DOI:
10.1039/D1TC05794D
(Paper)
J. Mater. Chem. C, 2022,
10, 4424-4433
Cross-conjugated isothianaphthene quinoids: a versatile strategy for controlling electronic structures†
Received
2nd December 2021
, Accepted 21st February 2022
First published on 22nd February 2022
Abstract
The elucidation of new structure–property relationships in π-conjugated molecules bearing quinoidal moieties is of relevance because of their use in organic electronics applications and their traditional assimilation as models of doped conducting polymers. Quinoidal oligothiophenes are ground state electronic hybrids between closed-shell Kekulé quinoidal and open-shell aromatic diradicaloid forms. The prominent contribution of the diradical character in longer oligomers beyond thiophene 4-mers results in a low stability, thereby limiting the ability to tune their properties. Thus, the control of these quinoidal/aromatic contributions is an important prerequisite to develop long quinoidal oligothiophenes. To address this problem, a series of quinoidal pentathiophenes with benzene-annelated isothianaphthene units were designed and successfully synthesized as stable structures. Combined molecular spectroscopies and theoretical modelling indicated that cross-conjugation appears upon the introduction of multiple benzene-annelated units, and that the number and position of the benzene-annelated units have a significant influence on the quinoidal/aromatic/cross-conjugated electronic structures. The newly developed quinoidal pentathiophenes functioned as organic semiconducting materials in transistor and near infrared phototransistor devices. This study demonstrates that modification of the cross-conjugated quinoidal structure is a promising strategy for fine-tuning electronic structures in π-extended quinoidal systems, which could help us to understand unique π-electronic features and to develop novel organic electronic materials.
Introduction
Considerable progress has been achieved in the development of π-conjugated oligomers and polymers for possible applications to organic semiconducting materials in organic electronics such as organic light-emitting diodes, organic field-effect transistors (OFETs), and organic solar cells (OSCs).1–3 Recently, growing efforts have been devoted to the development of π-conjugated systems with extended π-conjugation to acquire narrow energy gaps and red-shifted absorption, which has enabled the application of such systems in new electronic devices, such as near infrared (NIR)-type OSCs, photodetectors, and visible transparent electrodes.4,5 It has been documented that the carrier transport characteristics of such π-conjugated systems correlate with the formation of doped states of quinoidal structures.6 In this context, structurally well-defined quinoidal molecules represented by quinoidal oligothiophenes have been extensively investigated not only to understand their distinctive properties, but also to investigate their application to organic semiconducting materials.7–12 A unique feature of the electronic structures of quinoidal oligothiophenes includes the description of their ground electronic state as a competition between two resonant forms: closed-shell Kekulé quinoidal and open-shell aromatic diradicaloid forms13 (Scheme 1(a)).14–16 One co-author of the current article, Casado, revealed that short quinoidal oligothiophenes such as 2TQ and 3TQ dominantly contribute to the closed-shell quinoidal structure, while the diradical character becomes prominent upon increasing the number of thiophene rings for longer quinoidal oligothiophenes beyond thiophene 4-mer.15,16 This increased diradical character results in poorly stable quinoidal molecules. In fact, the precedent of quinoidal thiophene 5-mer is still limited to 5TQ,17 which hampers the fine-tuning of their molecular properties. Therefore, control of their quinoidal/aromatic electronic structures is of particular interest for the development of long quinoidal oligothiophenes with tuned electronic functions.
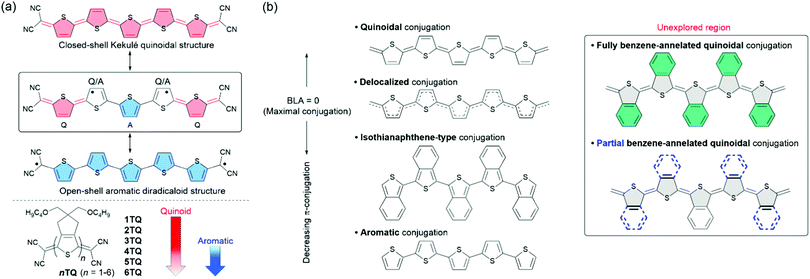 |
| | Scheme 1 (a) Closed-shell and open-shell diradical resonance in quinoidal pentathiophene with the balanced structure in the square highlighting the mixture of aromatic (A), quinoidal (Q), and intermediate (Q/A) characters from ref. 15. (b) π-Conjugation evolution as a function of the bond length alternation (BLA) pattern among families of aromatic and quinoidal oligothiophenes and isothianaphthene aromatic oligothiophenes that highlights the unexplored region of benzene-annelated quinoidal oligothiophenes. Benzene rings in blue color depict the different extent of benzene-annelation in quinoidal pentathiophenes under study. | |
The incorporation of the benzo[c]thiophene (isothianaphthene) unit into the polythiophene framework is a pioneering approach for increasing the quinoidal character and π-electron delocalization that exploits strengthening of the C–C single bond at the β position of the thiophene ring upon benzene-annelation.18 In Scheme 1(b), we summarize the scale of the π-conjugation degrees achieved in thiophene-based aromatic and quinoidal systems on which we place the isothianaphthene-based molecules between these two because of the mentioned quinoidization effect upon benzene-annelation of aromatic cores. The lacking family in this scheme is that of benzene-annelated quinoidal oligomers. A scientific interest in these compounds is the potential modulation of the diradical character in long quinoidal oligothiophenes caused by the inclusion of isothianaphthene unit. We have achieved some progress in this direction;19,20 we recently developed a quinoidal thiophene 3-mer bearing fully benzene-annelation; however, the effect of benzene-annelation on the electronic properties was found to be modest, since benzene-annelation accentuated the pre-existing quinoidal electronic structure.20 This result indicates that the scenario in longer quinoidal oligothiophenes upon benzene-annelation is completely different and challenging because pristine quinoidal molecules have mixed quinoidal/aromatic electronic structures attributed to the prominent contribution of the diradical character. For example, 5TQ has a central ring with an aromatic structure, while the two terminal rings are quinoidal with aromatic-quinoidal transition for the two in between (Scheme 1(a)).15,16 This phenomenon inspired us to consider that the partial benzene-annelation of the rings and positional isomerization in addition to full benzene-annelation are attractive in the context of molecular design for precisely controlling quinoidal/aromatic electronic structures.
