers were different. In contrast to the EL spectra of the OLEDs, the PL spectra of the UV irradiated neat thin films do not exhibit any emission peak at 640 nm. We propose that the origin of the emission at 640 nm in the degraded OLEDs arises from an electromer, which is an emissive charge-transfer state of two molecules, that can only form upon injection of charge carriers and not upon absorption of photons. Electromer emission is indicative of intermolecular interchromophore interactions.33 Electromer states do not generally quench phosphorescence to an appreciable extent, as described earlier, because the absorption of such charge-transfer states is very low, rendering resonant Förster or Dexter transfer inefficient. However, in our case we still measured some very weak PL from the electromer by UV-excitation (340 nm) of degraded Ir(SDTri)3- and Ir(DDTri)3-based OLEDs. This weak PL may stem from a small number of electromers that formed from UV-generated free charge carriers in the OLED at short circuit (0.016 mA cm−2). As free charge carriers react to an electrical field, we measured the PL of the OLEDs under electrical bias (ESI,† Fig. S1). By applying a voltage of +2 V, which is below the onset-voltage of the OLED, the internal electrical field is nullified. Consequently, both the photocurrent and the electromer emission were suppressed, providing strong evidence for the presence of electromers and by extension the increased interchromophore interactions (aggregation) caused by chemical degradation. Finally, dispersing the dendrimers at 5 wt% in an 3,3′-di(9H-carbazol-9-yl)-1,1′-biphenyl (mCBP) host led to the electroluminescence spectra being the same as the solution PL spectra. That is, no electromer emission was observed in the initial measurements, which can be assigned to the suppression of aggregation of the emissive species (ESI,† Fig. S7 and S8). To further support the theory of aggregation induced electromer formation, we fabricated Ir(SDTri)3-based OLEDs with different emitter concentrations in mCBP. At low emitter concentrations in the host (3.8 wt%), even after 10 min of electrical aging at strong current densities of J = 5 mA cm−2 we barely observed any electromer emission (ESI,† Fig. S9). At higher concentrations (19.2 wt%), a substantial amount of electromer emission became visible. The lifetimes of the Ir(SDTri)3-based OLEDs correlate with the electromer emission and are therefore higher for the OLEDs with host/guest architecture where the host ensures spacing between the emitter molecules.3. Conclusions
We compared the OLED efficiency and lifetime of two sky-blue emitting dendrimers that differed in the number of dendrons attached to each ligand of the irdium(III) complex core. The doubly dendronized emitter (two dendrons per ligand) showed an EQE of more than 10% in host-free OLEDs, which was over two times the EQE of the singly dendronized (one dendron per ligand) emitter. Likewise, the operational lifetime was strongly enhanced. The increased shielding of the doubly dendronized light-emitting core reduces the aggregation of the emitter molecules and hence quenching of the excited states and chemical decomposition. The degradation during OLED operation was not caused by exciton–polaron interactions, but by exciton-induced detachment of ligands and/or dendrons. The results show that the detachment of the ligands and/or dendrons leads to increased interchromophore interactions (aggregation) that can quench the PL and form red electromer emission. From this work we conclude that better shielding of the light-emitting core is not only beneficial for the performance of host-free dendrimer OLEDs but also for their operational stability.4. Experimental
OLED fabrication
Pre-structured ITO substrates (Dongguan Everest) were sonicated in acetone and 2-propanol (10 min each), followed by an oxygen-plasma treatment (Diener Atto, 2 min, 200 W). Then the samples were transferred into a nitrogen-glovebox for the remaining fabrication process. Poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS, VPAI 4083, Heraeus) was diluted in ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v:v) and spin-coated (4000 rpm, 45 s). After thermal annealing (10 min, 120 °C) the emission layer was spin-coated from tetrahydrofuran (3 g L−1, 3000 rpm, 45 s) and thermally annealed (10 min, 80 °C) to remove any residual solvents. TmPyPB (50 nm), Liq (2 nm) and Al (50 nm) were deposited in a home-built multi-chamber cluster-tool attached to the glovebox at a pressure <10−6 mbar. The light-emitting area (10.5 mm2) was defined by the overlap of the electrodes. Neat emitter layers were deposited on quartz substrates from solutions of the same concentration.
