
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and luminescence of Cs2HfCl6 micro- and Cs2HfF6 nanoparticles†
Madeleine
Fellner
 and
Alessandro
Lauria
*
and
Alessandro
Lauria
*
Laboratory for Multifunctional Materials, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland. E-mail: alessandro.lauria@mat.ethz.ch
First published on 21st February 2022
Abstract
Hafnium-based halide crystals are attractive wide-bandgap phosphor materials for scintillation applications due to their high density, low hygroscopicity and bright radioluminescence. Here, we describe synthetic approaches towards the formation of Cs2HfCl6 (CHC) microparticles and Cs2HfF6 (CHF) nanocrystals. The structure of the obtained materials was characterised by means of powder X-ray diffraction (XRD) and scanning electron microscopy (SEM). The surface chemistry was studied by attenuated total reflection infrared spectroscopy (ATR-IR) and dynamic light scattering (DLS) while thermal properties were determined with thermogravimetric analysis (TGA). The optical properties of the products were elucidated through absorption (UV-vis) and photoluminescence (PL/PLE) spectra. Intrinsically luminescent CHC can be synthesised as a micropowder through an emulsion synthesis. The synthesis of pure and Eu- or Mn-doped CHF nanopowders in organic solvents at 160 °C and atmospheric pressure is also disclosed. These materials might pave the way towards the versatile additive manufacturing of lead-free and dopant-free, particle-based scintillator materials.
Introduction
In recent years, heavy metal halide phosphor materials gained significant attention from both academic and industrial research. Intense work has been devoted to developing perovskite nanocrystals with an ABX3-stoichiometry, leading to great improvements in the control of their composition, particle shape, and optimisation of their optical properties.1 Due to their bright and highly colour-pure luminescence, they have been proposed and implemented as optically active materials in LEDs and photovoltaics.2,3 Double perovskites, using cations of a different valency than B2+ were proposed to access a wider range of possible materials.4–8 However, this change in stoichiometry also leads to structural changes in these materials compared to classic perovskite crystals, as the [BX6]-anion octahedra cease to be corner-sharing.9,10 By using a tetravalent cation, the stoichiometry of the halides changes and cubic double perovskite crystals of the K2PtCl6 (A2BX6) structure-type can be obtained as depicted in Fig. 1.11 Synthetic procedures to obtain such luminescent double perovskites Cs2TiX6 (X = Cl, Br) and Cs2ZrX6 (X = Cl, Br) as a micro- and nanopowder, respectively, have been recently reported.5,6 By replacing the Ti4+ and Zr4+ cations in these materials with Hf4+, materials relevant for scintillation can be accessed. Higher density and stopping power toward ionising radiation are expected in hafnium-based halides, due to the higher atomic weight of Hf. Scintillators materials find application in high energy physics, radiation detection, and diagnostics because of their ability to convert X-rays or gamma rays to visible light, which in turn can be transformed to a digital signal by common photodetectors.12,13 In this context, the cubic double perovskite Cs2HfCl6 (CHC) is known as a scintillator crystal with a strong, intrinsic fluorescence at 375 nm, high light yield and excellent energy resolution and proportionality.14–17 However, CHC has mostly been described as bulk single crystals in the literature, except for a report of CHC microparticles obtained via a precipitation synthesis in aqueous HCl at room temperature, and the synthesis of such materials by colloidal methods has not yet been extensively studied.18–21 This lack of synthetic strategies prevents the solution processing of nanomaterials with controlled surface chemistry, which would promise enhanced control on the resulting assemblies or composites in terms of more versatile geometries and microstructure.22 Indeed, micropatterned self-guiding scintillator layers could reduce the crosstalk between the active layer and the underlying detector arrays.23–26 Therefore, functional micro- or nanoparticles with idoneous surface chemistry could pave the way towards the additive manufacturing of such materials, making them possibly suitable for several applications including scintillator detectors with improved spatial resolution. Like for lead halide perovskites, the stability of halide scintillator materials is often affected by their hygroscopicity. By replacing Cl− with F− the water affinity of the crystal can be possibly reduced.27,28 However, the change of ionic radius of the halide leads to a change in the crystal structure causing Cs2HfF6 (CHF) to crystallise in the trigonal P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 space group (Fig. 1).29 Due to its higher band gap, the fluoride analogue of CHC does not exhibit a similarly strong intrinsic fluorescence.30,31 Nonetheless, CHF is a promising candidate for non-hygroscopic phosphors, if doped with Mn4+, as luminescence activator.32,33 In this work, we discuss two distinct synthetic approaches to obtain CHC microcrystals and CHF nanocrystals. Due to the chemical differences between the starting materials (CsCl and CsF), an emulsion synthesis is used to obtain fluorescent CHC microcrystals and a non-aqueous heating-up synthetic strategy to obtain surface functionalised, wide band gap pure and europium- or manganese-doped CHF nanocrystals is proposed. Products are studied with respect to their structure, their surface chemistry, and their luminescence.