In this study, we designed and conducted a full structural and electronic characterization of new bis(dicyanomethylene)-substituted quinoidal pentathiophenes with partial, positional isomerism and total benzene-annelation of their thiophene rings. These are 5TQ-B5 with full thiophene annelation and two different isomers (5TQ-B3 and 5TQ-BBB) with partial and positional isomerization three-fold annelation and a symmetric mono-annelated pentamer 5TQ-B (Fig. 1). A close comparison was performed with non-annelated quinoidal and aromatic oligothiophene derivatives; a selection of these were implemented as semiconducting materials in OFETs, and in the case of 5TQ-B5(EH), as NIR photosensitizers in organic phototransistors (OPTs).
 |
| | Fig. 1 Chemical structures of the benzene-annelated quinoidal pentathiophenes studied in this work. | |
Results and discussion
Synthesis
Synthetic details to prepare target compounds are summarized in Scheme 2. Based on the contribution of the ortho-quinoid electronic structure, benzo[c]thiophene is known as an unstable scaffold. Except for electrochemical polymerization,18a,c the π-extension of the isothianaphthene-repeated oligomer is limited up to thiophene 3-mer; these molecules possess end-capping with electron-withdrawing substituents to ensure chemical stability.21 Hence, we plan to synthesize 4,7-dihydro-4,7-ethanobenzo[c]thiophene-based oligomers and utilize a thermal retro Diels–Alder reaction in the final step of the synthetic route to prevent the transformation of benzo[c]thiophene-based thiophene 5-mers.21 Diiodo compound 1a was prepared according to our previous report.20 The Suzuki-coupling reaction of 1a and the boronic ester 2 afforded the π-conjugated framework of thiophene 5-mer; this reacted with N-iodosuccinimide (NIS) to provide compound 3a. The cross-coupling reaction of 3a with sodium dicyanomethanide, followed by oxidation using 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), afforded the quinoidal pentathiophene 5TQ-B(Ph) in 44% yield. The same synthetic protocol was adopted for the synthesis of 5TQ-B(EH), 6a, 6b, 9a, and 9b. Note that all the synthesized quinoidal molecules have enough ambient stability due to the fine-tuned diradical character.
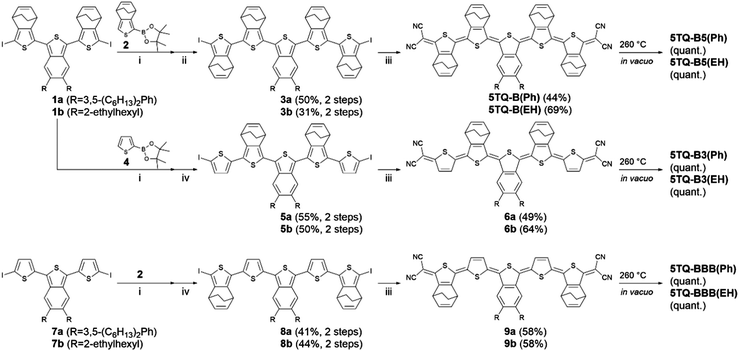 |
| | Scheme 2 Synthesis of quinoidal pentathiophenes. Reaction conditions: (i) Pd2(dba)3·CHCl3, SPhos, K3PO4, THF/H2O, 65 °C; (ii) NIS, DMF/CHCl3, 0 °C to room temperature; (iii) NaH, malononitrile, Pd(PPh3)4, dppf, THF, 75 °C; DDQ, HCl/CHCl3, 0 °C; (iv) LDA, THF, −78 °C to room temperature; I2, −78 °C. | |
Thermogravimetric analysis (TGA) was conducted to investigate if thermal retro Diels–Alder reactions can be applied to the precursor molecules (5TQ-B(EH), 6b, and 9b). As indicated in Fig. 2, these molecules presented the first weight loss of ca. 6.7% and 9.9% in the temperature range of 190–220 °C because of eliminating ethylene molecules. These results suggest that 5TQ-B(EH), 6b, and 9b were thermally converted to 5TQ-B5(EH), 5TQ-B3(EH), and 5TQ-BBB(EH). The TGA curves of the second weight loss correspond to the decomposition of 5TQ-B5(EH), 5TQ-B3(EH), and 5TQ-BBB(EH). The decomposition temperature was increased in the order of 5TQ-BBB(EH) (318 °C), 5TQ-B3(EH) (335 °C), and 5TQ-B5(EH) (353 °C). The TGA measurements of 5TQ-B(Ph), 6a, and 9a demonstrated similar behaviors (Fig. S1 in the ESI†), which indicates that the replacement of 2-ethylhexyl groups with 3,5-dihexylbenzene groups had little influence on the thermal properties of the molecules. These results suggest that the thermal conversion reactions of the synthesized precursors were performed at 260 °C under vacuum conditions to yield targets 5TQ-B5(EH), 5TQ-B3(EH), 5TQ-BBB(EH), 5TQ-B5(Ph), 5TQ-B3(Ph), and 5TQ-BBB(Ph). The detailed synthetic methods and characterization data are summarized in the ESI.†5TQ-B5(EH), 5TQ-B3(EH), and 5TQ-BBB(EH) showed very poor solubility, and moderate improvement (<1 mg mL−1 at 25 °C) in 5TQ-B5(Ph), 5TQ-B3(Ph), and 5TQ-BBB(Ph) in organic solvents such as chloroform and 1,1,2,2-tetrachloroethane (TCE), which allows physical measurements in the solution.
