
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating electronic and optical properties of monolayered MoS2 by covalent mono- and bisfunctionalization†
Kangli
Wang
 *,
Marco
Kapitzke
,
Lauren
Green
and
Beate
Paulus
*,
Marco
Kapitzke
,
Lauren
Green
and
Beate
Paulus
Institut für Chemie und Biochemie, Freie Universität Berlin, 14195 Berlin, Germany. E-mail: klwang0329@zedat.fu-berlin.de
First published on 18th March 2022
Abstract
By employing first-principles simulations, we present theoretical predictions regarding the modification of structural, electronic and optical properties of 2H- and 1T′-MoS2 monolayers by covalent mono- and bisfunctionalization. Specifically, non-aromatic groups (–F, –NH2, –CH3, –CH2CH2 CN and –CH2CH2 OH) and aromatic (–Ph, –PhNO2 and PhOH) groups are utilized for monofunctionalization, and –F/–NH2, –NH2/–CH3 and –CH3/–Ph for bisfunctionalization. The stability of functionalized 2H- and 1T′-MoS2 monolayers mainly depends on the bonded groups and their surface coverage. In particular, the mixed bisfunctionalization with –F/–CH3 and –NH2/–CH3 groups enhances the stability of 2H-MoS2 through the formation of intermolecular hydrogen bonds. Both 2H- and 1T′-MoS2 can serve not only as electron donors, but also as electron acceptors, subject to the charge transfer behavior of the attached groups. Furthermore, mono- and bisfunctionalization are predicted to be efficient approaches to control the electronic band gaps in 2H- and 1T′-MoS2, where the corresponding values can be tuned by varying the coverage of the absorbed groups. At the same time, the choice of the chemical groups and their coverage also effectively determines the optical adsorption range and intensity. Therefore, our work shows that chemical functionalization of 2D materials with varying coverage can be an important approach to extend the scope of 2D materials in specific electronic and optoelectronic applications.
1 Introduction
Monolayered MoS2 is one of the most common transition metal dichalcogenides (TMDCs) and finds a range of promising applications such as photodetectors,1–3 advanced catalysts,4,5 gas sensors,6,7 batteries8,9 and solar cells10,11 due to its unique features and excellent tunability of the electronic properties. Depending on the arrangement of S atoms on both sides of Mo atoms, the MoS2 monolayer has one of the three polytypic structures: the 2H, 1T and 1T′ (distorted 1T) phases.12–15 The Mo atom in the 2H and 1T phases is coordinated by six S atoms in an octahedral and trigonal prismatic arrangement, respectively. Generally, 1T phase is unstable under free-standing conditions and can be easily converted into the meta stable 1T′ phase that consists of zigzag Mo chains. Owing to the different localization behaviors of the d-band in Mo,16 2H-MoS2 is a semiconductor with a sizeable band gap and a photoluminophore, whereas 1T-MoS2 exhibits metallic character without photoluminescence.1,2,14–16 Interestingly, the 1T and 2H phases can also coexist in the same MoS2 monolayer,17 thereby allowing low contact resistance and holding high catalytic activity.18,19To further improve the applicability of TMDCs in diverse fields, many methods have been developed to tune their properties, such as adsorption of gas molecules20–22 and strain engineering.23,24 Among them, covalent functionalization offers the most compelling route. Since the basal plane of 1T-MoS2 is sensitive to functionalization, chemically exfoliated MoS2 can react with phenyl diazonium salts25,26 or organohalides (–I or –Br)27,28 to form new S–Mo bonds. Although 2H-MoS2 has a relatively inert basal plane, it is reported to be covalently functionalized by bonding through sulfur defects,29–31 coordinating to metal complexes,32 reacting with aryl diazonium33 and thiolate salts34 as well as maleimide derivatives.35 More impressively, a bisfunctionalized MoS2 hybrid structure bearing both alkyl and aryl groups has been recently demonstrated.36 On the other hand, the coverage of functional groups achieved after functionalization varies from 10% to 70%, depending on different methods and conditions.25,27,33,36,37 Given these experimental results, a theoretical perspective on how covalent functionalization alters the electronic and optical properties of 2H- and 1T-MoS2 (or 1T′-MoS2) is desirable. However, previous computational studies mainly focus on the monofunctionalization with small groups (–H, –O, –SH, –NH2, –CH3),38–41 while monofunctionalization with large groups as well as bisfunctionalization is far less explored.42,43