m1 space group (Fig. 1).29 Due to its higher band gap, the fluoride analogue of CHC does not exhibit a similarly strong intrinsic fluorescence.30,31 Nonetheless, CHF is a promising candidate for non-hygroscopic phosphors, if doped with Mn4+, as luminescence activator.32,33 In this work, we discuss two distinct synthetic approaches to obtain CHC microcrystals and CHF nanocrystals. Due to the chemical differences between the starting materials (CsCl and CsF), an emulsion synthesis is used to obtain fluorescent CHC microcrystals and a non-aqueous heating-up synthetic strategy to obtain surface functionalised, wide band gap pure and europium- or manganese-doped CHF nanocrystals is proposed. Products are studied with respect to their structure, their surface chemistry, and their luminescence.
 | ||
| Fig. 1 Crystal structures (a) model of trigonal Cs2HfF6 crystal structure (ICSD 25600), (b) model of cubic K2PtCl6-type Cs2HfCl6 crystal structure (ICSD 2582). Caesium ions are depicted in blue, hafnium fluoride distorted octahedra in red and golden, hafnium chloride octahedra in green and grey. | ||
Results and discussion
Synthesis
CHF nanoparticles were obtained via a hafnium oleate precursor in a bottom-up synthesis using octadecene and oleic acid as the reaction solvents. Hafnium oleate was previously reported in the literature, but directly used in further synthetic steps without isolation.34,35 Here, we isolated it and used it as a precursor instead of hafnium chloride, to avoid the presence of chloride ions in the reaction mixture (see ESI,† Fig. S1 for its IR-spectrum). Fluorination was achieved by adding CsF in methanol before heating the reaction mixture to 160 °C under N2 flux for one hour as shown in Scheme 1. | ||
| Scheme 1 Exchange reaction for the CHF-particle formation. | ||
The chemicals used in this procedure are less toxic than the aqueous HF solutions commonly employed for synthesising Cs2HfF6 in the literature.32,36,37 Moreover, using corrosive HF solutions leads to the formation of microparticles, while the synthesis route reported here yields oleic acid functionalised nanoparticles, as expected for non-aqueous syntheses. Nanoparticle doping is achieved by adding a solution of europium nitrate or manganese acetate in methanol prior to heating the reaction to 160 °C at a concentration of 2.5 mol% with respect to hafnium. The residual doping concentrations were evaluated using energy dispersive X-ray spectroscopy (EDX) (see ESI,† Fig. S2 and S3). According to this analysis the doping was 1.2 ± 0.33 and 1.1 ± 0.15 mol% for europium and manganese doped products, respectively (see ESI,† Tables S1 and S2), as often observed in wet chemical syntheses of inorganic materials. The presence of the dopants had a small effect on the lattice parameter a, increasing it to 6.42 Å in CHF-Eu and CHF-Mn from 6.4 Å in CHF (reference value 6.39) (see ESI,† Table S3).29
A similar synthetic strategy cannot be applied to obtain CHC crystals. Due to the very low solubility of CsCl in methanol, other alcohols or other organic solvents, even a hot-injection synthesis similar to the one reported by Abfalterer et al. for Cs2ZrCl6 did not enable the formation of CHC, with CsCl as the only precipitated product.6 This suggests, that in organic solvents, the difference in solubility of CsCl and HfCl4 is large enough, that a reaction to form CHC cannot proceed. Therefore, we designed an alternative synthetic strategy towards CHC, where HfCl4 and CsCl was dissolved in water, and an emulsion of this precursor solution was formed using Triton X-100 in toluene. Consequently, the water was evaporated leading to the precipitation of CHC microcrystals.
Here, a relative excess of HfCl4 was used, as hafnium has a higher affinity for the Triton X-100 than caesium. If stoichiometric quantities were used, a mixture of CHC and CsCl formed indicating a hafnium deficiency (Scheme 2).
 | ||
| Scheme 2 Proposed reaction for the formation of CHC in emulsion. | ||
Relatively small chemical differences, for example in the solubility of CsF and CsCl can dramatically change the viability of a synthetic pathway. This means that CHC cannot be obtained in a reaction in organic solvents like CHF. However, by drastically changing the synthetic conditions from an organic solvent system to an emulsion system containing toluene, surfactant, and water, the desired product CHC can be obtained.
