
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous enhancement of thermally activated delayed fluorescence and photoluminescence quantum yield via homoconjugation†
Stephanie
Montanaro‡
a,
Piotr
Pander‡
 bc,
Jai-Ram
Mistry
a,
Mark R. J.
Elsegood
a,
Simon J.
Teat
d,
Andrew D.
Bond
e,
Iain A.
Wright
*a,
Daniel G.
Congrave
*e and
Marc K.
Etherington
*f
bc,
Jai-Ram
Mistry
a,
Mark R. J.
Elsegood
a,
Simon J.
Teat
d,
Andrew D.
Bond
e,
Iain A.
Wright
*a,
Daniel G.
Congrave
*e and
Marc K.
Etherington
*f
aDepartment of Chemistry, Loughborough University, Epinal Way, LE11 3TU, UK. E-mail: i.a.wright@lboro.ac.uk
bDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK
cCentre for Organic and Nanohybrid Electronics, Silesian University of Technology, Konarskiego 22B, 44-100 Gliwice, Poland
dAdvanced Light Source, Lawrence Berkeley National Laboratory, Cyclotron Road, Berkeley, CA 94720, USA
eYusuf Hamied Department of Chemistry, Cambridge, CB2 1EW, UK. E-mail: dc704@cam.ac.uk
fDepartment of Mathematics, Physics and Electrical Engineering, Northumbria University, Ellison Place, Newcastle upon Tyne, NE1 8ST, UK. E-mail: marc.k.etherington@northumbria.ac.uk
First published on 25th March 2022
Abstract
A critical challenge facing thermally activated delayed fluorescence (TADF) is to facilitate rapid and efficient electronic transitions while ensuring a narrow singlet–triplet energy gap (ΔEST) in a single luminophore. We present a TADF-active iptycene that clearly demonstrates that homoconjugation can be harnessed as a viable design strategy towards answering this challenge. A homoconjugated analogue of an established quinoxaline-based TADF luminophore has been produced by fusing three of these luminophores together across a shared triptycene core. Homoconjugation was confirmed by electrochemistry, and as a direct consequence of this phenomenon we observed synergistic improvements to photoluminescence quantum yield (ΦPL), radiative rate of singlet decay (kSr), delayed fluorescence lifetime (τTADF), and rate of reverse intersystem crossing (krISC), all while narrowing the ΔEST. The enhancement is rationalised with TD-DFT calculations including spin–orbit coupling (SOC). A facile synthesis, the ubiquity of the pyrazine motif in state-of-the-art TADF materials of all colours, and the extent of the overall performance enhancement leads to a great potential for generality.
Introduction
Materials that exhibit thermally activated delayed fluorescence (TADF) are the emergent generation of emitters for organic light-emitting diodes (OLEDs). TADF materials require small singlet–triplet energy gaps (ΔEST) to enable triplet harvesting via reverse intersystem crossing (rISC). rISC is most typically observed in donor–acceptor compounds displaying intramolecular charge transfer (ICT) states which have a large spin–orbit coupling (SOC). The required high SOC is achieved through introducing a manifold of partially delocalised triplet states of both locally-excited (LE) and charge-transfer (CT) character.1–4Structurally, the need for a manifold of states of different characters has translated to ICT molecules built up of numerous weakly coupled chromophores, conjugated through twisted bonds and/or cross conjugation. Prominent examples are 5Cz-TRZ and TAT-3DBTO2 reported by Cui, Chen, Adachi, Friend et al.2 and by Bryce, Monkman and co-workers,5 respectively (Fig. 1a).
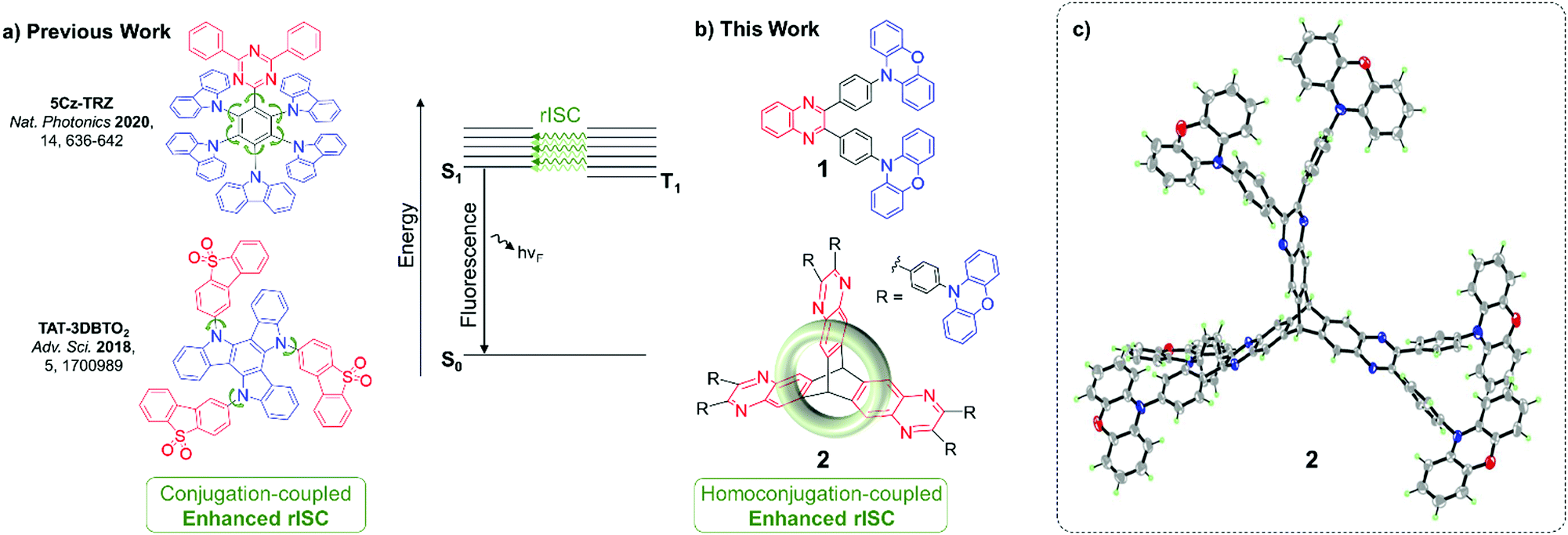 | ||
| Fig. 1 (a) Previous and (b) new designs for enhancing TADF through weakly coupled components; (c) X-ray crystal structure of 2, 7 CHCl3 of crystallisation omitted for clarity. | ||
To obtain efficient rISC a narrow ΔEST is necessary. The ΔEST can be reduced by decreasing the spatial overlap of the electron and hole wavefunctions, which is typically realised by introducing aromatic rings as π-spacers between the donor and acceptor moieties and/or increasing the dihedral angle between said moieties.1,6,7 Unfortunately, a rapid rISC rate (krISC) often comes at the expense of a high photoluminescence quantum yield (PLQY, ΦPL) and a fast radiative rate (kr),8 as reducing ΔEST is fundamentally accompanied by a significant reduction in oscillator strength.5 Hence, molecules capable of combining both a high PLQY and large krISC are prized in TADF materials development,2,3,5,9,10 and exploring new molecular design concepts which can synergistically enhance PLQY and kr alongside krISC is of vital importance.