In this work, we aim to give a comprehensive understanding towards the two-sided covalent functionalization of monolayered 2H- and 1T′-MoS2. We do not consider the functionalized T phase as this structure evolves to the T′ phase upon structural optimization. Non-aromatic groups (–F, –NH2, –CH3, –CH2CH2 CN and –CH2CH2 OH) and aromatic groups (–Ph, –PhNO2 and PhOH) are applied to monofunctionalize 2H- and 1T′-MoS2. Furthermore, bisfunctionalization of MoS2 is also considered in this study, including mixed bisfunctionalization (m-F/–NH2, m-NH2/–CH3 and m-CH3/–Ph) where each group is bonded to both sides of the MoS2 monolayer and separate bisfunctionalization (s-F/–NH2, s-NH2/–CH3 and s-CH3/–Ph) where each side of the monolayer is uniquely functionalized by a specie as illustrated in Fig. 1. It is well known that density functional theory (DFT) alone is ill-equipped to describe the electronic band structure, especially when predicting the size of the band gap for semiconductors or insulators. To accurately model the electronic band structure and optical absorption, we employ the GW (Green's function (G) and screened Coulomb (W) potential) approximation within the framework of many-body perturbation theory and Bethe–Salpeter equation based on the DFT method (DFT-GW-BSE).44–47 This method includes self-energy and excitonic effects, and thus has been successfully applied to a wide variety of materials.48–51
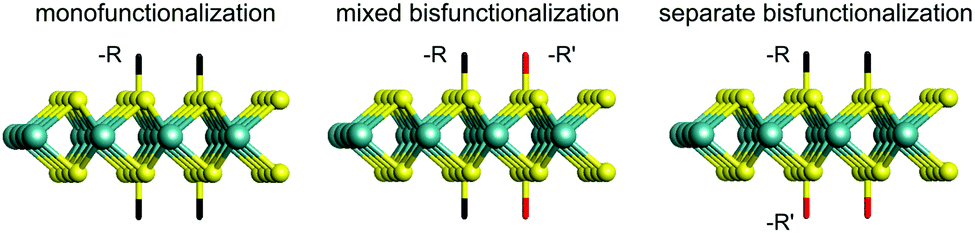 | ||
| Fig. 1 Schematic representation of mono- and bisfunctionalization considered in this work. | ||
2 Computational details
All of the calculations are performed with the GPAW code.52 The structure is relaxed until all forces are below 0.01 eV Å−1 using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional53 with 450 eV plane wave cutoff, and more than 15 Å between periodically repeated layers. A 4 × 4 supercell (contains 32 adsorption sites) is considered to bond with 2, 4,…, 14 groups corresponding to the functionalized coverages varying from 6.25% to 43.75%; a 2 × 2 supercell with 2 groups corresponds to a 50% coverage. A Γ-centered Monkhorst–Pack k-grid scheme of 6 × 6 × 1 and 12 × 12 × 1 are used for 4 × 4 and 2 × 2 supercells, respectively. The charge transfer between the group and the substrate is discussed by means of Bader analysis.54The DFT-GW-BSE calculations are performed according to following protocol. Firstly, standard Kohn–Sham DFT calculations are performed to obtain the ground state Kohn–Sham orbitals. The same parameters as for the structural relaxation calculations are adopted. Then, non-self-consistent G0W0 is employed to calculate the quasiparticle (QP) band structures, which involves the calculation of QP energy using the input DFT orbitals. To avoid spurious interactions between neighboring supercells, a 2D Coulomb truncation is applied with 8 Å vacuum ensuring the convergence of the QP band gap. The dielectric matrix and correlation self-energy is evaluated using a cutoff of 50 eV on a nonlinear grid with Δω0 = 0.25 eV frequency spacing. At ω2 = 10 eV, this spacing increases to 2Δω0. Finally, the BSE spectrum is obtained by applying the Tamm–Dancoff approximation.55,56 The 24 highest valence and 48 lowest conduction bands are utilized as a basis for excitonic eigenstates, which is enough to attain converged optical absorption spectrum. A Lorentzian broadening of 0.05 eV is employed for all the optical calculations. The convergence tests for the DFT-GW-BSE treatment are in ESI† (see Fig. S1, ESI†).