Structural characterisation
The crystal structures of CHF and CHC are elucidated using XRD. The observed diffraction peaks match the references for Cs2HfF6 and Cs2HfCl6 and show no crystalline impurities (Fig. 2(d) and (g)).29,38 Europium doping at 2.5 mol% nominal concentration leads to an impurity corresponding to a cubic double perovskite lattice (K2PtCl6-type), while 2.5 mol% nominally manganese doped CHF is obtained without impurity (Fig. 2(a)). SEM and TEM are used to characterise the morphology of the products (Fig. 2(b), (c), (e), (f), (h), (i) and ESI,† Fig. S4). CHF, CHF-Eu and CHF-Mn consist of nanopowders with particle sizes ranging from 50 to 100 nm. According to Scherrer analysis the average sizes of CHF, CHF-Eu and CHF-Mn crystallites are 60, 25 and 40 nm, respectively. The microparticles of CHC are in the μm range and agglomerates of up to 10 μm are observed. This corresponds to a size difference of one order of magnitude comparing CHF and CHC particle sizes. Scanning transmission electron microscopy (STEM) images of CHF particles seen in Fig. 2F further confirms the particle morphology and size, which agrees well to the size obtained by the Scherrer equation (60 nm). Imaging the crystal structure directly using TEM is not possible, as the lattice decomposes at high magnifications.39,40 CHC particles are also imaged using TEM (Fig. 2(i)). No large crystalline areas can be detected at the particle edge. Since large crystallites are present in the samples (see XRD Fig. 2(g)), amorphous regions in the outer shell of highly crystalline cores are possible.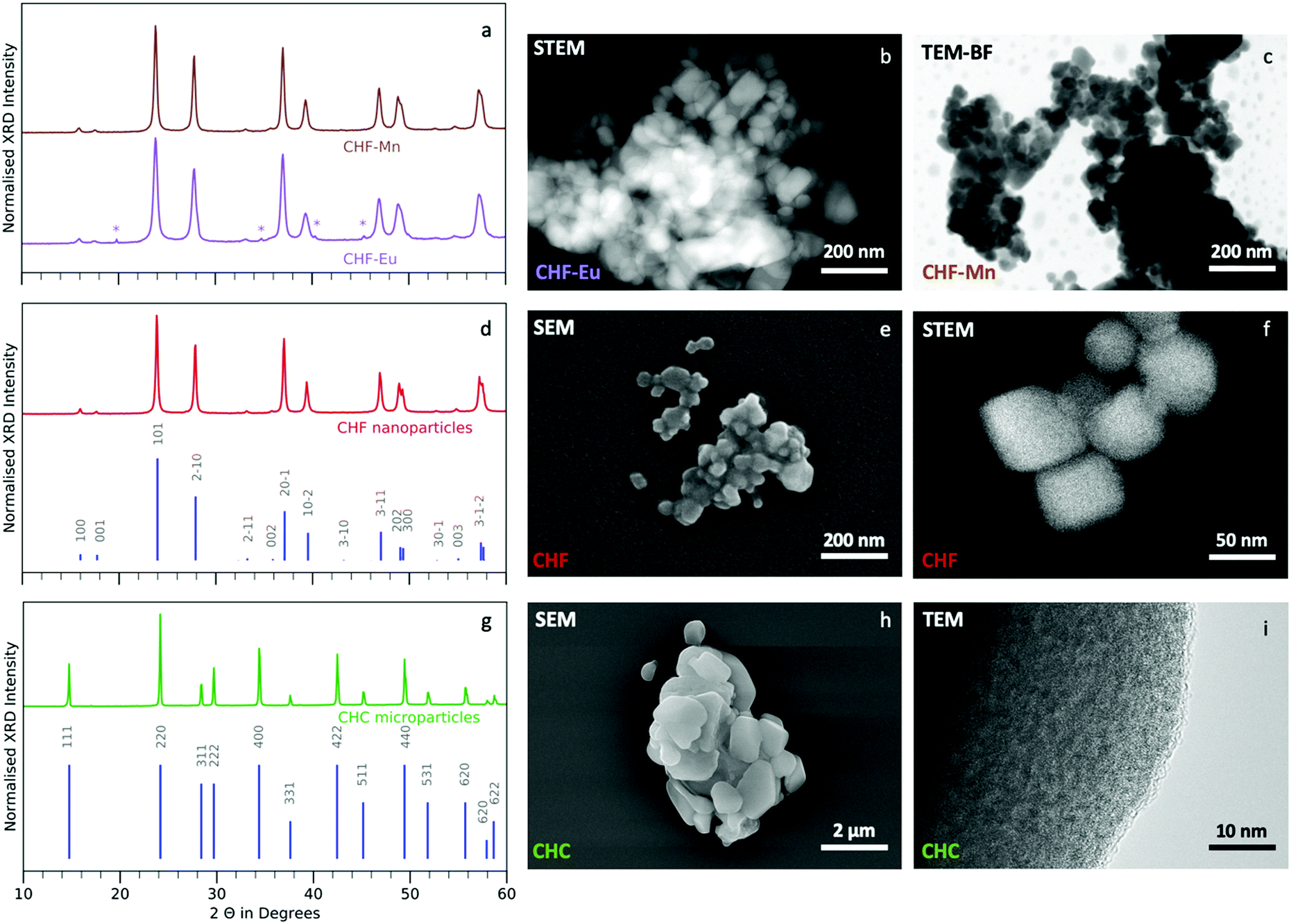 | ||
| Fig. 2 Structural characterisation (a) XRD of CHF-Eu and CHF-Mn, the peaks corresponding to a cubic impurity (K2PtCl6-type) in CHF-Eu are marked by an asterisk (*), (b) STEM image of CHF-Eu, (c) bright field TEM (TEM-BF) image of CHF-Mn, (d) CHF diffractogram with reference ICSD Coll. Code 25600, (e) SEM image of CHF, (f) STEM image of CHF nanoparticles, (g) CHC diffractogram with PDF reference 00-032, (h) SEM image of CHC microcrystals, (i) TEM image of CHF microcrystal. | ||
Surface chemistry
The surface chemistry of the nano- and microparticles presented are evaluated using IR-spectroscopy. Dispersions and solids are also studied using UV-visible spectroscopy and DLS.CHF nanoparticles are functionalised with oleic acid surface groups in the synthesis. This is confirmed by comparing their IR-spectrum with a sodium oleate reference (Fig. 3(a)). The strong peak at 467.5 cm−1 matches the energy range in which Lane et al. reported a ν3 transition of CHF (478 cm−1 and a shoulder at 491 cm−1), reported as well by Forrest et al. in a similar range (490 and 498 cm−1).41,42 Moreover, this peak is significantly higher than the allylic hydrogen stretches at 2900 cm−1 originating from the tails of the oleic acid surface groups. The content of water and organics was determined using thermogravimetric analysis (TGA, Fig. 4a) to be only 1.4%, based on the weight loss at 240 °C. This explains the higher IR absorption by the inorganic part of the sample, which is the major component by weight, with respect to the organic one.