:3 v:v) and spin-coated (4000 rpm, 45 s). After thermal annealing (10 min, 120 °C) the emission layer was spin-coated from tetrahydrofuran (3 g L−1, 3000 rpm, 45 s) and thermally annealed (10 min, 80 °C) to remove any residual solvents. TmPyPB (50 nm), Liq (2 nm) and Al (50 nm) were deposited in a home-built multi-chamber cluster-tool attached to the glovebox at a pressure <10−6 mbar. The light-emitting area (10.5 mm2) was defined by the overlap of the electrodes. Neat emitter layers were deposited on quartz substrates from solutions of the same concentration.
OLED characterization
All optoelectronic characterization was carried out in a glovebox in a nitrogen atmosphere. The OLEDs were driven with a programmable source measurement unit (Keithley 2400). The luminous flux and the spectrum of the OLEDs were measured in an integrating sphere (Gigahertz Optik, diameter: 19 cm) in a 2π-configuration. The integrating sphere was equipped with a photometer (Gigahertz Optik VL-1101-2) and coupled to a spectrometer (Instrument Systems CAS140CT-151). The substrate edges were concealed to detect only light that was emitted from the surface of the OLED. The measurement setup was calibrated with a halogen luminance standard with a known luminous flux and spectrum traceable to the Physikalisch Technische Bundesanstalt (PTB). A halogen aiding lamp was used to correct for reflections of the calibration lamp and the OLEDs in the sphere. The respective calculations are detailed in the ESI.† The external quantum efficiency (EQE) was calculated from the luminous flux, the spectrum and the driving current. The luminance was calculated from the luminous flux assuming Lambertian emission. The measurement uncertainty is 5.8% (95% confidence interval). The lifetime of the OLEDs was assessed in the same setup under constant current operation.Transient electroluminescence (EL)
Transient EL was performed with a spectrofluorometer (Edinburgh Instruments FS5) flushed with nitrogen. The OLEDs were pulsed with an arbitrary function generator (Keysight 81150A) with an output impedance of 5 Ohm to reduce RC-effects.Optical spectroscopy
All optical spectroscopy was performed in the same spectrofluorometer continually flushed with nitrogen. Solid state photoluminescence quantum yields (PLQYs) were measured in an integrating sphere with excitation from a xenon lamp (340 nm). Solution PLQYs were measured in degassed tetrahydrofuran (3 freeze-pump thaw cycles) using 9,10-diphenylanthracene in ethanol as the standard (PLQY = 95%).34 Samples were excited with a pulsed LED (340 nm, pulse width: 1 ns) for the transient photoluminescence measurements.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
P. L. B. acknowledges the Helmholtz International Fellow Award under which the exchange of scientists and this collaborative work was made possible. The synthesis of the materials was supported by the Australian Research Council Laureate Fellowship awarded to P. L. B. (FL160100067). The fluorometer was made available under grant no. 03EK3571 (project TAURUS2), funded by the German Ministry for Education and Research (BMBF). We thank Alexander Deniz Schulz for AFM measurements.Notes and references
- OLED-info, https://www.oled-info.com/joled-supplies-its-printed-oleds-lgs-upcoming-315-inch-4k-monitor.
- Display Supply Chain Consultants, OLED-info, https://www.oled-info.com/dscc-ink-jet-printing-could-lead-17-cost-reduction-55-oled-tv-production.
- S. Höfle, M. Pfaff, H. Do, C. Bernhard, D. Gerthsen, U. Lemmer and A. Colsmann, Org. Electron., 2014, 15, 337–341 CrossRef.
- Q. Zhang, D. Tsang, H. Kuwabara, Y. Hatae, B. Li, T. Takahashi, S. Y. Lee, T. Yasuda and C. Adachi, Adv. Mater., 2015, 27, 2096–2100 CrossRef CAS.
- X. Ban, W. Jiang, K. Sun, B. Lin and Y. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 7339–7346 CrossRef CAS.
- J. Li, X. Liao, H. Xu, L. Li, J. Zhang, H. Wang and B. Xu, Dyes Pigm., 2017, 140, 79–86 CrossRef CAS.
- F. Ma, X. Zhao, H. Ji, D. Zhang, K. Hasrat and Z. Qi, J. Mater. Chem. C, 2020, 8, 12272–12283 RSC.
- X. Ban, Y. Liu, J. Pan, F. Chen, A. Zhu, W. Jiang, Y. Sun and Y. Dong, ACS Appl. Mater. Interfaces, 2020, 12, 1190–1200 CrossRef CAS.
- S.-C. Lo, R. N. Bera, R. E. Harding, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2008, 18, 3080–3090 CrossRef CAS.