Through-space CT (TSCT) can occur in molecules where donor and acceptor moieties are adjacent and proximal, allowing orbital overlap between the donor and the acceptor through the space between them. This contrasts with through-bond CT (TBCT) observed in traditional organic ICT systems. TSCT has recently gained significant attention towards improving TADF parameters, most notably SOC, as it can support multiple CT states and a large manifold of near-degenerate excited states. Molecules can be designed to show solely TSCT11–20 or a combination of TBCT and TSCT.21,22
Homoconjugation is another non-traditional form of π-electron delocalisation which arises when two conjugated π-systems are separated by a single non-conjugated group, typically an sp3 carbon atom.23 It can allow the two π-systems to continue to communicate despite a formal break in conjugation. Triptycene is an archetypal homoconjugated hydrocarbon.24–29 The unsaturated π-systems of its three phenylene “fins” sit 120° apart, enforced by the two saturated sp3 bridgehead carbon atoms. Such a rigid conformation forces a trans-annular, through-space overlap between the π-clouds of each phenylene ring (Fig. 1b), resulting in homoconjugated electronic delocalisation which is distinct from conventional through-bond conjugation.30–33
Homoconjugation has recently received significant attention in the context of novel luminophores. It has been shown to mediate intramolecular charge transfer between donor and acceptor moieties to introduce efficient TSCT–TADF including spirocyclic34 and triptycene-based structures.35–38 A few other TSCT–TADF systems have been synthesised using spirocycles12,14,18 and triptycene20 as the scaffolds which link the donor and acceptor moieties. However, in these previous examples the homoconjugated fragments are spectating rather than being actively involved in the TADF process.
A promising phenomenon attributed to homoconjugation in symmetrical iptycenes is an enhancement of π–π* oscillator strength due to cooperative interactions between fins. Experimentally this is observed as a significantly greater than factor of three increase in extinction coefficient (ε) compared with a single fin congener, and is most pronounced when the LUMO is localised over the region of homoconjugation.39–41 Recently, we extended this phenomenon to ICT transitions through trimerization of a carbazole-based fluorescent TBCT luminophore which had six N-phenylcarbazole moieties linked by a shared, electron-deficient tris(pyrazino)triptycene.42 The design can be viewed as a core–shell type structure with the HOMO residing over peripheral, electronically isolated donor heterocycles which, upon excitation, push electrons in a TBCT fashion into the LUMO which is localised over the shared homoconjugated triptycene-based acceptor at the centre of the molecule. The design resulted in large enhancements in ICT parameters such as ε, kr, and PLQY (i.e. radiative transitions). Such an approach to harnessing homoconjugation is distinctly different to the previously mentioned existing examples where TSCT takes place between individual donor and acceptor functionalised fins of asymmetric triptycenes or either side of a spirocyclic carbon atom.
To directly address the challenge of sustaining a high PLQY and rapid krISC we now employ a TADF luminophore in the same fashion to exploit two precedents: (1) the increase of TADF efficiency afforded by weakly coupling numerous chromophores,2,5 and (2) the enhancement of ICT radiative processes via LUMO homoconjugation.39–42 Simultaneous improvements in both radiative decay and krISC are realised.
Results and discussion
Design and synthesis