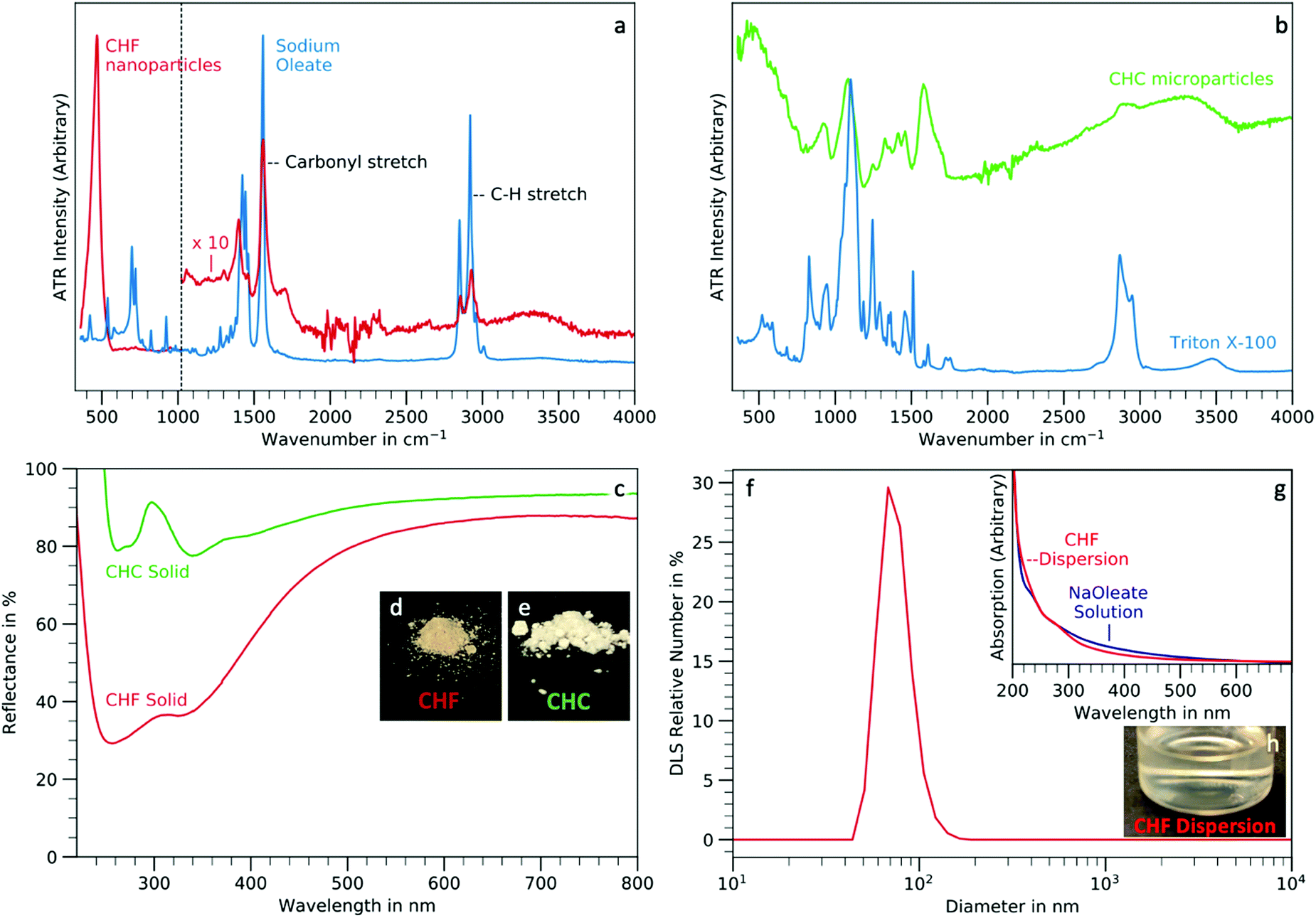 | ||
| Fig. 3 Surface chemistry. (a) ATR-IR-spectra of CHF nanoparticles and sodium oleate, the CHF spectrum (red) was multiplied by 10 in the range 1020–4000 cm−1, (b) ATR-IR-spectra of CHC microparticles and Triton X-100 reference, (c) reflection spectra of CHF powder and CHC powder, (d) photo of solid CHF powder, (e) photo of solid CHC powder, (f) DLS number distribution of a CHF dispersion in hexane, (g) absorption spectrum of a CHF nanoparticle dispersion in hexane and sodium oleate in water as reference, (f) dispersion of CHF in hexane (12 mg mL−1). | ||
 | ||
| Fig. 4 Heat treatment (a) TGA (dashed line) and DSC (solid line) of CHF nanoparticles in air (orange) and N2 (dark red), after being stored in atmospheric conditions for more than 4 weeks, (b) TGA (dashed line) and DSC (solid line) of CHC nanoparticles in air (light green) and N2 (dark green), after being stored in atmospheric conditions for more than 4 weeks, (c) XRD of CHF, CHF heated to 240 °C and 600 °C for 1 h, (d) XRD of CHC, CHC heated to 240 °C and 600 °C for 1 h, peaks corresponding to CsCl were marked with a circle (o), peaks corresponding to HfO2 were marked with an asterisk (*), (e) ATR-IR-spectra of CHF as synthesised, heated to 240 °C and to 600 °C for 1 h, spectra between 1020 and 4000 cm−1 were multiplied by a factor of 10, (f) ATR-IR-spectra of CHC as synthesised, heated to 240 °C and to 600 °C for 1 h. | ||
On the other hand, CHC microparticles are obtained in the presence of Triton X-100. A comparison of the IR-spectra of the CHC microparticles and pure Triton X-100 shows the presence of the surfactant in the product, together with water (Fig. 3(b)). The IR-spectrum also shows, that the CHC product contains more water than CHF after both samples were stored at room temperature in air for several weeks, even though it has a smaller surface area due to its larger particle size. This is also confirmed by TGA analysis (Fig. 4(a)): the CHF sample only loses 0.5% mass when heated to 200 °C, where weight is primarily due to the evaporation of physisorbed water, while the CHC microparticles lose 1.9% mass in the same range.