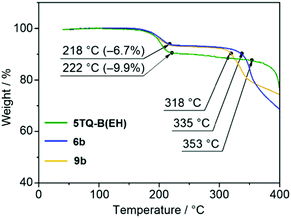 |
| | Fig. 2 TGA curves for 5TQ-B(EH) (green), 6b (blue), and 9b (yellow) with a heating rate of 10 °C min−1 in an N2 atmosphere. | |
UV-Vis-NIR electronic absorption spectra
The UV-Vis-NIR absorption spectra of 5TQ-B5(Ph), 5TQ-B3(Ph), 5TQ-BBB(Ph), and 5TQ-B(Ph) in TCE solutions are presented in Fig. 3(a), and the photophysical data are summarized in Table 1. The absorption maximum (λmax) of 5TQ-B(Ph) is observed in the near-infrared region at λmax = 1021 nm, red-shifted with λmax = 913 nm in 5TQ. Compared to 5TQ-B(Ph), 5TQ-B5(Ph) showed a remarkable blue-shift absorption with λmax at 757 nm. In addition, the λmax values for 5TQ-B3(Ph) and 5TQ-BBB(Ph) were located close to 830 nm between those of 5TQ-B5(Ph) and 5TQ-B(Ph). Fig. 3(b) compares the chain length dependence of the energies of the band maxima of the present compounds compared to those of the non-annelated quinoidal thiophene analogues (nTQ),16 and of the most representative aromatic oligothiophene analogues (nTA).22 In the nTQ series from the monomer to the trimer, the quinoidal contribution is 100%, based on which the extrapolation of this behavior (see dotted lines in Fig. 3(b)) for longer compounds discloses their expected band energy maxima (i.e., it would be entirely quinoidal). The same is shown for the aromatic oligothiophene nTA behavior in Fig. 3(b). Between these two extreme quinoidal/aromatic behaviors (highlighted as black solid lines), oligothiophenes with mixed quinoidal/aromatic electronic composition are recognized in the graph as transiting from one regime to another; this occurs incipiently for 4TQ and 5TQ and 6TQ because of the progressive aromatization of the quinoidal core attributed to the stabilization of the open-shell diradical aromatic structure.16 In this quinoidal-aromatic map/plane in Fig. 3(b), 5TQ-B(Ph) is placed closer to the quinoidal line than 5TQ, which reveals a greater quinoidal character in the latter. According to the progression on 5TQ → 5TQ-B(Ph) upon benzene-annelation, 5TQ-B3(Ph) with three benzene-annelated thiophenes are expected to gain more quinoidal character; however, the energy peak maxima, which is unlike 5TQ → 5TQ-B(Ph), separate from the quinoidal area; this behavior is more marked in 5TQ-B5(Ph). This evolution on 5TQ-B(Ph) → 5TQ-B3(Ph) → 5TQ-B5(Ph) is not observed as a progressive gain of aromatic character (such as on 4TQ → 5TQ → 6TQ); instead, it emerges from the effect of cross-conjugation.23 Cross-conjugated π-electronic paths produce blue shifts of the absorption peak maxima (opening the optical energy gap) compared to homologues with linear π-conjugation. Hence, the studied benzene-annelated compounds cannot be represented by a simple quinoidal-aromatic competition, but a third cross-conjugation factor by benzene-annelation enters into play. This cross-conjugation might impart electron wavefunction draining from the linear-conjugated path, and it has a significant impact on the electronic, optical, and molecular properties.
 |
| | Fig. 3 (a) UV-Vis-NIR absorption spectra of 5TQ-B5(Ph) (green), 5TQ-B3(Ph) (blue), 5TQ-BBB(Ph) (yellow), and 5TQ-B(Ph) (red) in TCE. Gray line indicates the λmax of 5TQ. (b) Representation of the evolution of the energy peak maxima of the lowest energy band of absorption spectra of nTQ16 and nTA22 compounds from the monomer to the hexamer as representative cases both of linear conjugations. In the vertical axis is shown the evolution of the energy maxima of the absorption bands in the series of benzene-annelated quinoidal pentamers which on increasing the number of benzene-annelating units cross-conjugation increases starting in 5TQ-B(Ph) (red bar), 5TQ-B3(Ph) (blue bar), 5TQ-BBB(Ph) (yellow bar), and 5TQ-B5(Ph) (green bar). Cross-conjugation evolution in the vertical axis is thus out of the plane of linear aromatic and quinoidal conjugations. | |
Table 1 Photophysical and electrochemical properties of molecules
| Compounds |
λ
max
/nm |
ΔEoptg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/eV a/eV |
E
oxb/V |
E
redb/V |
E
HOMO
/eV |
E
LUMO
/eV |
ΔEDPVge/eV |
|
In TCE.
In TCE containing 0.1 M TBAPF6.V vs. Fc/Fc+.
E
HOMO = −Eox − 4.8.
E
LUMO = −Ered − 4.8.
ΔEgDPV=ELUMO − EHOMO.
|
|
5TQ-B5(Ph)
|
757 |
1.48 |
0.30 |
−1.06 |
−5.10 |
−3.74 |
1.36 |
|
5TQ-B3(Ph)
|
829 |
1.34 |
0.35 |
−0.76 |
−5.15 |
−4.04 |
1.11 |
|
5TQ-BBB(Ph)
|
830 |
1.34 |
0.21 |
−0.81 |
−5.01 |
−3.99 |
1.02 |
|
5TQ-B(Ph)
|
1021 |
0.95 |
n.d. |
n.d. |
n.d. |
n.d. |
n.d. |
Solid-state Raman spectra
The 1064 nm FT-Raman spectra in the solid state of 5TQ-B(Ph) compared with that of 5TQ are shown in Fig. 4(a). The Raman spectrum of 5TQ displays two main bands of similar intensity at 1462 and 1386 cm−1, which are associated with the CC bond stretching modes placed at rather different places of the conjugated skeleton; that at 1462 cm−1 is attributed to the CC inter-ring bond stretching modes [i.e., ν(CC)inter in Fig. 4(b)] with the main contribution from the CC bond connecting to the dicyano groups; that at 1386 cm−1 belongs to the CC stretching modes of the two intra-ring α–β CC bonds of the thiophene [i.e., ν(CC)intra in Fig. 4(b)]. The 1386 cm−1 wavenumber of the ν(CC)intra Raman band in 5TQ is associated with the aromatization of the central three rings (more marked in the central ring) because of the formation of a diradical structure in 5TQ, as depicted in Scheme 1(a) and Fig. 4(c). This description of 5TQ with a central aromatic part and the outermost thiophenes with more quinoidal character is pivotal for understanding the role of benzene annelation. The Raman spectrum of 5TQ-B(Ph) is similar to that of 5TQ but with wavenumber downshifts of the two CC stretching bands, in particular the 1386 → 1335 cm−1 for the ν(CC)intra band. This indicates a weakening of the α−β bonds consistent with a decrease in the overall aromatic character of the thienyl rings, or conversely, a gain of quinoidal character (see Fig. 4(c)), as also revealed in the UV-Vis-NIR discussion.