3 Results and discussion
Average binding energy
We first employ DFT to determine the most favorable adsorption sites and calculate the thermodynamic stability of the functionalized MoS2 monolayer by computing the average binding energy per group as a function of the adsorbate coverages:| ΔE = (Etot − EMoS2 − nEgroup)/n, | (1) |
 | ||
| Fig. 2 Ball-and-stick model of the optimized structures of the monofunctionalized (6.25% coverage) and bisfunctionalized (12.5% coverage) 2H- and 1T′-MoS2 by different groups (side and perspective views). The hydrogen, carbon, nitrogen, oxygen, fluorine, sulfur and molybdenum are in color white, gray, blue, red, green, yellow and turquoise, respectively. | ||
 | ||
| Fig. 3 Average binding energy as a function of the group coverage with respect to the 2H-MoS2 and 1T′-MoS2 monolayers. | ||
Fig. 3(b) and (d) illustrate the average binding energy of the functionalized 1T′-MoS2 at varying coverage. For the 1T′ phase, the average binding energy of each system reaches a minimum at 12.5% coverage, which is smaller than that of 2H-MoS2. Furthermore, a considerable stability range of the functionalized 1T′ phase can be noticed, exhibiting a negative average binding energy even at 50% coverage. This suggests the possibility of high functional group coverage of functionalized 1T′-MoS2, which is consistent with previous experimental observations.37 In addition, the average binding energy of 1T′-MoS2 is much smaller than that of 2H-MoS2 for any given coverage, indicating that the reaction of chemical functionalization of the 1T′ phase is thermodynamically more favorable. This is attributed to the well-paired electrons in 2H-MoS2, whereas the electronic states around the Fermi level are only partially filled in 1T′-MoS2.39
By comparing the total energy between 2H- and 1T′-MoS2, we investigate the phase stability (see Fig. S3, ESI†). At low coverage, the functionalized 2H phase is more energetically stable regardless of mono- or bisfunctionalization. As the coverage increases, the energy difference becomes smaller. In particular, at high coverage, the functionalized 1T′ phase is more stable than 2H case. This indicates that the selective tuning of the coverage of covalent functionalization enables the phase transformation of MoS2.
To gain an insight into the bonding nature of the considered systems, we further execute the Bader charge analysis to investigate the charge transfer between the MoS2 monolayer and the attached groups. The corresponding results are displayed in Fig. S4 (ESI†). When –F, –NH2, m-F/–CH3 and s-F/–CH3 are bonded to the surface of both 2H- and 1T′-MoS2, the electron is transferred to the groups from the substrate which acts as an electron-donor. In contrast, an inverted electron flow can be recognized for other functional groups, for instance –CH3, –CH2CH2 CN, –CH2CH2 OH, –Ph, –PhNO2 and –PhOH. Here the substrate gains electrons despite bare MoS2 is an n-type semiconductor. The direction of charge transfer exhibits a clear dependence on the electron donating or withdrawing ability of the adsorbed species. Correlation of the binding energy with the electron donating or withdrawing properties of the functional group shows that when the 2H- or 1T′-MoS2 serves as an electron-donor, the average binding energy becomes more negative for strongly electron-withdrawing groups (–F vs. –NH2). It implies that the stronger electron-withdrawing effect could enhance the stability of the functionalized MoS2 monolayer in this case. On the other hand, the attached groups with a stronger electron donating-ability show more negative binding energies, advocating that MoS2 monolayers are more stable under this condition. Examples include the monofunctionalization with –CH2CH2 OH vs. –CH2CH2 CN or –PhOH vs. –PhNO2.