The UV-visible absorption of the dry nano- and micropowders is estimated by means of diffuse reflectance spectra (Fig. 3(c)). The spectrum obtained from the CHf nanopowder shows a broad double peak between 200 and 400 nm. The higher absorption of the CHF nanopowder in the blue region, peaking at 260 and 340 nm, explains the yellow-orange colour of the sample (Fig. 3(d)). CHC, on the other hand, is a white powder, as displayed by the high reflectance between 400 and 800 nm (Fig. 3(e)). Weak absorption bands at 260 and 340 nm are observed also in this material. Below 220 nm the reflectance exceeded 100%. This effect is due to the sample fluorescing in this range, leading to a conversion of 220 nm light to 375 nm (Fig. 4(b)).
The CHF nanoparticles are well dispersible in hexane after the synthesis due to their oleate ligand shell as demonstrated by DLS (Fig. 3(f) and ESI,† Fig. S5). The DLS number distribution peaked at 68.2 nm, which is in good agreement with the particle size obtained by SEM (50–100 nm) and by Scherrer analysis (60 nm). CHF particles in dispersion were studied by UV-visible spectroscopy (Fig. 3(h)). The pale-yellow colour may derive from oleate groups as sodium oleate in water at similar concentration shows a similar absorption profile as the CHF nanoparticle dispersion (Fig. 3(g)).
Thermal stability
To predict the processability of the powder products, their thermal evolution was studied using TGA. Calcinations at 240 °C and 600 °C were carried out to study the thermal stability of CHC and CHF.TGA curves of CHF in air and N2 show that in air, CHF loses 1.4% of its mass at 240 °C, while in N2 a similar loss of 1.5% is observed at 420 °C. The mass loss in air corresponds to an exothermic peak in differential scanning calorimetry (DSC) at 240 °C. A CHF sample was calcined in air at 240 °C, after this treatment the inorganic portion of the sample remains unchanged (XRD Fig. 4(c)). However, as shown by IR-spectroscopy, the allylic C–H stretches at 2900 cm−2 disappear, while the rest of the spectrum remains unchanged (Fig. 4(e)). This suggests that the organic tails of the oleate surface groups oxidise at this temperature. Similar observations have been reported for oleic acid functionalised nanoparticles in the literature.43,44 The mass loss in N2 is due to the oxygen-free decomposition of the organic ligands in the CHF nanoparticles and is accompanied by a broad endothermic peak in the DSC. In the TGA of CHF in Fig. 4(a) the sample weight does not significantly change between 420 and 600 °C in N2 and 240 and 600 °C in air. However, the endothermic peak in the DSC at 500 °C shows that the material changes at this temperature in both atmospheres possibly due to eutectic melting of Cs2HfF6 which has been reported for CHC (Fig. 4(b)).45–47 Nikl et al. attribute the eutectic melting to the formation of gaseous HfCl4 in the presence of the solids Cs2HfCl6 and CsCl. This phenomenon is independent of the presence of oxygen and can be ascribed solely to the components of the inorganic crystal. The eutectic melting of Cs2ZrF6, which is chemically very similar to Cs2HfF6 and crystallises in the same space group, has been reported at 646 °C.29,48 The thermal dissociation of Cs2HfF6 has been reported at 1076 °C.49 Compared to this, both the eutectic melting (500 °C) and the decomposition (700 °C) occur at much lower temperatures in the CHF nanopowders presented here. This is possibly due to nanometric particle sizes occurring in the present study. When the samples are heated to 600 °C in air, a decomposition of the inorganic crystal takes place, accompanied by broad endothermic peaks in the DSC. The decomposition could be confirmed using XRD, where all the peaks are altered after heating to 600 °C, showing that the components of the nanocrystals form a variety of oxide species. The complete change of the material is also shown in the IR-spectrum (Fig. 4(c) and (e)). Other similar halide materials have been studied with respect to their thermal decomposition in N2 and decomposition temperatures have been reported at 670 to 800 °C for Cs2SiF6 and 775 °C for Cs2HfCl6.46,47,50 This decomposition is ascribed to the formation of CsF and SiF4 and CsCl and HfCl4 respectively.