- S.-C. Lo, R. E. Harding, C. P. Shipley, S. G. Stevenson, P. L. Burn and I. D. W. Samuel, J. Am. Chem. Soc., 2009, 131, 16681–16688 CrossRef CAS PubMed.
- D. Xia, B. Wang, B. Chen, S. Wang, B. Zhang, J. Ding, L. Wang, X. Jing and F. Wang, Angew. Chem., 2014, 126, 1066–1070 CrossRef.
- Y. Wang, S. Wang, J. Ding, L. Wang, X. Jing and F. Wang, Chem. Commun., 2016, 53, 180–183 RSC.
- X.-L. Chen, J.-H. Jia, R. Yu, J.-Z. Liao, M.-X. Yang and C.-Z. Lu, Angew. Chem., 2017, 129, 15202–15205 CrossRef.
- K. Wu, Z. Wang, L. Zhan, C. Zhong, S. Gong, G. Xie and C. Yang, J. Phys. Chem. Lett., 2018, 9, 1547–1553 CrossRef CAS.
- L. Graf von Reventlow, W. Jiang, D. M. Stoltzfus, S. M. Russell, P. L. Burn and A. Colsmann, Adv. Opt. Mater., 2020, 8, 2001289 CrossRef.
- D. M. Stoltzfus, W. Jiang, A. M. Brewer and P. L. Burn, J. Mater. Chem. C, 2018, 6, 10315–10326 RSC.
- R. C. Little and R. Y. Ting, J. Chem. Eng. Data, 1976, 21, 422–423 CrossRef CAS.
- P. L. Burn, R. Beavington, M. J. Frampton, J. Pillow, M. Halim, J. M. Lupton and I. D. W. Samuel, Mater. Sci. Eng., B, 2001, 85, 190–194 CrossRef.
- J. Y. Liew, S.-C. Lo, P. L. Burn, E. R. Krausz, J. D. Hall, E. G. Moore and M. J. Riley, Chem. Phys. Lett., 2015, 641, 62–67 CrossRef CAS.
- S.-C. Lo, R. E. Harding, E. Brightman, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2009, 19, 3213 RSC.
- M. Tanaka, H. Noda, H. Nakanotani and C. Adachi, Adv. Electron. Mater., 2019, 5, 1–8 Search PubMed.
- J.-H. Lee, S. Lee, S.-J. Yoo, K.-H. Kim and J. Kim, Adv. Funct. Mater., 2014, 24, 4681–4688 CrossRef CAS.
- S. Schmidbauer, A. Hohenleutner and B. König, Adv. Mater., 2013, 25, 2114–2129 CrossRef CAS PubMed.
- X. Zhou, P. L. Burn and B. J. Powell, Inorg. Chem., 2016, 55, 5266–5273 CrossRef CAS PubMed.
- R. Seifert, I. Rabelo De Moraes, S. Scholz, M. C. Gather, B. Lüssem and K. Leo, Org. Electron., 2013, 14, 115–123 CrossRef CAS.
- C. Jeong, C. Coburn, M. Idris, Y. Li, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Org. Electron., 2019, 64, 15–21 CrossRef CAS.
- Q. Wang, Y. Luo and H. Aziz, J. Appl. Phys., 2010, 107, 84506 CrossRef.
- J. A. McEwan, A. J. Clulow, A. Nelson, N. R. Yepuri, P. L. Burn and I. R. Gentle, ACS Appl. Mater. Interfaces, 2017, 9, 14153–14161 CrossRef CAS PubMed.
- X. Zhou, J. He, L. S. Liao, M. Lu, X. M. Ding, X. Y. Hou, X. M. Zhang, X. Q. He and S. T. Lee, Adv. Mater., 2000, 12, 265–269 CrossRef CAS.
- Q. Wang, B. Sun and H. Aziz, Adv. Funct. Mater., 2014, 24, 2975–2985 CrossRef CAS.
- Q. Wang and H. Aziz, Adv. Opt. Mater., 2015, 3, 967–975 CrossRef CAS.
- H. Yu and H. Aziz, J. Phys. Chem. C, 2019, 123, 16424–16429 CrossRef CAS.
- Y. J. Cho, S. Taylor and H. Aziz, ACS Appl. Mater. Interfaces, 2017, 9, 40564–40572 CrossRef CAS PubMed.
- J. V. Morris, M. A. Mahaney and J. R. Huber, J. Phys. Chem., 1976, 80, 969–974 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc00602b |
| This journal is © The Royal Society of Chemistry 2022 |