 |
| | Fig. 4 (a) FT-Raman spectra recorded with the 1064 nm excitation in solid state at room temperature of 5TQ-B(Ph) and 5TQ. (b) Vibrational dynamics of the main Raman bands associated with the CC stretching modes of the quinoidal (red) and aromatic aromatic (blue) rings. (c) Structural evolution of the quinoidal rings upon benzene-annelation. | |
The FT-Raman spectra of the benzene-annelated quinoidal pentathiophenes are shown in Fig. 5(a). From 5TQ-B(Ph) to 5TQ-B5(Ph), the two discussed Raman band patterns disappear, and one main Raman band emerges at 1468 cm−1. The theoretical Raman spectrum of 5TQ-B5(H) is calculated and shown in Fig. 5(b), which reveals a good resemblance with the experimental spectrum with a very strong theoretical Raman band at 1457 cm−1, this correlates with the experimental results at 1468 cm−1. According to a theoretical vibrational eigenvector, this 1457 cm−1 band can be described as the ν(CC)inter mentioned above; It indicates that the full benzene annelation of the quinoidal pentathiophene core changes the vibrational dynamics of the π-conjugated system, and it results in the disappearance of the ν(CC)intra. Raman band of 5TQ and 5TQ-B(Ph). This can be understood by considering that benzene-annelation/cross-conjugation breaks the π-electron delocalization and transmission through the intra-ring CC bonds, which results in less efficient linear π-conjugation that weakens the ν(CC)intra Raman intensity to the point of clearance (Fig. 5(c)). In 5TQ-B3(Ph), the ν(CC)inter Raman band is located at 1442 cm−1, which agrees with an intermediate scenario between 5TQ-B(Ph) and 5TQ-B5(Ph). In 5TQ-BBB(Ph), the main band at 1404 cm−1 between the wavenumbers of 5TQ-B(Ph) at 1335 cm−1 and 5TQ-B3(Ph) at 1442 cm−1 was measured, as accompanied by another medium-large intensity Raman band at 1420 cm−1 that comprises the two-band pattern featured by 5TQ-B(Ph) and 5TQ; it indicates that the alternant mode of non-annelated and annelated thiophenes produces a smaller interference in the linear conjugation path by cross-conjugation than that in the isomeric 5TQ-B3(Ph). The multiple connectivity bonding pattern on the β carbons of thiophenes in the presence of benzenes originates from the emergence of cross-conjugation. However, we wonder why single benzene-annelation 5TQ-B(Ph) strengthens quinoidization but not cross-conjugation, which is operative in compounds with multiple benzene annelations. Benzene annelation in the central ring of 5TQ-B(Ph) acts on an aromatic ring (see 5TQ in Scheme 1(a) and Fig. 4(c)), whereas in the case of multiannelation, benzenes are fused with thiophenes that are mostly quinoidal (Scheme 1(a) and Fig. 4(c) for the distribution of aromatic and quinoidal rings in 5TQ) in 5TQ-BBB(Ph) in 5TQ-B3(Ph) and fully quinoidal in 5TQ-B5(Ph). Thus, the spectroscopic behavior in 5TQ-B(Ph) reveals that the effect of the electronic structure of the mono-benzene-annelation of 5TQ is identical to that of the benzene-annelation of aromatic oligothiophenes (i.e., in the case of isothianaphthene aromatic oligomers) in which benzene-annelation produces the quinoidization of aromatic thiophenes. Conversely, multiple benzene-annelation units of quinoidal rings of 5TQ in 5TQ-B3(Ph), 5TQ-BBB(Ph), and 5TQ-B5(Ph) produces cross-conjugation.
 |
| | Fig. 5 (a) 1064 nm FT-Raman spectra of benzene-annelated quinoidal pentathiophenes in the solid state at room temperature from the bottom: 5TQ-B(Ph), 5TQ-BBB(Ph), 5TQ-B3(Ph), and 5TQ-B5(Ph). (b) Comparison of B3LYP/6-31G(d,p) theoretical Raman spectrum of a model compound of 5TQ-B5(H) (top) compared with the experimental Raman spectrum of 5TQ-B5(Ph) (bottom). (c) B3LYP/6-31G(d,p) vibrational eigenvector of the 1457 cm−1 Raman band of the theoretical spectrum of 5TQ-B5(H). The inset is the comparison with the previously described ν(CC)inter. | |
Quantum chemical calculations
Density functional theory (DFT) calculations were performed at the UB3LYP/6-31G(d,p) level (Fig. 6(a) and Fig. S2, ESI†) to gain insights into the effects of benzene-annelation on the electronic structures and frontier orbital energies. All solubilizing groups were replaced with hydrogen atoms to reduce the computational work. The number of benzene-annelation affects the lowest unoccupied molecular orbital (LUMO) energy level (Fig. 7 where the impact of the confluence of ortho-conjugation, benzo-aromatic, and para-quinoid structures arise on benzene-annelation is in the LUMO orbital) by which decreasing the number of benzene annelation leads to a decrease in the highest occupied molecular orbital (HOMO)–LUMO energy gap in line with the experimental optical gap (ΔEoptg) reduction from the onset and peak maxima of the optical absorption spectra as 1.48 eV in 5TQ-B5(Ph), 1.34 eV in 5TQ-B3(Ph) and 5TQ-BBB(Ph), and 0.95 eV in 5TQ-B(Ph).