The thermal study of CHC in air and N2 shows a mass loss due to the evaporation of water observed below 150 °C (Fig. 4(b)). The content of organics starts to be depleted at 240 °C in air, as was visible in the IR-spectra of samples calcined at 240 °C, which lack the C–H stretches at 2900 cm−1. Moreover, the XRD (Fig. 4(d)) of the sample calcined at 240 °C shows peaks corresponding to CsCl. The most pronounced mass loss is observed at 390 °C accompanied by endothermic peaks in both air and N2. This possibly derives from the evaporation of HfCl4 and Triton X-100 surface groups.51 Triton X-100 is not oxidised, since this would lead to an exothermic feature and a CO stretch in the IR-spectrum at 1700 cm−1 as observed by Mitsuda et al.51 The sharp endothermic peak, which occurs at 597 °C in both air and N2, is attributed to the eutectic melting of Cs2HfCl6 leading to the formation of CsCl(l) and HfCl4(g). The value of 597 °C is in a similar range as the values reported by Nikl et al. (586 °C) and Asvestas et al. (591 °C) for experiments done in N2 and argon respectively.45,47 The decomposition of the samples occurs from 700 °C onwards, forming CsCl and HfCl4. In air, the DSC indicates an exothermic process, due to the oxidation of hafnium. This is further confirmed by XRD of CHC calcined at 600 °C in air, showing the presence of CsCl and monoclinic HfO2 crystallites (Fig. 4(d)). In N2 atmosphere, the DSC shows an endothermic peak at 760 °C, which is due to the melting of Cs2HfCl6 and the decomposition of the sample to liquid CsCl and gaseous HfCl4. This effect is observed at higher temperatures by Nikl et al. (775 and 801 °C), which is most probably due to the higher purity of their samples.47
The thermal stability of CHF nanopowders and CHC micro-powders is studied using TGA/DSC and calcination experiments. It is shown that CHF contains less water than CHC even though it has a higher surface area due to its smaller particle size. This could be an indication that CHF is less hygroscopic than CHC. Moreover, CHF is more thermally stable than CHC, retaining its crystal structure after calcining at 240 °C in air, while CHC transforms to a mixture of CHC and CsCl after a heat treatment at 240 °C. IR-spectroscopy shows the partial decomposition of organic ligands at 240 °C in CHF, leading to the loss of C–H stretches at 2900 cm−1. In CHC the organic ligand is lost due to evaporation. Both samples undergo eutectic melting at 500 and 597 °C for CHF and CHC respectively. This is well known for CHC in the literature, however has not been described extensively for CHF.47 In fact, the temperature of eutectic melting is expected at higher temperatures for CHF, as it was reported at 646 °C for Cs2ZrF6, which is chemically very similar to CHF.48 Calcination at 600 °C in air leads to a total destruction of the halide lattices in both cases, forming various oxide species in the case of CHF, while CsCl and HfO2 are formed in the case of CHC. By studying the thermal evolution of the CHF and CHC particle powders obtained, CHF proved to be less hygroscopic, and showed superior thermal stability up to 240 °C, which was not the case for CHC micropowders.
Optical characterisation
The optical properties of the phosphor powders were studied by PL-PLE. CHF nanoparticles exhibit defect related luminescence, while CHC shows an intrinsic band to band luminescence.Undoped CHF nanoparticles showed weak and broad fluorescence, with emission centred at around 489 nm (Fig. 5(a)) while exciting at 395 nm. Doping with manganese(II) leads to an increase of intensity of the broad emission between 420 and 600 nm without significant shift. However, the sample had to be heated in a solvent mixture of octadecene and oleic acid up to 280 °C for 1 h, since no luminescence could be observed in as synthesised samples. This is likely due to their dark brown colour, leading to strong absorption of the excitation radiation and of the emitted light. The heat treatment in the solvent mixture leads to a loss of the colouration, while the crystal structure of the sample does not change after this treatment (see ESI,† Fig. S6). The stronger luminescence (around double) of Mn(II) doped samples compared to the undoped sample could be due to an increase in defects thanks to the difference in charge between Hf(IV) and Mn(II) (see ESI,† Fig. S7). However, since the luminescence of Mn(II) and Mn(IV) is expected between 575–600 and 600–650 nm, respectively, based on the literature, this seems to suggest that the emission observed in the CHF-Mn powder studied here is not derived from the Mn centres.52,53 Moreover, the PL excitation spectrum does not show any manganese related absorptions between 300 and 550 nm, which could be expected for Mn(II)-dopant based luminescence in a wide band gap material.32,54 Similarly, when doping with europium, an enhanced fluorescence (this time four-fold with respect to the undoped material), similar to the one observed in CHF and CHF-Mn centred at 512 nm is measured (see ESI,† Fig. S7). As for manganese, the difference of oxidation state between Eu(III) and Hf(IV) could lead to an increase in optically active defects in the particles. However, the sharp emission of Eu(III) centres is not observed. Underwood et al. reported the very weak radioluminescence of CHF around 400 nm.30 Even though the size of crystals is not specified in this work, it can be assumed to be larger than the nanocrystals discussed here, as they were also used for single crystal X-ray diffraction. Due to larger crystal sizes, the defect concentration could be lower, leading to much weaker luminescence. In fact, similar broad defect related emission bands have been reported for different fluoride materials.55–57 Hence, the luminescence we observe in CHF irrespective of doping might, originate from optically active defect sites leading to broad emissions in the visible range. After calcining at 600 °C for 1 h, a complete transformation of the fluoride lattice is observed, likely due to the formation of oxide species (Fig. 4(c) and (d)). This is also observed by looking at the fluorescence of the samples: CHF and CHF-Mn cease showing significant emissions after the heat treatment at 600 °C in air, while CHF-Eu shows a characteristic Eu3+ emission at 615 nm (see ESI,† Fig. S8) observed in some oxides.58 The europium emission could also be observed by exciting through the charge transfer band at 273 nm, indicating that it was directly bound to oxygen (see ESI,† Fig. S9). The lack of the expected luminescence from Mn and Eu dopants cannot be easily explained. However, their effect on the broad luminescence of CHF suggests that the dopant incorporation acts as the primary cause of a lattice perturbation which is responsible for the emission, in which the dopant itself does not directly participate as recombination centre.