To test our hypothesis the iptycene 2 was synthesised. Three factors led to the selection of this system: (1) the opportunity for comparison with the homologous fin 1, a known TBCT–TADF emitter,43 (2) the facile synthesis (Scheme S2.1, ESI†) of quinoxaline-based iptycenes, and (3) the ubiquity of the pyrazine motif in TADF materials.44–50 The NMR characterisation of the compounds can be found in the ESI† (Fig. S3.1–S3.5). The structure of 2·7CHCl3 was confirmed by single crystal X-ray diffraction (Fig. 1c and Fig. S4.7, Table S4.3, ESI†). 2 crystallised as a CHCl3 heptasolvate, presumably due to the large internal free volume of iptycene structures.51 The interfin angles and distances observed for 2 are essentially identical to those observed for triptycene,52,53 supporting homoconjugation in 2. The structure of two, polymorphic, CDCl3 mono-solvates of 1 were also determined using synchrotron radiation (Tables S4.1, S4.2 and Fig. S4.1–S4.6, ESI†). 2 has good thermal stability with a decomposition temperature of 544 °C (Td corresponding to 5% weight loss) as demonstrated by thermal gravimetric analysis (Fig. S2.1, ESI†). This compares with a Td of 421 °C reported previously for single fin 1.43Computational chemistry
1 and 2 were investigated using TD-PBE0/def2-svp (Fig. 2 and Fig. S6.1–S6.34, ESI†).54 The lowest energy singlet and triplet states for 1 and 2 are predominantly represented by ICT transitions between the phenoxazine donor and quinoxaline acceptor moieties involving the HOMO and LUMO as expected (Fig. S6.1, S6.3, S6.6, S6.9, S6.33 and S6.34, ESI†).43 For 2 the S1 and S2 states are near-degenerate (Fig. 2a) – a consequence of the localisation of the LUMO/LUMO+1 orbitals of the iptycene core of 2 due to through-space π-system overlap between fins. Such a MO structure is in agreement with established experimental data for triptycene,49,55,56 and indicates that homoconjugation should influence the photophysical properties of 2. Furthermore, IRI-π analysis57 indicates weak π-interactions between fins, consistent with homoconjugation (Fig. S6.35, ESI†). Narrow ΔEST values are predicted for 1 and 2 (0.01 and 0.03 eV, respectively). Calculations predict a notably larger oscillator strength (f) for the S0 → S1 transition of 2 than for 1 (f = 0.051 vs. 0.003). f is similarly large for the near-degenerate S0 → S2 transition of 2 (ƒ = 0.051). Larger f for the lowest lying singlet states of 2 compared with 1 are promising for enhancements in ε and the radiative rate of singlet decay (kSr) (confirmed experimentally below). Furthermore, within <0.4 eV of T1 a manifold of 26 higher energy singlet and triplet states are predicted for the iptycene 2 (Fig. S6.7–S6.32, ESI†), in contrast to only 4 for 1 (Fig. S6.2–S6.5, ESI†). This collection of energetically close excited states (Fig. 2b), comprising numerous CT states spread across the different fins of 2 and a larger number of 3LE states than for 1, is highly encouraging for accelerated delayed fluorescence in 2.2,3,5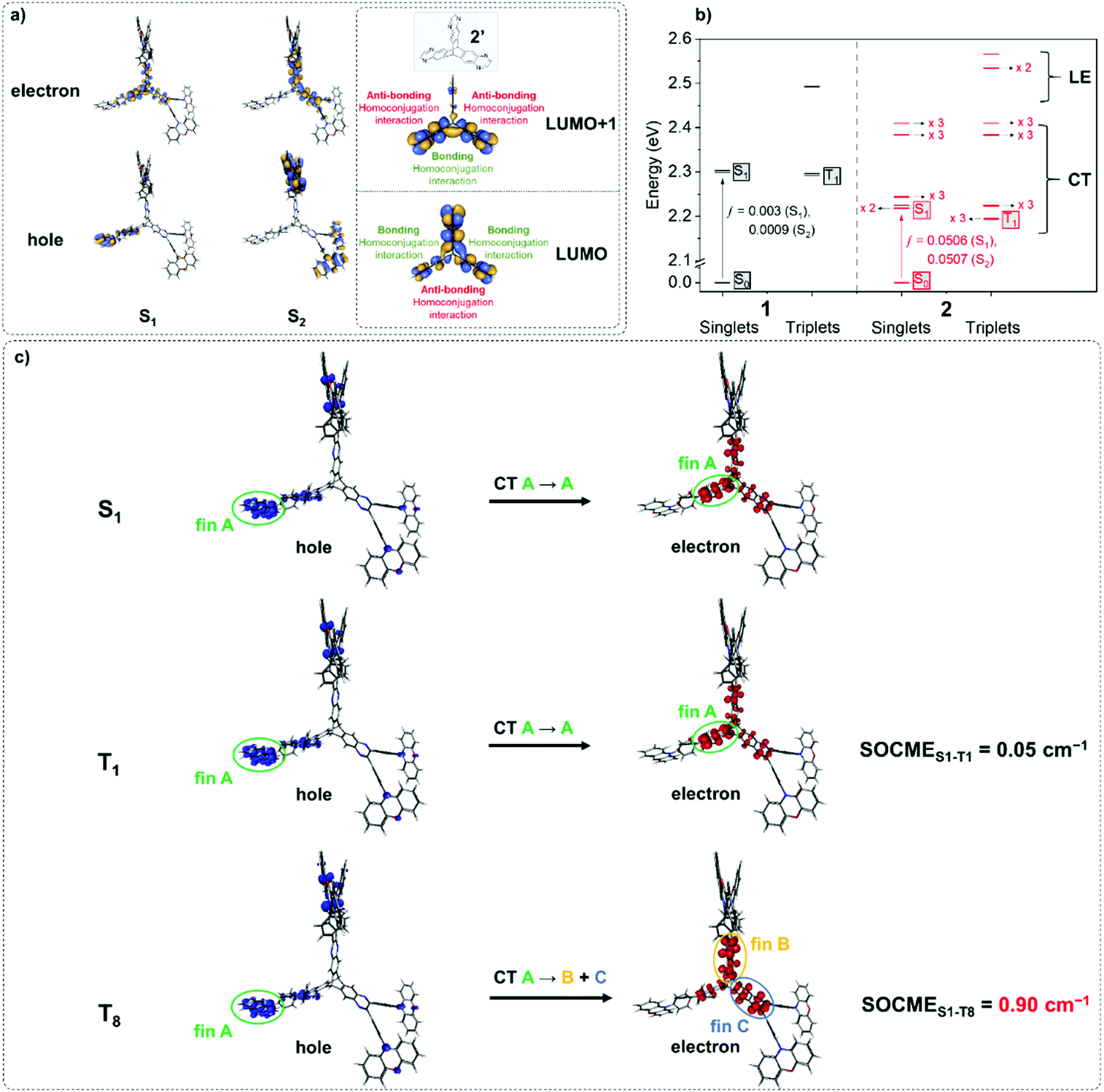 | ||
| Fig. 2 (a) Natural transition orbitals for S1 and S2 states of 2. The LUMO and LUMO+1 orbitals of the core 2’ are included to highlight the effect of homoconjugation within the LUMO manifold; (b) Jablonski diagram for 1 and 2; (c) natural transition orbitals for the S1 and appropriate T states of 2 to highlight spin–orbit coupling. | ||
Spin–orbit coupling (SOC) is essential to permit the formally forbidden spin-flip associated with rISC. Hence, the merit of the manifold of triplet states predicted for 2 was investigated by calculating the spin–orbit coupling matrix elements (SOCMEs) for the ground state optimized structure (TD-PBE0/def2-svp and PySOC script58). The relevant states were probed via electron–hole analysis with the Multiwfn program to determine their CT or LE character.59–61 Detailed electron–hole analysis and assignment of excited state character are presented in the ESI† (Section S6 and Table S6.1). Some relevant natural transition orbitals (NTOs) for 2 are shown in Fig. 2c. The fins of 2 in Fig. 2c are denoted A, B and C to aid discussion of the spatial distribution of states. Data for S1, T1, and triplet states with SOCME > 0.1 cm−1 to S1 within <0.4 eV of T1 are summarized in Table 1 for 1 and 2.