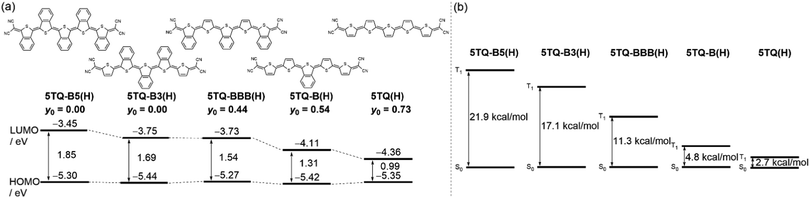 |
| | Fig. 6 (a) Calculated energy levels and y0 values and (b) the energy differences between S0 and T1 of 5TQ-B5(H), 5TQ-B3(H), 5TQ-BBB(H), 5TQ-B(H), and 5TQ(H). | |
 |
| | Fig. 7 HOMO and LUMO orbitals of 5TQ-B(H) (top) and of 5TQ(H) (bottom) together with the inter-atomic interactions (arrows) among vicinal atoms in these two orbitals that reveal the presence of wavefunction bifurcation atoms in the benzene-annelated ring of 5TQ-B(H) compared to 5TQ(H). Valence bond structures equivalent to ortho-conjugation, benzo- and thio-aromatic, and para-quinoid are shown for clarity. | |
Quinoidal systems with narrow HOMO–LUMO energy gaps and aromaticity recovery induce diradical contribution in the ground state.24 The diradical character (y0) defined in terms of the occupation number of the lowest unoccupied natural orbital for optimized model structures is calculated by the spin-unrestricted Hartree–Fock method at the 6-31G(d) level for the studied compounds.25 The y0 value becomes zero when the π-system is dominated by a closed-shell quinoid structure, whereas it approaches 1 when increasing the contribution of the open-shell diradical structure. For quinoidal oligothiophenes, y0 increases with increasing molecular length because of the increasing number of quinoidal rings, which increases the aromatic stabilization of the diradical form. For instance, 5TQ(H) discloses y0 = 0.73, and its ground electronic state is featured by a central part (i.e., spreading approximately three central rings, see Scheme 1(a) and Fig. 4(c)) with aromatic-like character flanked by the two radical centers. For 5TQ-B(H), this amounts to y0 = 0.54, which is smaller than that of 5TQ(H). This behavior is in good agreement with the removal of the aromatic character in 5TQ(H) upon benzene-annelation on the central thiophene. The extension of benzene-annelation of quinoidal oligothiophenes to 5TQ-B3(H) and 5TQ-B5(H) provokes the complete closure of the electronic shell with y0 = 0.00, which agrees with the complete removal of aromatic character upon full thiophene benzene-annelation. On the other hand, 5TQ-BBB(H), which is a positional isomer of 5TQ-B3(H), showed moderate diradical character of y0 = 0.44. This result indicates that the alternant mode of non-annelated and annelated thiophenes leads to increasing the radical character. This trend is in good agreement with the discussions based on 5TQ-BBB(Ph) and 5TQ-B3(Ph) in the Raman spectroscopy measurements. Finally, the energy differences between S0 and T1 of 5TQ-B5(H), 5TQ-B3(H), 5TQ-BBB(H), 5TQ-B(H), and 5TQ(H) were calculated to be 21.9, 17.1, 11.3, 4.8, and 2.7 kcal mol−1, respectively, in line with the previous discussion of y0 (Fig. 6(b)).
NICS(1.7)zz values are obtained for the benzene and thiophene rings of the studied compounds and are summarized in Table S1 (ESI†). For the central benzene rings, the NICS values are more negative for 5TQ-B(H) → 5TQ-BBB(H) → 5TQ-B3(H) → 5TQ-B5(H), which is consistent with the gain of aromatic character with increasing cross-conjugation. This produces electronic isolation between the benzene and the linear-conjugated path. This is accompanied by a decrease in the aromatic character of the fused thiophene, and it is similar to that of benzene in 5TQ-B(H) and it progressively diminishes its aromatic character, eventually becoming nonaromatic in 5TQ-B5(H).
As shown in Fig. S3 (ESI†), the 1H NMR spectra of 5TQ-B5(Ph) and 5TQ-B3(Ph) in a solution of 1,1,2,2-tetrachloroethane-d2 exhibited sharp peaks in the aromatic region even at 130 °C, which indicated their closed-shell electronic structure. In contrast, the 1H NMR spectrum of 5TQ-BBB(Ph) showed broadened signals for the protons of the thiophene rings, which is explained by the thermal population of the excited triplet species. In the case of 5TQ-B(Ph), the 1H NMR spectrum in the solution exhibited broadened signals, besides those for phenyl substituents, even at 25 °C, which indicates the triplet state is dominant for 5TQ-B(Ph).
Electrochemical measurements
The influence of the position and number of benzene-annelations on redox behavior is determined via differential pulse voltammetry (DPV) in 5TQ-B5(Ph), 5TQ-B3(Ph), and 5TQ-BBB(Ph) in TCE containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte. Fig. 8(a) shows that these compounds exhibit oxidation (Eox) and reduction processes (Ered) that exchange the same number of electrons, which indicates that they are one electron or single-electron processes in contrast with nTQ quinoidal oligothiophenes, which display two-electron reductions. Further, the distinctive behavior of annelated compounds is revealed in the electrochemical potential Eox and Ered values (Table 1), which are negatively displaced relative to 5TQ at −0.16 V.16 In addition, those of 5TQ-B5(Ph) is negatively shifted in comparison with those of 5TQ-B3(Ph), in agreement with the larger deviation of the quinoidal linear π-conjugation behavior in the cases with more benzenes. Compared to 5TQ-B3(Ph), the alternating introduction of benzene-annelation for 5TQ-BBB(Ph) resulted in slight changes in the reduction potentials. The disruption of the electronic communication in the thienyl chain by cross-conjugation alters the role of the external electron-withdrawing cyano groups, which further affects the oxidation potentials (Table 1). The HOMO and LUMO energy levels of these compounds were estimated from the equations EHOMO = −Eox − 4.8 and ELUMO = −Ered − 4.8 presented in Table 1, which follow the same trend as the optical gaps from the absorption spectra.