 | ||
| Fig. 5 Luminescence (a) photoluminescence excitation and emission spectra of CHF, CHF-Eu and CHF-Mn (heated to 280 °C in an octadecene, oleic acid mixture post-synthesis for 1 h) emission at 515 nm, excitation at 395 nm, the photos show the glow of the samples under 366 nm UV-irradiation, the excitation spectra were corrected with MgO powder as an optically inactive reference, (b) CHC photoluminescence excitation and emission spectra, the emission was fixed at 375 nm and the excitation at 223 nm respectively, the photo shows the CHC powder under 366 nm UV-irradiation, the emission spectrum was corrected with MgO powder as an optically inactive reference. | ||
The fluorescence observed in the CHC micropowders replicates well the one reported for CHC bulk monocrystals.59 In agreement with previous reports on CHC single crystal luminescence, CHC powders show an emission centred at 375 nm (3.3 eV) with a maximum of excitation at 223 nm (5.6 eV) (Fig. 5(b)).18,19
Materials and methods
Chemicals
Hafnium tetrachloride, 99.9% (Abcr, Karlsruhe), sodium oleate, >97% (Tokyo Chemicals Industry, Tokyo), caesium chloride, 99.5% (CsCl, Fluka, Buchs), octadecene, 90%, (Acros, Pittsburgh), oleic acid, 90% (Sigma-Aldrich, Buchs), caesium fluoride, 99.9% (CsF, Sial, Buchs), europium nitrate hexahydrate, 99.9% (Eu(NO3)2·6H2O, Abcr, Karlsruhe), manganese acetate, 98%, (Sigma Aldrich, Buchs), iron(II) stearate (Tokyo Chemicals Industry, Tokyo), methanol (Sigma-Aldrich, Buchs), hexane (Sigma-Aldrich, Buchs), toluene (VWR, Radnor), ethanol (Fisher Scientific, Pittsburgh), 2-propanol (Sigma-Aldrich, Buchs), Triton X-100 (Sigma-Aldrich, Buchs) and diethyl ether (VWR, Pittsburgh) were used as received without further purification.Synthesis
Characterisation
Conclusions
A synthetic pathway for the controlled formation of CHF nanoparticles in organic solvents without the use of HF is demonstrated here. The synthesis led to CHF nanocrystals through fluorination by CsF in milder and safer conditions. On the other hand, CHC was synthesised as a microcrystalline powder by an emulsion approach, leading to powder products in an oil phase. As predicted, while CHC starts to decompose at 240 °C, CHF is less hygroscopic and more thermally stable in air.CHF nanocrystals showed a relatively weak and broad emission in the visible range. The doping with Mn and Eu did not lead to the expected dopant-related luminescence. Instead, the presence of dopants only led to a significant increase of the luminescence observed in the undoped material. CHC microparticles showed optical properties like those reported for the single crystals, with a bright emission centred at 375 nm, while exciting at 223 nm.
The syntheses proposed here enrich the chemical toolbox towards particle-based Cs Hf halide materials. Importantly, these results also demonstrate the possibility to replicate the optical properties of CHC single crystals in a micropowder, enabling the manufacturing of CHC-based scintillator materials by powder/ink processing.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge ETH Zurich for financial support. The authors are grateful to Dr Elena Tervoort and Dr Alla Sologubenko, ScopeM at ETH Zurich for TEM measurements. Moreover, we thank Viola Vogler-Neuling for DLS measurements and Prof. Lucio Isa of the Department of Materials at ETH for providing access to the instrument.Notes and references
- K. Hong, Q. Van Le, S. Y. Kim and H. W. Jang, J. Mater. Chem. C, 2018, 6, 2189–2209 RSC.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- N. R. Wolf, B. A. Connor, A. H. Slavney and H. I. Karunadasa, Angew. Chem., Int. Ed., 2021, 60, 2–17 CrossRef PubMed.