| Compound | State | ΔES1–Tna (eV) | SOCMES1–Tnb (cm−1) | Assignmentc |
|---|---|---|---|---|
| a Calculated energy gap between S1 and Tn. b Calculated spin–orbit coupling matrix element between S1 and Tn. c A, B and C fin nomenclature is consistent with Fig. 2c; CT = charge transfer, LE = locally excited. | ||||
| 1 | S1 | — | — | CT |
| T1 | 0.01 | 0.05 | CT | |
| T3 | −0.19 | 0.23 | LE | |
| 2 | S1 | — | — |
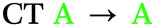
|
| T1 | 0.03 | 0.05 |
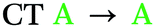
|
|
| T7 | −0.17 | 0.29 |

|
|
| T8 | −0.17 | 0.90 |

|
|
| T13 | −0.32 | 0.11 | LE | |
| T14 | −0.32 | 0.08 | LE | |
| T15 | −0.35 | 0.12 | LE | |
In agreement with what is intuitive upon visual inspection of the NTOs (Fig. 2a and c), the S1 state of 2 is assigned as CT in nature. The electron–hole analysis for T1 is near-identical to that of S1, meaning both states are the same in configuration (CT) with the same spatial distribution. Hence, as expected from El-Sayed's rule,62 the calculated S1–T1 SOCME is very small (0.05 cm−1), and rISC should be very inefficient between these states. A similar case is observed for the S1 and T1 states of 1 (Table 1 and Fig. S6.1, S6.3, ESI†).
It has been accepted that in TADF materials vibronic coupling can facilitate rISC between a CT singlet such as S1 and upper LE triplet states, for which there should be greater SOC due to a change in excited state configuration.1–4,10 Indeed, the SOCME values between S1 and upper LE triplet states (T3 for 1; T13, T14 and T15 for 2) are of the order of 0.1–0.25 cm−1.
We note for 1 that there is only one LE triplet state (T3) within 0.4 eV of T1 with a greater SOCME to S1 than T1. This is in stark contrast to what is observed for 2.
We can explain the more efficient rISC observed experimentally for 2 through the larger presence of states with high SOC. Firstly, 2 has three LE triplets (T13, T14 and T15) that exhibit appreciable SOCMEs with S1. Secondly, due to electronic coupling between the fins of 2, it is possible to localise CT states across neighbouring fins (Fig. 2c). This is apparent for the T8 state which is localised differently to S1. T8 is a CT state with the hole predominantly localised on the phenoxazine moieties of fin A, and the electron mainly on the quinoxaline heterocycles of fins B and C with some bridging A contribution. Conversely, for S1 both the electron and hole are predominantly localised over fin A with some electron density extending onto the quinoxalines of B and C. These distinct differences in the localisation of S1 and T8, can translate into a change in orbital angular momentum between the two states, but they crucially retain some spatial overlap over the homoconjugated core of 2. This enables a large SOCME value of 0.9 cm−1 between the CT states S1 and T8, which are predicted to be only 0.17 eV apart in energy (Table 1). A significant SOCME is also predicted between S1 and T7 (0.29 cm−1), which is a CT state degenerate with T8 also spread across multiple fins (Table 1 and Fig. S6.18, ESI†).
While the exact rISC mechanism in 2 may be complex, from simply considering the SOCME calculations it is abundantly clear that homoconjugation in 2 can facilitate strong SOC interactions between TADF-relevant states, which are not predicted for the non-homoconjugated single chromophore 1.
Electrochemistry
1 and 2 display identical first oxidation potentials (Eox) (Fig. 3 and Table S5.1, ESI†), consistent with localisation of the HOMO of 2 on the peripheral phenoxazine donors. Conversely, 2 displays a less negative first reduction potential (Ered) than 1, and hence a more accessible LUMO, which slightly increases its acceptor strength. This can be attributed to the LUMO of 2 being delocalized in a trans-annular fashion over the three quinoxaline rings and provides further evidence of homoconjugation in the LUMO. Additionally, while the first reduction wave for 1 corresponds to what is typically expected for a single electrochemically reversible process, the wave for 2 is clearly a superposition of three electrochemical reductions. This electrochemical behavior supports weak electronic coupling between the multiple fins of 2 through the lower lying unoccupied molecular orbitals, consistent with homoconjugation and the DFT predictions above.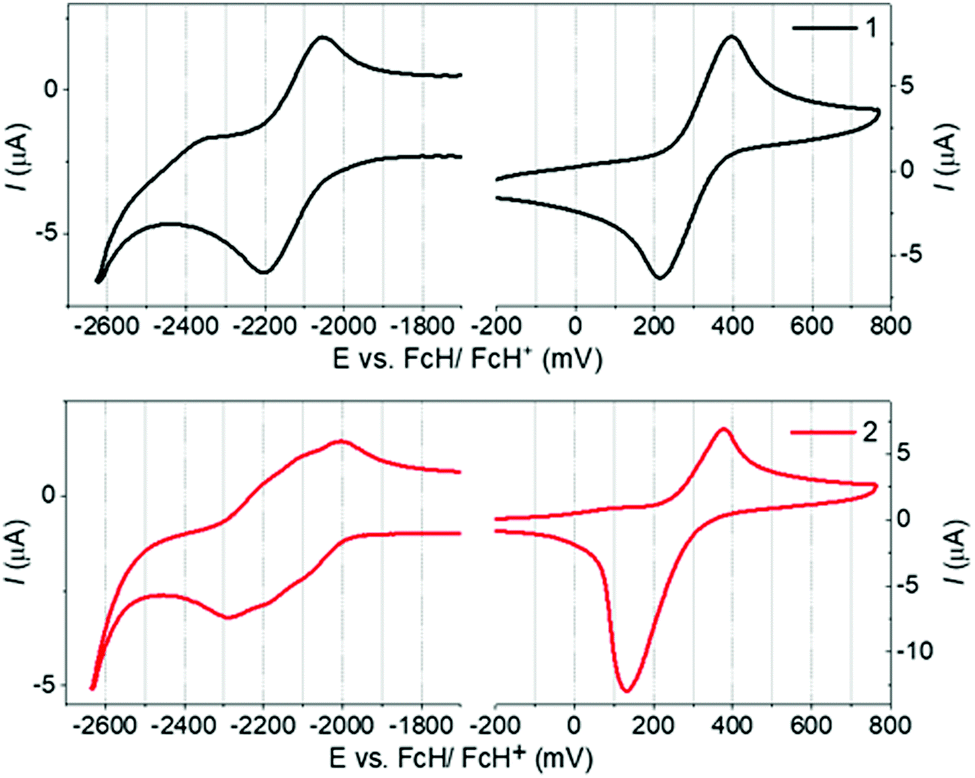 | ||
| Fig. 3 Cyclic voltammograms for 1 and 2 measured in 0.1 M n-Bu4PF6 in 1,2-dichlorobenzene as the supporting electrolyte. Scan rate = 100 mV s−1. | ||
Photophysics