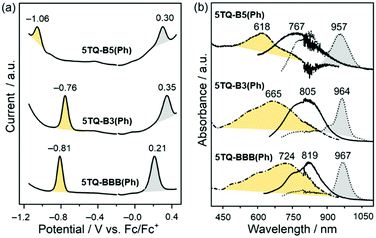 |
| | Fig. 8 (a) DPV of 5TQ-B5(Ph), 5TQ-B3(Ph) and 5TQ-BBB(Ph) in TCE containing 0.1 M TBAPF6. (b) Spectroelectrochemical UV-Vis-NIR spectra of the reduced (broken lines, yellow shaded) and oxidized (dotted lines, grey shaded) species obtained in the same electrochemical conditions than those use in DPV together with the spectra of the neutrals (solid lines). | |
The UV-Vis-NIR absorption spectra of redox species found in the electrochemical oxidation and reduction processes are obtained by UV-Vis-NIR spectroelectrochemistry; the spectra are shown in Fig. 8(b) (Step-by-step UV-Vis-NIR followed redox processes are shown in Fig. S4, ESI†). The spectra of the reduced species of the three compounds are always blue-shifted compared with the absorption bands of the neutral compounds. In contrast, for the oxidized species, the relevant absorption bands are red-shifted with respect to the neutrals. Interestingly, 5TQ-B3(Ph) and 5TQ-BBB(Ph) display smaller differences between the wavelength maxima of the reduced and oxidized species, which indicates that these two redox species of different charge signs are stabilized in similar ways with moderate or smaller structural changes from one to the other. This is important because 5TQ-B3(Ph) and 5TQ-BBB(Ph) show ambipolar semiconducting behavior, whereas 5TQ-B5(Ph) with larger differences in the optical spectra between the reduced and oxidized species reveals a preferential stabilization of one of the two, which indicates unipolar semiconducting behavior.
Device applications
We fabricated thin films of 5TQ-B5(EH), 5TQ-B3(EH), and 5TQ-BBB(EH) using the drop-cast method from a hot TCE solution. The X-ray diffraction (XRD) profiles of the drop-cast films of all molecules showed first-order diffraction peaks with 2θ = 4.8–6.1°, which correspond to the d-spacing of 18.4–14.5 Å (Fig. S5, ESI†). This length is approximately close to the calculated molecular lengths of quinoidal thiophene 5-mer in the long axis direction (ca. 20 Å), and it implies that these molecules are arranged in an edge-on orientation against the SiO2 substrates. Atomic force microscopy (AFM) images showed distinct crystal-shaped microstructures (Fig. S6, ESI†). We investigated the charge carrier characteristics using bottom-gate bottom-contact OFET devices. The transfer and output characteristics of these devices are shown in Fig. S7 and S8 (ESI†); key device parameters such as mobility (μ), threshold voltage (Vth), and current on/off ratio (Ion/Ioff) are extracted and summarized in Table 2. 5TQ-B5(EH) demonstrated typical hole-transporting characteristics with a mobility of 10−5 orders. In contrast, 5TQ-B3(EH) and 5TQ-BBB(EH) exhibited ambipolar characteristics with well-balanced mobilities of 10−3 and 10−4 orders; these values indicate that these compounds possess the appropriate HOMO and LUMO energy levels for hole and electron injection and structural stability of the redox species, as mentioned above. The ambipolar characteristics of 5TQ-B3(EH) and 5TQ-BBB(EH) resulted in low Ion/Ioff ratios due to the accumulation of opposite carriers. To examine the stability of these molecules under the biased conditions, the OFETs were repeatedly biased by sweeping the gate voltage. As shown in Fig. S9 (ESI†), all the devices showed small hysteresis even after 100 cycles. These results indicate that these quinoidal molecules showed enough stability for OFET materials.
Table 2 Field-effect transistor characteristics
| Compounds |
Carrier type |
μ/cm2 V−1 s−1 |
V
th/V |
I
on/Ioff |
|
5TQ-B5(EH)
|
p |
6.8 × 10−5 |
−9 |
103 |
|
5TQ-B3(EH)
|
p |
1.8 × 10−3 |
−27 |
101 |
|
|
n |
1.7 × 10−3 |
19 |
101 |
|
5TQ-BBB(EH)
|
p |
2.0 × 10−4 |
−31 |
102 |
|
|
n |
4.3 × 10−4 |
13 |
102 |
The UV-Vis-NIR absorption spectrum of the 5TQ-B5(EH) film showed an absorption band in the NIR region with an absorption maximum at 806 nm (Fig. S10, ESI†). Thus, we investigated the photoresponses of 5TQ-B5(EH) in the architecture of the OPTs (Fig. 9(a)). We adopted binary systems based on organic semiconductor and NIR sensitizer to construct the active layers of NIR photodetectors.26 The HOMO energy level of 5TQ-B5(EH) in thin film was determined to be −5.5 eV from the photoelectron spectroscopy measurements (Fig. S11, ESI†). We chose a porphyrin derivative 2CF3BP as the counterpart of an organic semiconductor to reveal the OPT characteristics of 5TQ-B5(EH) as the NIR sensitizer in the binary system because 2CF3BP fulfills good energy matching of the HOMO level (−5.5 eV) and weak absorbance at a wavelength of 810 nm.27 The bulk heterojunction configuration of the 5TQ-B5(EH)/2CF3BP films were prepared by thermal conversion at 200 °C for 20 min in N2 atmosphere from the 5TQ-B(EH)/2CF3BP-pre (0.05/0.25 wt%) solutions to ascertain the area of semiconductor/NIR sensitizer interfaces. The OPT characteristics were measured in the dark and under NIR light illumination of 810 nm at an intensity of 143 mW cm−2 using light-emitting diodes. Fig. 9(b) shows that the transfer characteristics of the 5TQ-B5(EH)/2CF3BP-based OPTs under the bias voltage of the hole-transporting mode exhibits an apparent increase in the drain current (ISD) when the active layer of the OPTs is exposed to NIR light. Photosensitivity (P), as estimated from the equation P = (Ilight − Idark)/Idark, is calculated to be P = 103. The specific detectivity (D*) of this device is calculated to be 3.8 × 108 Jones according to the equation D* = R·S1/2/(2qIdark)1/2, where R is the photoresponsivity, S is the channel area, and q is the electron charge. Fig. 9(c) shows that the time-dependent photoresponses were measured under NIR light on/off switching at 5 s intervals. The gate and drain voltages were set to −5 and −50 V, respectively. This device showed stable photoswitching behavior under repeated light on/off switching conditions. We considered that the hole generated by NIR light absorption of 5TQ-B5(EH) efficiently passes across the interface of 5TQ-B5(EH)/2CF3BP and is collected at the drain electrode, which leads to the appearance of photosensitivity (Fig. 9(d)).28 These results indicate that π-extended quinoidal oligothiophenes with tuned closed-shell structures have the potential for use as an organic NIR-sensitive semiconducting material.