For bisfunctionalized 2H- and 1T′-MoS2, the average binding energy can be compared to that of the corresponding monofunctionalized case. In particular, when –F and –CH3 are functionalized separately on both sides of 2H-MoS2, the average binding energy is more negative than the average between the two corresponding monofunctionalized 2H-MoS2. This is because the cooperative effect of electron withdrawing and donating groups could promote the charge transfer and consequently enhances its stability. Intriguingly, the average binding energy for the 1T′-MoS2 in the case is almost equal to the monofunctionalized ones. This is attributed to the distorted octahedral structure and non-equivalent S atoms of 1T′-MoS2, which attenuates this cooperative effect. Concerning the mixed bisfunctionalization with –F and –CH3, this combined effect is offset for both 2H- and 1T′-MoS2. As for the bisfunctionaliation of –CH3 and –Ph including the separate and mixed functionalization, the average binding energy is also close to average between the two corresponding monofunctionalized MoS2 because both of these two groups tend to donate electrons to the substrate. In summary, the investigation of the average binding energy reveals that the coverage, stereostructure and electron doping of the groups drive the stability of both functionalized 2H- and 1T′-MoS2 monolayers.
Electronic properties
To provide a benchmark for our computational scheme, we initially apply the DFT-GW-BSE method to the bare MoS2 monolayer. The calculated electronic band gaps with the DFT method at PBE level are 1.65 eV and 0.04 eV for 2H- and 1T′-MoS2, respectively, whereas the self-energy correction considered in G0W0 method opens the band gaps to 2.51 eV and 0.08 eV, respectively. The G0W0 results are consistent with reported experimental values of 2.50 eV and 0.08 eV for 2H-MoS2 and 1T′-MoS2, respectively.57,58 Next, we turn our attention to the functionalized 2H- and 1T′-MoS2 monolayers. Fig. 4 depicts the variation of the electronic band gaps calculated by the G0W0 method with different adsorbates and coverages. The corresponding DFT results are presented in Fig. S5 (ESI†). At 6.25% coverage, the monofunctionalized and bisfunctionalized 2H-MoS2 display semiconducting characteristics with varying QP band gaps from 0.99 eV to 2.22 eV for the considered systems. The reduced band gap is associated with the donor or acceptor state inside the gap of bare 2H-MoS2 (see Fig. S6, ESI†). The introduced state primarily originates from the Mo 4d band with additional contributions from the 3p orbitals of S and the sp orbitals of the adsorbate. This suggests that the 4d of Mo atoms will make a main contribution to the electronic transport in all cases although groups bind directly to the S atoms of MoS2. As adsorbate coverage increases from 12.5% to 25%, the band gap tends to decrease. At even higher coverage (31.25–50%), the band gap fluctuates, except in the case of –NH2/CH3 and –F/CH3. We note that the band gaps of both mono- and bisfunctionaliation are highly sensitive to the adsorption site, which is also found in other materials.59–61 Therefore, the modification of the band gap for mixed bisfunctionalization of –NH2/–CH3 and –F/–CH3 differs due to the difference in adsorption sites which arises from the intermolecular hydrogen bonds. With respect to the introduced donor or acceptor state around the Fermi level, the contribution of both S 3 p and adsorbate sp states increases with the increase of coverage for both mono- and bisfunctionalization, implying the influence of group coverage on electronic transport (Fig. S6, ESI†). | ||
| Fig. 4 QP band gaps as a function of the group coverages with respect to the functionalized 2H-MoS2 and 1T′-MoS2 monolayers. | ||
Compared with the 2H phase, the 1T′-MoS2 exhibits different behaviors after covalent functionalization. It is obvious from Fig. 4 that at 6.25% coverage, both mono- and bisfunctionalized 1T′-MoS2 exhibit metallic character, in contrast to 2H-MoS2. As the coverage increases, the QP band gaps first increase and then oscillate at high coverage. Furthermore, the variation of QP band gaps of m-NH2/–CH3 and m-F/–CH3 displays a similar trend as that of monofunctionalized 1T′-MoS2. This is caused by the distorted structure of the 1T′ phase, which inhibits the formation of intermolecular hydrogen bonds and thereby enables the mixed groups to adsorb at the same or similar site with that of monofunctionalization. This also means the interaction between the adsorbate and the substrate in case of bisfunctionalized 1T′-MoS2 is strong. Overall, our calculations reveal that the variation in the band gaps of both 2H- and 1T′-MoS2 provides promising results of the utility of the chemical mono-/bisfunctionalization and coverages to tune the electronic properties of 2D materials.