Photophysical measurements (Table 2) in toluene solution (Fig. 4) provide a platform to accurately compare pertinent rates, as the data can be considered intrinsic, with strictly monoexponential decay for both prompt and delayed fluorescence in the absence of any solid-state phenomena. The absorption (and PL) spectrum of 2 is bathochromically shifted compared with that of 1 due to homoconjugation in the LUMO of 2 facilitating a more extended π-structure and thereby a slightly reduced optical band gap. This is consistent with electrochemical and TD-DFT results. The absorption spectra for 1 and 2 consist of intense transitions below ca. 350 nm, assigned to π–π* excitations, and lower energy broad ICT transitions ≥400 nm.63 Similarly, the PL spectra of 1 and 2 are also ascribed to ICT, being broad and featureless and exhibiting positive solvatochromism (Fig. S8.1, ESI†).| Solvent/matrix | λ abs (nm) [ε × 103 M−1 cm−1]a | λ em (nm) | ΔESTc (eV) [S1/T1] | Φ PL | Φ PF | Φ DF | τ PF (ns) | τ TADF (μs) | k Sr (107 s−1) | k rISC (104 s−1)f | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a sh = shoulder. b Steady state PL maxima. c The S1 energy is taken from delayed fluorescence (and not prompt fluorescence) spectra due to its relevance for the TADF mechanism. d Total PLQY in degassed solution or under N2 in film. e Radiative rate of the S1 state, kSr = ΦPF/τPF. f For calculation details refer to the ESI. All values determined at room temperature except for T1 energy which is taken from phosphorescence spectra at 80 K. | |||||||||||
| 1 | Toluene | 332 [26], 398 sh [3.5] | 574 | — | 0.50 | 0.35 | 0.15 | 19.6 | 27.3 | 1.8 | 2.5 |
| Zeonex | — | 504 | 0.26 [2.74/2.48] | — | — | — | 8.9 | 656 | — | — | |
| PVK:PBD | — | 548 | 0.13 [2.54/2.41] | 0.63 | 0.41 | 0.22 | 16.4 | 26.2 | 2.5 | 3.4 | |
| 2 | Toluene | 338 [62], 366 [69], 420 sh [12] | 583 | — | 0.61 | 0.48 | 0.13 | 20.5 | 5.8 | 2.4 | 8.7 |
| Zeonex | — | 517 | 0.21 [2.65/2.44] | — | — | — | 12.4 | 90.1 | — | — | |
| PVK:PBD | — | 556 | 0.06 [2.47/2.41] | 0.75 | 0.49 | 0.26 | 24.0 | 16.7 | 2.1 | 6.5 | |
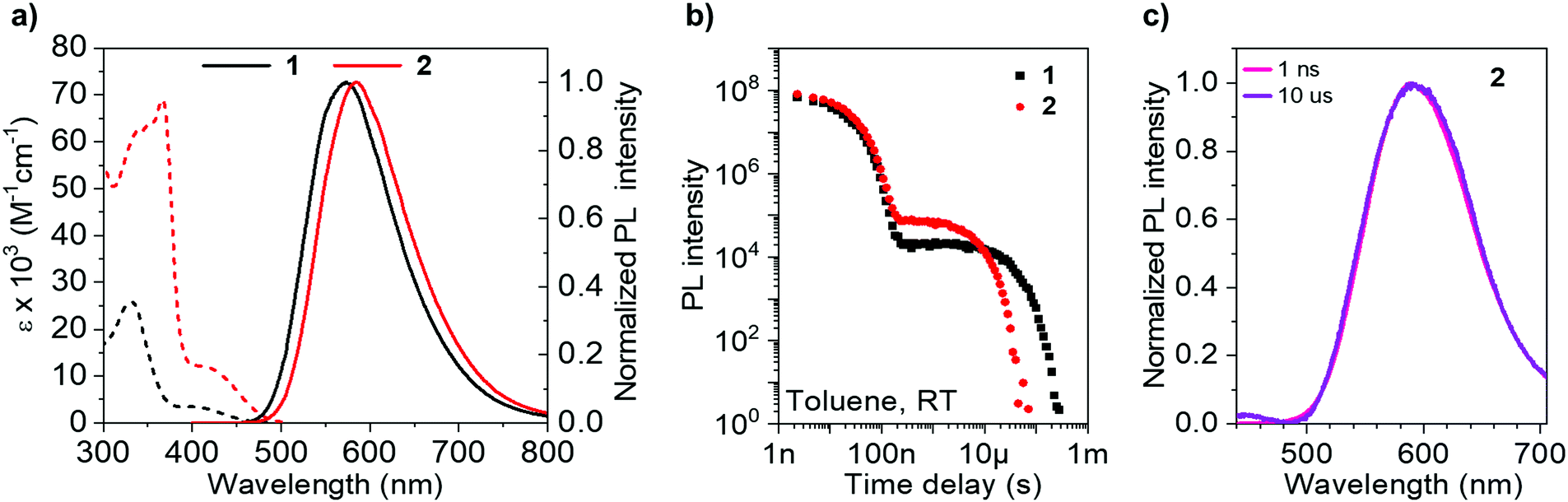 | ||
| Fig. 4 (a) Extinction coefficient and steady state PL spectra (1 × 10−5 M) of 1 and 2 in toluene; (b) PL decays of 1 and 2 in degassed toluene; (c) prompt and delayed time-resolved spectra for 2 in degassed toluene. | ||
As 2 can be considered a molecular trimer of the monomer unit 1, the maximum ICT absorption extinction coefficient for 2 is expected to be three times that of 1 in the absence of any electronic communication between fins. However, our experimental values, obtained from triplicate measurements on rigorously dried samples, yield a ratio of 3.4 (Table 2) (for 1ε = 3.5 × 103 M−1 cm−1 at 398 nm; for 2ε = 12 × 103 M−1 cm−1 at 420 nm). An expansion of the ICT region of the UV-vis spectra for 1 and 2 is provided in Fig. S8.2 (ESI†). TD-DFT results indicate that the singlet ICT states of 2 have significant localisation over the homoconjugated iptycene core, while electrochemical measurements also indicate trans-annular orbital overlap between fins. Therefore, the larger than factor of three increase in ICT extinction coefficient for 2 compared with 1 is ascribed to a cooperative effect between the fins of 2 through homoconjugation. Concomitant with this increase, the solution values for ΦPL and kSr, are considerably enhanced for 2 compared with 1 (1 − ΦPL = 50%, kSr = 1.8 × 107 s−1; 2 − ΦPL = 63%, kSr = 2.4 × 107 s−1). Notably, for 2 the value of kSr is increased by a third compared with 1. Therefore, there is a clear enhancement in the probability of a radiative transition both to and from the ICT state in 2, crucially observed despite the narrower ΔEST of 2 compared with 1 (1 − ΔEST = 0.26 eV; 2 − ΔEST = 0.21 eV), as measured in a zeonex host (Table 2 and Fig. S9.1–S9.9, S10.1, ESI†). The acceleration of radiative transitions in 2 is very likely linked to homoconjugation, in-line with the computational predictions and electrochemical experiments above. We note that electron–hole analysis (Fig. S6.3 and S6.9, ESI†) vaguely suggests that the homoconjugation in 2 leads to an increase in π–π* character of the S1 state for 2 compared with 1, which may help explain the improved ε and kSr values. Such an enhancement of ICT through homoconjugation has only been noted once previously, in a structure that does not display TADF.42 Notably, S1 is also significantly more stabilised than T1 upon trimerization of compound 1 to 2, suggesting the iptycene core can be employed to fine-tune the ΔEST of TADF compounds.