 |
| | Fig. 9 (a) Chemical structures of 2CF3BP-pre and 2CF3BP and OPT device architecture. (b) Transfer characteristics and (b) time-dependent switching response profile of the 5TQ-B5(EH)/2CF3BP-based NIR OPTs in the dark and under NIR light illumination. (d) Energy diagram of 2CF3BP and 5TQ-B5(EH). | |
Conclusions
To control the quinoidal/aromatic contributions of long quinoidal oligothiophenes, a series of quinoidal pentathiophenes end-capped with dicyanomethylene units and bearing isothianaphthene units was designed. These molecules were successfully synthesized as stable structures by a thermal retro Diels–Alder reaction in the final step of the synthetic route. Theoretical calculations together with UV-Vis-NIR electronic absorption and vibrational FT-Raman spectroscopies showed that cross-conjugation between the annelating benzene moieties and the thiophene oligomer chain emerges upon increasing the number of benzene units. Concurrently, cross-conjugation imparts an increasing degree of the closed-shell Kekulé form, resulting in a new mode of tuning this conjugation in long quinoidal oligomers. More specifically, 5TQ-B(Ph) increased/decreased the contribution of quinoidal/aromatic conjugation compared to 5TQ, whereas 5TQ-B5(Ph), which comprised fully of isothianaphthene units, exhibited significant cross-conjugation to further suppress the diradical character. Our elucidation of the competition between quinoid/aromatic/cross-conjugation points to new structure–property relationships, which inspired us to fabricate OFETs based on the newly synthesized quinoidal pentathiophenes. These OFETs exhibited unipolar and ambipolar electrical semiconducting behaviors, which were further ascribed to the preferential stabilization of the reduced and/or oxidized species. Furthermore, 5TQ-B5(EH) exhibited NIR OPT characteristics that benefited from the relatively high current on/off ratio and NIR absorption behavior. Our results demonstrated that the combination of an extended quinoidal structure (providing NIR absorption) with isothianaphthene units (providing energy gap modulation and stability by cross-conjugation) is a promising design strategy for fine-tuning electronic structures in π-extended quinoidal oligothiophenes. Since we are able to further extend the molecular lengths of quinoidal oligothiophenes based on the results presented herein, the emergence of such strategies will help in the design of improved organic semiconductor materials that may pioneer novel electronic applications.
Author contributions
The manuscript was written through contributions of all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI (20H02814, 20K21224, 20H05841, 20KK0123, 19K15505, 20H04639, 20K15352, 21K05213, 20H00379, 20H05833, and 20K15261), CREST (J205101030), NEDO (21500248-0), and “Dynamic Alliance for Open Innovation Bridging Human, Environmental and Materials” from The Ministry of Education, Culture, Sports, Science and Technology, Japan. We are grateful to Prof. Toshihiro Ohnishi for helpful discussion. The authors thank the Spanish Ministry of Science, Innovation and Universities MCIU and MINECO/FEDER of the Spanish Government (project PGC2018-098533-B-100), the Ministry of Science and Technology of the Spanish Government (project RED2018-102626-T) and the Junta de Andalucía, Spain (UMA18FEDERJA057). We also thank the Research Central Services (SCAI) of the University of Málaga.
Notes and references
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Adv. Mater., 2020, 32, 1905909 CrossRef CAS PubMed.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- K. J. Baeg, M. Binda, D. Natali, M. Caironi and Y. Y. Noh, Adv. Mater., 2013, 25, 4267–4295 CrossRef CAS PubMed.
- S. R. González, Y. Ie, Y. Aso, J. T. López Navarrete and J. Casado, J. Am. Chem. Soc., 2011, 133, 16350–16353 CrossRef PubMed.
- S. Handa, E. Miyazaki, K. Takimiya and Y. Kunugi, J. Am. Chem. Soc., 2007, 129, 11684–11685 CrossRef CAS PubMed.
- J. Casado, R. P. Ortiz and J. T. López Navarrete, Chem. Soc. Rev., 2012, 41, 5672–5686 RSC.
- R. P. Ortiz, J. Casado, S. R. González, V. Hernández, J. T. López Navarrete, P. M. Viruela, E. Ortí, K. Takimiya and T. Otsubo, Chem. – Eur. J., 2010, 16, 470–484 CrossRef PubMed.
- K. Yamamoto, S. I. Kato, H. Zajaczkowska, T. Marszalek, P. W. M. Blom and Y. Ie, J. Mater. Chem. C, 2020, 8, 3580–3588 RSC.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC.
- C. Zhang, Y. Zang, E. Gann, C. R. McNeill, X. Zhu, C. A. Di and D. Zhu, J. Am. Chem. Soc., 2014, 136, 16176–16184 CrossRef CAS PubMed.
-
(a) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed;
(b) J. J. Dressler, A. C. Valdivia, R. Kishi, G. E. Rudebusch, A. M. Ventura, B. E. Chastain, C. J. Gómez-Garcia, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Chem, 2020, 6, 1353–1368 CrossRef CAS.
- C. Zhang and X. Zhu, Acc. Chem. Res., 2017, 50, 1342–1350 CrossRef CAS PubMed.
- R. P. Ortiz, J. Casado, S. R. González, V. Hernández, J. T. López Navarrete, P. M. Viruela, E. Ortí, K. Takimiya and T. Otsubo, Chem. – Eur. J., 2010, 16, 470–484 CrossRef PubMed.