Optical properties
Next, we examine the optical properties of the functionalized 2H- and 1T′-MoS2, which are crucial for MoS2-based optoelectronic devices. As a result of the low stability at high coverage, only low group coverages are considered. The corresponding optical absorption spectra of 2H phase are presented in Fig. 5. Compared to bare 2H-MoS2, the optical absorption upon mono- and bisfunctionalization is red-shifted at 6.25% coverage. The red-shift is attributed to the interplay of two joint effects: (1) the reduced electronic band gap upon functionaliation as we discussed above and (2) the variation of the exciton binding energy (see Table S1, ESI†). Our calculation shows that at 6.25% coverage, monofunctionalition with each group and bisfunctionalization with –NH2/–CH3 lowers the excitonic binding energy slightly; whereas the bisfunctionaliation with –F/–CH3 or –CH3/–Ph tends to strengthen the Coulomb interaction between the electron and hole of an exciton, promoting the formation of tight excitonic states. As the reduction of the electronic band gap is dominant upon functionalization, the optical spectrum is strongly red-shifted and the absorbance is enhanced in the 0–2 eV regime. It is worth mentioning that the optical band gap varies significantly from 0.18 eV to 1.49 eV at 6.25% coverage, indicating the high sensitivity of optical properties of 2H phase towards the different groups. As coverage increases to 18.75%, the overall optical absorbance for each functionalization remains red-shifted with a reduced optical band gap. At the same time, the intensity of the optical absorption is remarkably enhanced, especially in the spectral range of 0–1 eV. | ||
| Fig. 5 Optical absorption spectra of the functionalized 2H-MoS2 monolayer. | ||
In the case of 1T′-MoS2, we observe an obvious blue-shift of the optical spectrum and an increase in optical absorption as the coverage increases from 6.25% to 18.75% (see Fig. S7, ESI†), which can be explained by the opening of the electronic band gap of 1T′-MoS2 upon functionalization. Our study regarding the optical properties of the functionalized MoS2 offers the prospect of artificially tuning the optical properties of MoS2 by controlling the type and coverage of the functional groups.