The TADF parameters of 1 and 2 were evaluated in toluene solution (Fig. 4). Both luminophores exhibit a delayed fluorescence (DF) component that spectrally matches the prompt fluorescence (PF) (Fig. 4c and S10.1, ESI†) and contributes similarly to the total ΦPL (1 − ΦDF = 15%; 2 − ΦDF = 13%). However, the lifetime of the delayed fluorescence (τTADF) for the iptycene 2, is nearly five times shorter than that observed for 1 (Fig. 4b), in line with a greater than threefold increase in krISC (1 − τTADF = 27.3 μs, krISC = 2.5 × 104 s−1; 2 − τTADF = 5.8 μs, krISC = 8.7 × 104 s−1). This is consistent with the dense manifold of energetically close excited states and enhanced SOC in 2 predicted by TD-DFT (Fig. 2), a consequence of intrafin coupling via homoconjugation.
To summarise, considered alongside electrochemical and computational studies, the photophysical data obtained for 2 in solution indicate that it is possible to simultaneously amplify the radiative transition probability and krISC of a TADF luminophore by exploiting homoconjugation. Similar results are obtained in the PVK:PBD blend used for OLED fabrication below i.e. a larger PLQY, shorter τTADF, and faster krISC for 2 compared with 1 (Fig. S11.1–S11.6, ESI†).
Electroluminescence
Solution-processed OLED devices were fabricated using 1 and 2 as emitters, denoted Dev 1 and Dev 2, respectively. Pertinent data and the device structure are shown in Fig. 5. Additional data are presented in the ESI† (Fig. S12.1 and Table S12.1). The device structure offers low VON and large current densities, desirable for high device luminance, but also high external quantum efficiency (EQE) due to the presence of an additional electron blocking PVKH layer.64Dev 1 and Dev 2 display EQEs reaching maximum values of 9.7% and 11.9%, respectively, in agreement with ΦPL in film, demonstrating a ∼20% increase in efficiency due to homoconjugation. Most crucially, Dev 2 displays smaller efficiency roll-off than Dev 1, thanks to the shorter τTADF and larger krISC of 2 in the OLED host (Table 2). The difference in roll-off between Dev 1 and Dev 2 is manifested by their diverging characteristics of EQE vs. current density in Fig. 5b. The electroluminescence (EL) spectra match the PL in the same host (Table 2). The 11.9% figure is larger than that reported earlier for a similar device structure.64 While the larger efficiency of Dev 2 contributes to its greater maximum luminance (Dev 1 – 8900 cd m−2; Dev 2 – 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 cd m−2), the other significant factor is the higher current density (Dev 1 – 120 mA cm−2; Dev 2 – 220 mA cm−2). Both OLEDs show a similar VON = 5.5–6 V as 1 and 2 have nearly identical HOMO and LUMO energies.
800 cd m−2), the other significant factor is the higher current density (Dev 1 – 120 mA cm−2; Dev 2 – 220 mA cm−2). Both OLEDs show a similar VON = 5.5–6 V as 1 and 2 have nearly identical HOMO and LUMO energies.
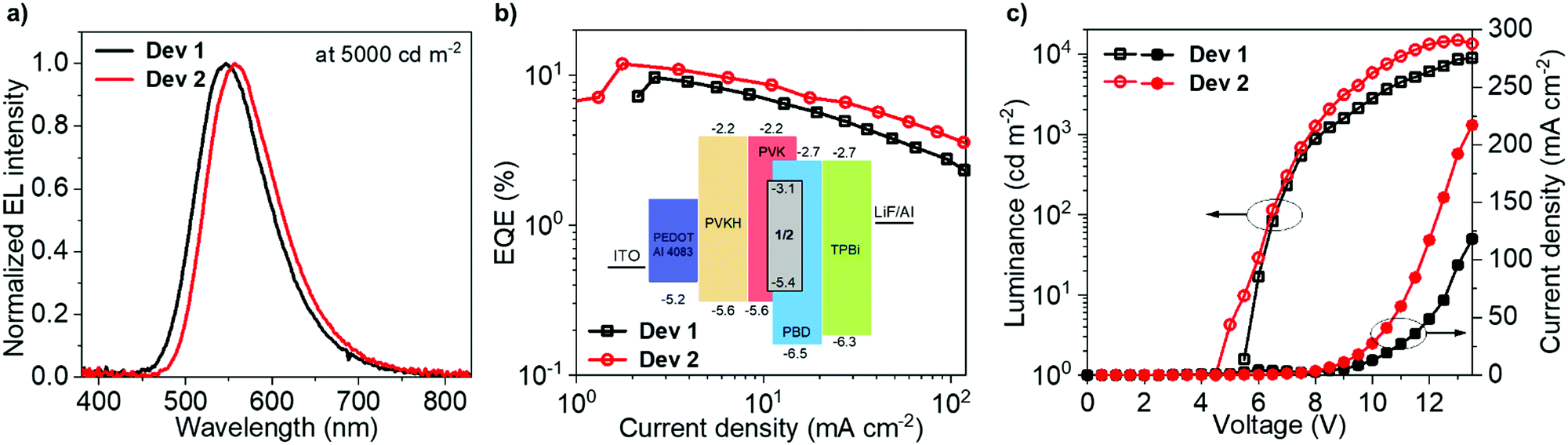 | ||
| Fig. 5 Electroluminescence characteristics for Dev 1 and Dev 2. | ||
Conclusions
The simultaneous realization of high oscillator strength alongside efficient rISC is essential in the design of next generation TADF materials. For the first time we have proven that it is possible to harness homoconjugation as a viable strategy towards this objective. By synthesizing a trimer of the known TADF luminophore 1 where the three monomers share an iptycene core, homoconjugation was induced in 2, which was observed electrochemically. As a direct consequence of the homoconjugation in 2, simultaneous improvements to PLQY, kSr, τTADF, krISC, and ΔEST were achieved, in-line with TD-DFT calculations which predicted a synergistic enhancement of oscillator strength (f) and spin–orbit coupling (SOC). Owing to the facile synthesis of this new system, and the ubiquity of the pyrazine moiety in state-of-the-art TADF materials across the electromagnetic spectrum from the deep blue to the near-infrared (NIR),44–49 this concept has great promise for generality. NIR TADF emitters in particular are poised to benefit from the enhancement in radiative transitions offered by homoconjugation and will be explored in future work.Author contributions