- R. Ponce Ortiz, J. Casado, V. Hernandez, J. T. Lopez Navarrete, P. M. Viruela, E. Orti, K. Takimiya and T. Otsubo, Angew. Chem., Int. Ed., 2007, 46, 9057–9061 CrossRef PubMed.
- T. Takahashi, K. I. Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127, 8928–8929 CrossRef CAS PubMed.
-
(a) F. Wudl, M. Kobayashi and A. J. Heeger, J. Org. Chem., 1984, 49, 3382–3384 CrossRef CAS;
(b) J. L. Bredas, A. J. Heeger and F. Wudl, J. Chem. Phys., 1986, 85, 4673–4678 CrossRef CAS;
(c) Y. Ikenoue, F. Wudl and A. J. Heeger, Synth. Met., 1991, 40, 1–12 CrossRef CAS;
(d) H. Neugebauer, C. Kvarnström, C. Brabec, N. S. Sariciftci, R. Kiebooms, F. Wudl and S. Luzzati, J. Chem. Phys., 1999, 110, 12108–12115 CrossRef CAS;
(e) J. D. Douglas, G. Griffini, T. W. Holcombe, E. P. Young, O. P. Lee, M. S. Chen and J. M. J. Fréchet, Macromolecules, 2012, 45, 4069–4074 CrossRef CAS;
(f) G. Long, X. Wan, J. Zhou, Y. Liu, Z. Li, G. He, M. Zhang, Y. Hou and Y. Chen, Macromol. Chem. Phys., 2012, 213, 1596–1603 CrossRef CAS;
(g) W. A. Braunecker, Z. R. Owczarczyk, A. Garcia, N. Kopidakis, R. E. Larsen, S. R. Hammond, D. S. Ginley and D. C. Olson, Chem. Mater., 2012, 24, 1346–1356 CrossRef CAS.
-
(a) K. Yamamoto, Y. Ie, M. Nitani, N. Tohnai, F. Kakiuchi, K. Zhang, W. Pisula, K. Asadi, P. W. M. Blom and Y. Aso, J. Mater. Chem. C, 2018, 6, 7493–7500 RSC;
(b) S. Moles Quintero, J. L. Zafra, K. Yamamoto, Y. Aso, Y. Ie and J. Casado, J. Mater. Chem. C, 2021, 9, 10727–10740 RSC.
- K. Yamamoto, S. Jinnai, T. Takehara, T. Suzuki and Y. Ie, Org. Lett., 2020, 22, 547–551 CrossRef CAS PubMed.
- Y. Shimizu, Z. Shen, S. Ito, H. Uno, J. Daub and N. Ono, Tetrahedron Lett., 2002, 43, 8485–8488 CrossRef CAS.
-
Handbook of Thiophene-Based Materials: Applications in Organic Electronics and Photonics, ed. I. F. Perepichka, D. F. Perepichka, John Wiley & Sons, Ltd., Chichester, 2009 Search PubMed.
-
(a) N. F. Phelan and M. Orchin, J. Chem. Educ., 1968, 45, 633–637 CrossRef CAS;
(b) R. Ponce, A. Facchetti, T. J. Marks, J. Casado, M. Z. Zgierski, M. Kozaki, V. Hernández and J. T. López Navarrete, Adv. Funct. Mater., 2009, 19, 386–394 CrossRef;
(c) H. Hayashi, J. E. Barker, A. Cárdenas, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142, 20444–20455 CrossRef CAS PubMed;
(d) K. Kawabata, I. Osaka, M. Sawamoto, J. L. Zafra, P. Mayorga-Burrezo, J. Casado and K. Takimiya, Chem. – Eur. J., 2017, 23, 4579–4589 CrossRef CAS PubMed;
(e) C. Zhang, S. M. Rivero, W. Liu, D. Casanova, X. Zhu and J. Casado, Angew. Chem., Int. Ed., 2019, 58, 11291–11295 CrossRef CAS PubMed;
(f) K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. R. Delgado, H.
Y. Woo, R. P. Ortiz and X. Guo, J. Am. Chem. Soc., 2020, 142, 4329–4340 CrossRef CAS PubMed.
- Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- R. Kishi, S. Ochi, S. Izumi, A. Makino, T. Nagami, J.-y. Fujiyoshi, N. Matsushita, M. Saito and M. Nakano, Chem. – Eur. J., 2016, 22, 1493–1500 CrossRef CAS PubMed.
-
(a) X. Ren, F. Yang, X. Gao, S. Cheng, X. Zhang, H. Dong and W. Hu, Adv. Energy Mater., 2018, 8, 1801003 CrossRef;
(b) N. Li, Z. Lan, L. Cai and F. Zhu, J. Mater. Chem. C, 2019, 7, 3711–3729 RSC;
(c) H. Xu, J. Li, B. H. K. Leung, C. C. Y. Poon, B. S. Ong, Y. Zhang and N. Zhao, Nanoscale, 2013, 5, 11850–11855 RSC;
(d) S. Park, S. J. Kim, J. H. Nam, G. Pitner, T. H. Lee, A. L. Ayzner, H. Wang, S. W. Fong, M. Vosgueritchian, Y. J. Park, M. L. Brongersma and Z. Bao, Adv. Mater., 2015, 27, 759–765 CrossRef CAS PubMed.
- E. Jeong, K. Takahashi, S. K. Rajagopal, T. Koganezawa, H. Hayashi, N. Aratani, M. Suzuki, T. Q. Nguyen and H. Yamada, J. Org. Chem., 2020, 85, 168–178 CrossRef CAS PubMed.
- Y. Peng, W. Lv, B. Yao, G. Fan, D. Chen, P. Gao and M. Zhou, Org. Electron., 2013, 14, 1045–1051 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, UV-Vis-NIR, Raman, NICS, XRD, AFM, CV, FET, OPT, PESA results, synthetic detail, NMR charts, and calculated details. See DOI: 10.1039/d1tc05794d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Eunjeong
Jeong
d,
Kyohei
Matsuo
a,
Eunjeong
Jeong
d,
Kyohei
Matsuo