4 Conclusion
In this work, we systemically study the stability, electronic band gap and optical absorption spectrum of both chemically functionalized 2H- and 1T′-MoS2 for various adsorbates by means of the DFT-GW-BSE method. The chemical groups studied include –F, –NH2, –CH3, –CH2CH2 CN, –CH2CH2 OH, –Ph, –PhNO2 and PhOH for monofunctionalization and m-F–CH3, m-NH2 –CH3, m-CH3 -Ph, s-F–CH3, s-NH2 –CH3 and s-CH3 –Ph for bisfunctionalization. We find when no intermolecular interaction is observed, the average binding energy falls at first and rises again as the coverage is elevated, reaching a minimum at 25% and 12.5% for 2H and 1T′-MoS2, respectively. The bisfunctionalization of m-F–CH3 and m-NH3–CH3 on 2H-MoS2 can promote the formation of intermolecular hydrogen bonds and thus improve the stability. When –F, –NH2, m-F/–CH3 and s-F/–CH3 are attached on the surface, both of 2H- and 1T′-MoS2 serve as electron-donors; in other cases, both of 2H- and 1T′-MoS2 act as electron acceptors. At low coverage, the functionalized 2H-MoS2 generally exhibits semiconducting character, whereas the functionalized 1T′-MoS2 is predicted to be metallic; as the coverage increases, the variation of band gaps of both 2H-and 1T′-MoS2 shows an oscillatory behavior. The changes in the electronic structure as well as the excitonic binding energy of the functionalized 2H- and 1T′-MoS2 lead to strong modulations of their optical response with respect to the bare monolayers. In conclusion, our work demonstrates that mono- and bisfunctionalization of 2H- and 1T′-MoS2 are vigorous tools for tuning the electronic and optical properties.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
K. Wang acknowledges the China Scholarship Council for the financial support. The computations were performed with resources provided by the North-German Supercomputing Alliance (HLRN) and computer facilities of the Freie Universität Berlin (ZEDAT). Authors are also thankful to Liangliang Zhang and Jianliang Low for their assistance in the proofreading of the manuscript.References
- Z. Yin, H. Li, H. Li, L. Jiang, Y. Shi, Y. Sun, G. Lu, Q. Zhang, X. Chen and H. Zhang, ACS Nano, 2012, 6, 74–80 Search PubMed.
- W. Zhang, C.-P. Chuu, J.-K. Huang, C.-H. Chen, M.-L. Tsai, Y.-H. Chang, C.-T. Liang, Y.-Z. Chen, Y.-L. Chueh, J. He-Hau, M.-Y. Chou and L.-J. Li, Sci. Rep., 2014, 4, 3826 Search PubMed.
- Y. Xin, X. Wang, Z. Chen, D. Weller, Y. Wang, L. Shi, X. Ma, C. Ding, W. Li, S. Guo and R. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 15406–15413 Search PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 Search PubMed.
- G. Li, Z. Chen, Y. Li, D. Zhang, W. Yang, Y. Liu and L. Cao, ACS Nano, 2020, 14, 1707–1714 Search PubMed.
- H. Tabata, H. Matsuyama, T. Goto, O. Kubo and M. Katayama, ACS Nano, 2021, 15, 2542–2553 Search PubMed.
- B. Zong, Q. Li, X. Chen, C. Liu, L. Li, J. Ruan and S. Mao, ACS Appl. Mater. Interfaces, 2020, 12, 50610–50618 Search PubMed.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 Search PubMed.
- J. Xiao, D. Choi, L. Cosimbescu, P. Koech, J. Liu and J. P. Lemmon, Chem. Mater., 2010, 22, 4522–4524 Search PubMed.
- G. Eda and S. A. Maier, ACS Nano, 2013, 7, 5660–5665 Search PubMed.
- M.-L. Tsai, S.-H. Su, J.-K. Chang, D.-S. Tsai, C.-H. Chen, C.-I. Wu, L.-J. Li, L.-J. Chen and J.-H. He, ACS Nano, 2014, 8, 8317–8322 Search PubMed.
- F. Wypych and R. Schöllhorn, J. Chem. Soc., Chem. Commun., 1992, 1386–1388 Search PubMed.
- J. A. Wilson and A. D. Yoffe, Adv. Phys., 1969, 18, 193–335 Search PubMed.
- I. Song, C. Park and H. C. Choi, RSC Adv., 2015, 5, 7495–7514 Search PubMed.
- X. Qian, J. Liu, L. Fu and J. Li, Science, 2014, 346, 1344–1347 Search PubMed.
- R. Bissessur, M. G. Kanatzidis, J. L. Schindler and C. R. Kannewurf, J. Chem. Soc., Chem. Commun., 1993, 1582–1585 Search PubMed.
- G. Eda, T. Fujita, H. Yamaguchi, D. Voiry, M. Chen and M. Chhowalla, ACS Nano, 2012, 6, 7311–7317 Search PubMed.