S. M. conceptualization, formal analysis, investigation, writing – original draft, writing – review & editing, visualization; P. P. conceptualization, formal analysis, investigation, writing – original draft, writing – review & editing, visualization; J.-R. M. formal analysis (thermal methods), investigation (thermal methods); M. R. J. E. formal analysis (X-ray data sets), investigation (X-ray crystallography), funding acquisition (ALS beam time), validation (of the structures), visualisation (X-ray structure figures), writing – review & editing; S. J. T. formal analysis (X-ray data sets at the ALS); A. D. B. formal analysis (X-ray data sets), investigation (X-ray Crystallography), validation (of the structures), visualisation (X-ray structure figures). I. A. W. conceptualization, funding acquisition, project administration, supervision, writing – review & editing; D. G. C. conceptualization, project administration, software, visualization, writing – original draft, writing – review & editing; M. K. E. project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. thanks Loughborough University for a PhD Studentship. P. P. acknowledges the EPSRC (EP/S012788/1) for support. J.-R. M. thanks the EPSRC Sustainable Hydrogen CDT (EP/S023909) and Loughborough University for a PhD Studentship. I. A. W. thanks the EPSRC (EP/T028688/1) and RSC (RF19-2751) for support. D. G. C. acknowledges the Herchel Smith fund for an early career fellowship, and H. Bronstein (University of Cambridge) in a mentoring capacity and for providing laboratory space. M. K. E. thanks the Royal Society of Chemistry (R20-1668) for support. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. We would like to thank A. P. Monkman and F. B. Dias for access to their experimental setups. We thank Sobereva (USTB Beijing) for providing the Multiwfn program and providing instruction on how to run the software. We thank C. Zhong (Wuhan University) and W. Zheng (University of Cambridge) for help and advice with the PySOC script and running SOCME calculations.Notes and references
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- L. S. Cui, A. J. Gillett, S. F. Zhang, H. Ye, Y. Liu, X. K. Chen, Z. Sen Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J. L. Bredas, C. Adachi and R. H. Friend, Nat. Photonics, 2020, 14, 636–642 CrossRef CAS.
- H. Noda, X. K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J. L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- P. L. dos Santos, J. S. Ward, D. G. Congrave, A. S. Batsanov, J. Eng, J. E. Stacey, T. J. Penfold, A. P. Monkman and M. R. Bryce, Adv. Sci., 2018, 1700989 CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- X.-K. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J.-L. Brédas, Adv. Mater., 2017, 29, 1702767 CrossRef PubMed.
- X. K. Chen, S. F. Zhang, J. X. Fan and A. M. Ren, J. Phys. Chem. C, 2015, 119, 9728–9733 CrossRef CAS.
- K. L. Woon, C. L. Yi, K. C. Pan, M. K. Etherington, C. C. Wu, K. T. Wong and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 12400–12410 CrossRef CAS PubMed.
- X. Tang, L. S. Cui, H. C. Li, A. J. Gillett, F. Auras, Y. K. Qu, C. Zhong, S. T. E. Jones, Z. Q. Jiang, R. H. Friend and L. S. Liao, Nat. Mater., 2020, 19, 1332–1338 CrossRef CAS PubMed.
- H. Tsujimoto, D.-G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS PubMed.
- S. Y. Yang, Y. K. Wang, C. C. Peng, Z. G. Wu, S. Yuan, Y. J. Yu, H. Li, T. T. Wang, H. C. Li, Y. X. Zheng, Z. Q. Jiang and L. S. Liao, J. Am. Chem. Soc., 2020, 142, 17756–17765 CrossRef CAS PubMed.
- X. Wang, S. Wang, J. Lv, S. Shao, L. Wang, X. Jing and F. Wang, Chem. Sci., 2019, 10, 2915–2923 RSC.
- Q. Li, J. Hu, J. Lv, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2020, 59, 20174–20182 CrossRef CAS PubMed.
- X. L. Chen, J. H. Jia, R. Yu, J. Z. Liao, M. X. Yang and C. Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 15006–15009 CrossRef CAS PubMed.
- X. Q. Wang, S. Y. Yang, Q. S. Tian, C. Zhong, Y. K. Qu, Y. J. Yu, Z. Q. Jiang and L. S. Liao, Angew. Chem., Int. Ed., 2020, 2, 5213–5219 Search PubMed.
- H. Miranda-Salinas, Y.-T. Hung, Y.-S. Chen, D. Luo, H.-C. Kao, C.-H. Chang, K.-T. Wong and A. Monkman, J. Mater. Chem. C, 2021, 9, 8819–8833 RSC.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643–649 CrossRef CAS.
- T. Huang, Q. Wang, S. Xiao, D. Zhang, Y. Zhang, C. Yin, D. Yang, D. Ma, Z. Wang and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 23771–23776 CrossRef CAS PubMed.
- X. Zheng, R. Huang, C. Zhong, G. Xie, W. Ning, M. Huang, F. Ni, F. B. Dias and C. Yang, Adv. Sci., 2020, 7, 1–7 Search PubMed.
- IUPAC. Compendium of Chemical Terminology, Gold Book. Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997, ISBN 0-9678550-9-8, DOI: 10.1351/goldbook.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef CAS.