- R. Kappera, D. Voiry, S. E. Yalcin, B. Branch, G. Gupta, A. Mohite and M. Chhowalla, Nat. Mater., 2014, 13, 1128–1134 Search PubMed.
- Y. Yao, K. Ao, P. Lv and Q. Wei, Nanomaterials, 2019, 9, 844 Search PubMed.
- T. Venanzi, H. Arora, A. Erbe, A. Pashkin, S. Winnerl, M. Helm and H. Schneider, Appl. Phys. Lett., 2019, 114, 172106 Search PubMed.
- K. Wang and B. Paulus, Phys. Chem. Chem. Phys., 2020, 22, 11936–11942 Search PubMed.
- S. Mouri, Y. Miyauchi and K. Matsuda, Nano Lett., 2013, 13, 5944–5948 Search PubMed.
- P. Gant, P. Huang, D. Pérez de Lara, D. Guo, R. Frisenda and A. Castellanos-Gomez, Mater. Today, 2019, 27, 8–13 Search PubMed.
- S. Wang, M. S. Ukhtary and R. Saito, Phys. Rev. Res., 2020, 2, 033340 Search PubMed.
- K. C. Knirsch, N. C. Berner, H. C. Nerl, C. S. Cucinotta, Z. Gholamvand, N. McEvoy, Z. Wang, I. Abramovic, P. Vecera, M. Halik, S. Sanvito, G. S. Duesberg, V. Nicolosi, F. Hauke, A. Hirsch, J. N. Coleman and C. Backes, ACS Nano, 2015, 9, 6018–6030 Search PubMed.
- E. E. Benson, H. Zhang, S. A. Schuman, S. U. Nanayakkara, N. D. Bronstein, S. Ferrere, J. L. Blackburn and E. M. Miller, J. Am. Chem. Soc., 2018, 140, 441–450 Search PubMed.
- D. Voiry, A. Goswami, R. Kappera, C. d. C. C. e Silva, D. Kaplan, T. Fujita, M. Chen, T. Asefa and M. Chhowalla, Nat. Chem., 2015, 7, 45–49 Search PubMed.
- X. Chen, D. McAteer, C. McGuinness, I. Godwin, J. N. Coleman and A. R. McDonald, Chem. – Eur. J., 2018, 24, 351–355 Search PubMed.
- S. S. Chou, M. De, J. Kim, S. Byun, C. Dykstra, J. Yu, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2013, 135, 4584–4587 Search PubMed.
- Q. Ding, K. J. Czech, Y. Zhao, J. Zhai, R. J. Hamers, J. C. Wright and S. Jin, ACS Appl. Mater. Interfaces, 2017, 9, 12734–12742 Search PubMed.
- S. Bertolazzi, S. Bonacchi, G. Nan, A. Pershin, D. Beljonne and P. Samorì, Adv. Mater., 2017, 29, 1606760 Search PubMed.
- C. Backes, N. C. Berner, X. Chen, P. Lafargue, P. LaPlace, M. Freeley, G. S. Duesberg, J. N. Coleman and A. R. McDonald, Angew. Chem., Int. Ed., 2015, 54, 2638–2642 Search PubMed.
- X. S. Chu, A. Yousaf, D. O. Li, A. A. Tang, A. Debnath, D. Ma, A. A. Green, E. J. G. Santos and Q. H. Wang, Chem. Mater., 2018, 30, 2112–2128 Search PubMed.
- I. K. Sideri, R. Arenal and N. Tagmatarchis, ACS Mater. Lett., 2020, 2, 832–837 Search PubMed.
- M. Vera-Hidalgo, E. Giovanelli, C. Navío and E. M. Pérez, J. Am. Chem. Soc., 2019, 141, 3767–3771 Search PubMed.
- X. Chen, C. Bartlam, V. Lloret, N. Moses Badlyan, S. Wolff, R. Gillen, T. Stimpel-Lindner, J. Maultzsch, G. S. Duesberg, K. C. Knirsch and A. Hirsch, Angew. Chem., Int. Ed., 2021, 60, 13484–13492 Search PubMed.
- E. X. Yan, M. Cabán-Acevedo, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, ACS Mater. Lett., 2020, 2, 133–139 Search PubMed.
- Q. Tang and D.-e. Jiang, ACS Catal., 2016, 6, 4953–4961 Search PubMed.
- Q. Tang and D.-e. Jiang, Chem. Mater., 2015, 27, 3743–3748 Search PubMed.
- M. Palummo, A. N. D’Auria, J. C. Grossman and G. Cicero, J. Phys.: Condens. Matter, 2019, 31, 235701 Search PubMed.
- Y. Linghu, N. Li, Y. Du and C. Wu, Phys. Chem. Chem. Phys., 2019, 21, 9391–9398 Search PubMed.
- A. Gul, C. Bacaksiz, E. Unsal, B. Akbali, A. Tomak, H. M. Zareie and H. Sahin, Mater. Res. Express, 2018, 5, 036415 Search PubMed.
- L. O. Jones, M. A. Mosquera, M. A. Ratner and G. C. Schatz, ACS Appl. Mater. Interfaces, 2020, 12, 4607–4615 Search PubMed.
- M. S. Hybertsen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 5390–5413 Search PubMed.
- M. Rohlfing and S. G. Louie, Phys. Rev. Lett., 1998, 81, 2312–2315 Search PubMed.
- L. X. Benedict, E. L. Shirley and R. B. Bohn, Phys. Rev. Lett., 1998, 80, 4514–4517 Search PubMed.
- S. Albrecht, L. Reining, R. Del Sole and G. Onida, Phys. Rev. Lett., 1998, 80, 4510–4513 Search PubMed.
- D. Y. Qiu, F. H. da Jornada and S. G. Louie, Phys. Rev. Lett., 2013, 111, 216805 Search PubMed.
- L. Yang, J. Deslippe, C.-H. Park, M. L. Cohen and S. G. Louie, Phys. Rev. Lett., 2009, 103, 186802 Search PubMed.
- C. D. Spataru, S. Ismail-Beigi, L. X. Benedict and S. G. Louie, Phys. Rev. Lett., 2004, 92, 077402 Search PubMed.
- M. L. Tiago and J. R. Chelikowsky, Solid State Commun., 2005, 136, 333–337 Search PubMed.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 Search PubMed.
- I. Tamm, in Selected Papers, Springer, 1991, pp. 157–174 Search PubMed.
- S. M. Dancoff, Phys. Rev., 1950, 78, 382–385 Search PubMed.
- A. R. Klots, A. K. M. Newaz, B. Wang, D. Prasai, H. Krzyzanowska, J. Lin, D. Caudel, N. J. Ghimire, J. Yan, B. L. Ivanov, K. A. Velizhanin, A. Burger, D. G. Mandrus, N. H. Tolk, S. T. Pantelides and K. I. Bolotin, Sci. Rep., 2014, 4, 6608 Search PubMed.
- X. Yin, Q. Wang, L. Cao, C. S. Tang, X. Luo, Y. Zheng, L. M. Wong, S. J. Wang, S. Y. Quek, W. Zhang, A. Rusydi and A. T. S. Wee, Nat. Commun., 2017, 8, 486 Search PubMed.
- K. Wang, J. Shao and B. Paulus, J. Chem. Phys., 2021, 154, 104705 Search PubMed.
- R. Nechache, C. Harnagea, L. Cardenas, W. Huang, J. Chakrabartty and F. Rosei, Nat. Photonics, 2015, 9, 61–67 Search PubMed.
- C. Shao, C. Rui, J. Liu, T. Wang, Q. Shao and F. Chen, Diamond Relat. Mater., 2020, 106, 107824 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2tc00391k |
| This journal is © The Royal Society of Chemistry 2022 |