- K. Baumgärtner, M. Hoffmann, F. Rominger, S. M. Elbert, A. Dreuw and M. Mastalerz, J. Org. Chem., 2020, 85, 15256–15272 CrossRef PubMed.
- N. Harada, H. Uda, K. Nakasuji and I. Murata, J. Chem. Soc., Perkin Trans. 2, 1989, 1449–1453 RSC.
- T. Nakazawa and I. Murata, J. Am. Chem. Soc., 1977, 99, 1996–1997 CrossRef CAS.
- N. Harada, Y. Tamai, Y. Takuma and H. Uda, J. Am. Chem. Soc., 1980, 102, 501–506 CrossRef CAS.
- H.-D. Martin and B. Mayer, Angew. Chem., Int. Ed. Engl., 1983, 22, 283–314 CrossRef.
- J. Li, P. Shen, Z. Zhao and B. Z. Tang, CCS Chem., 2019, 1, 181–196 CAS.
- L. T. Scott, Pure Appl. Chem., 1986, 58, 105–110 CrossRef CAS.
- A. Braendle, A. Perevedentsev, N. J. Cheetham, P. N. Stavrinou, J. A. Schachner, N. C. Mösch-Zanetti, M. Niederberger and W. R. Caseri, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 707–720 CrossRef CAS.
- L. T. Scott and M. M. Hashemi, Tetrahedron, 1986, 42, 1823–1830 CrossRef CAS.
- Y. K. Wang, C. C. Huang, H. Ye, C. Zhong, A. Khan, S. Y. Yang, M. K. Fung, Z. Q. Jiang, C. Adachi and L. S. Liao, Adv. Opt. Mater., 2020, 8, 1–7 CAS.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- Y. Gao, T. Su, Y. Wu, Y. Geng, M. Zhang and Z. M. Su, Chem. Phys. Lett., 2016, 666, 7–12 CrossRef CAS.
- C. C. A. Voll, J. U. Engelhart, M. Einzinger, M. A. Baldo and T. M. Swager, Eur. J. Org. Chem., 2017, 4846–4851 CrossRef CAS.
- P. Lei, S. Zhang, N. Zhang, X. Yin, N. Wang and P. Chen, ACS Omega, 2020, 5, 28606–28614 CrossRef CAS PubMed.
- L. Lv, W. Sun, Z. Jia, G. Zhang, F. Wang, Z. Tan and L. Zhang, Mater. Chem. Front., 2020, 4, 3539–3545 RSC.
- T. Kodama, Y. Hirao, T. Nishiuchi and T. Kubo, ChemPlusChem, 2017, 82, 1006–1009 CrossRef CAS PubMed.
- T. Kodama, S. Miyazaki and T. Kubo, ChemPlusChem, 2019, 84, 599–602 CrossRef CAS PubMed.
- S. Montanaro, D. G. Congrave, M. K. Etherington and I. A. Wright, J. Mater. Chem. C, 2019, 7, 12886–12890 RSC.
- L. Yu, Z. Wu, C. Zhong, G. Xie, Z. Zhu, D. Ma and C. Yang, Adv. Opt. Mater., 2017, 5, 1700588 CrossRef.
- J. Liu, K. Zhou, D. Wang, C. Deng, K. Duan, Q. Ai and Q. Zhang, Front. Chem., 2019, 7, 312 CrossRef CAS PubMed.
- T. Huang, D. Liu, J. Jiang and W. Jiang, Chem. – Eur. J., 2019, 25, 10926–10937 CrossRef CAS PubMed.
- P. Rajamalli, D. Chen, S. M. Suresh, Y. Tsuchiya, C. Adachi and E. Zysman-colman, J. Subst. Abuse Treat., 1996, 13, 287–288 CrossRef.
- K. Zhang, F. Yang, Y. Zhang, Y. Ma, J. Fan, J. Fan, C.-K. Wang and L. Lin, J. Phys. Chem. Lett., 2021, 12, 1893–1903 CrossRef CAS PubMed.
- U. Balijapalli, R. Nagata, N. Yamada, H. Nakanotani, M. Tanaka, A. D’Aléo, V. Placide, M. Mamada, Y. Tsuchiya and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 8477–8482 CrossRef CAS PubMed.
- P. Meti, H. H. Park and Y. D. Gong, J. Mater. Chem. C, 2020, 8, 352–379 RSC.
- L. Salah, M. K. Etherington, A. Shuaib, A. Danos, A. A. Nazeer, B. Ghazal, A. Prlj, A. T. Turley, A. Mallick, P. R. McGonigal, B. F. E. Curchod, A. P. Monkman and S. Makhseed, J. Mater. Chem. C, 2021, 9, 189–198 RSC.
- T. M. Long and T. M. Swager, Adv. Mater., 2001, 601–604 CrossRef CAS.
- R. G. Hazell, G. S. Pawley and C. E. Lund Petersen, J. Cryst. Mol. Struct., 1971, 1, 319–324 CrossRef CAS.
- W. F. Sanjuan-Szklarz, A. A. Hoser, M. Gutmann, A. Ø. Madsen and K. Woźniak, IUCrJ, 2016, 3, 61–70 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. V. Ivanov, S. A. Reid and R. Rathore, J. Phys. Chem. Lett., 2018, 9, 3978–3986 CrossRef CAS PubMed.
- M. R. Talipov, T. S. Navale and R. Rathore, Angew. Chem., Int. Ed., 2015, 54, 14468–14472 CrossRef CAS PubMed.
- T. Lu and Q. Chen, Chem. Methods, 2021, 1, 231–239 CrossRef.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2011, 33, 580–592 CrossRef PubMed.
- W. Zeng, T. Zhou, W. Ning, C. Zhong, J. He, S. Gong, G. Xie and C. Yang, Adv. Mater., 2019, 1901404 CrossRef PubMed.
- W. Zhuang, S. Wang, Q. Tao, W. Ma, M. Berggren, S. Fabiano, W. Zhu and E. Wang, Macromolecules, 2021, 54, 970–980 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., Int. Ed., 2016, 55, 5739–5744 CrossRef CAS PubMed.
- R. Pashazadeh, P. Pander, A. Lazauskas, F. B. Dias and J. V. Grazulevicius, J. Phys. Chem. Lett., 2018, 9, 1172–1177 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2092859–2092861. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2tc00460g |
| ‡ These authors have contributed equally to the experimental work. |
| This journal is © The Royal Society of Chemistry 2022 |