- D. Kong, D. Cheng, X. Wang, K. Zhang, H. Wang, K. Liu, H. Li, X. Sheng and L. Yin, J. Mater. Chem. C, 2020, 8, 1591–1597 RSC.
- A. Abfalterer, J. Shamsi, D. J. Kubicki, C. N. Savory, J. Xiao, G. Divitini, W. Li, S. Macpherson, K. Gałkowski, J. L. Macmanus-Driscoll, D. O. Scanlon and S. D. Stranks, ACS Mater. Lett., 2020, 2, 1644–1652 CrossRef CAS PubMed.
- S. Liu, Y. Bin, J. Chen, W. Donghui, D. Zheng, K. Qingkun and K. Han, Angew. Chem., Int. Ed., 2020, 59, 21925–21929 CrossRef CAS PubMed.
- G. Xiong, L. Yuan, Y. Jin, H. Wu, Z. Li, B. Qu, G. Ju, L. Chen, S. Yang and Y. Hu, Adv. Opt. Mater., 2020, 8, 2000779 CrossRef CAS.
- Q. A. Akkerman and L. Manna, ACS Energy Lett., 2020, 5, 604–610 CrossRef CAS PubMed.
- J. Breternitz and S. Schorr, Adv. Energy Mater., 2018, 8, 1802366 CrossRef.
- P. Scherrer and P. Stoll, Z. Anorg. Allg. Chem., 1922, 121, 319–320 CrossRef CAS.
- M. Nikl and A. Yoshikawa, Adv. Opt. Mater., 2015, 3, 463–481 CrossRef CAS.
- M. Nikl, Meas. Sci. Technol., 2006, 17, R37–R54 CrossRef CAS.
- A. Burger, E. Rowe, M. Groza, K. Morales Figueroa, N. J. Cherepy, P. R. Beck, S. Hunter and S. A. Payne, Appl. Phys. Lett., 2015, 107, 143505 CrossRef.
- K. Saeki, Y. Fujimoto, M. Koshimizu, T. Yanagida and K. Asai, Appl. Phys. Express, 2016, 9, 042602 CrossRef.
- S. Kurosawa, S. Kodama, Y. Yokota, T. Horiai, A. Yamaji, Y. Shoji, R. Král, J. Pejchal, Y. Ohashi, K. Kamada, M. Nikl and A. Yoshikawa, J. Instrum., 2017, 12, C02042–C02042 CrossRef.
- R. Král, V. Babin, E. Mihóková, M. Buryi, V. V. Laguta, K. Nitsch and M. Nikl, J. Phys. Chem. C, 2017, 121, 12375–12382 CrossRef.
- E. Rowe, W. B. Goodwin, P. Bhattacharya, G. Cooper, N. Schley, M. Groza, N. J. Cherepy, S. A. Payne and A. Burger, J. Cryst. Growth, 2019, 509, 124–128 CrossRef CAS.
- V. Vanecek, R. Kral, J. Paterek, V. Babin, V. Jary, J. Hybler, S. Kodama, S. Kurosawa, Y. Yokota, A. Yoshikawa and M. Nikl, J. Cryst. Growth, 2020, 533, 125479 CrossRef CAS.
- S. Lam, C. Guguschev, A. Burger, M. Hackett and S. Motakef, J. Cryst. Growth, 2018, 483, 121–124 CrossRef CAS.
- R. Liu, W. Zhang, W. Liu and G. Li, Inorg. Chem., 2021, 60, 10451–10458 CrossRef CAS PubMed.
- J. Zhu and M. C. Hersam, Adv. Mater., 2017, 29, 1603895 CrossRef PubMed.
- J. Liu, B. Liu, Z. Zhu, L. Chen, J. Hu, M. Xu, C. Cheng, X. Ouyang, Z. Zhang, J. Ruan, S. He, L. Liu, M. Gu and H. Chen, Opt. Lett., 2017, 42, 987–990 CrossRef CAS PubMed.
- B. K. Cha, S. J. Lee, P. Muralidharan, D. K. Kim, J. Y. Kim, G. Cho, S. Jeon and Y. Huh, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 652, 717–720 CrossRef CAS.
- B. K. Cha, J. Hyung Bae, C. H. Lee, S. Chang and G. Cho, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 633, S297–S299 CrossRef CAS.
- A. Knapitsch, E. Auffray, C. W. Fabjan, J. L. Leclercq, P. Lecoq, X. Letartre and C. Seassal, Nucl. Instrum. Methods Phys. Res., 2011, 628, 385–388 CrossRef CAS.
- H. Wei, V. Martin, A. Lindsey, M. Zhuravleva and C. L. Melcher, J. Lumin., 2014, 156, 175–179 CrossRef CAS.
- C. Gumiński, H. Voigt and D. Zeng, Monatsh. Chem., 2011, 142, 211–218 CrossRef PubMed.
- H. Bode and G. Teufer, Z. Anorg. Allg. Chem., 1956, 283, 18–25 CrossRef CAS.
- C. C. Underwood, C. D. McMillen, H. Chen, J. N. Anker and J. W. Kolis, Inorg. Chem., 2013, 52, 237–244 CrossRef CAS PubMed.
- X. Mao, L. Sun, T. Wu, T. Chu, W. Deng and K. Han, J. Phys. Chem. C, 2018, 122, 7670–7675 CrossRef CAS.
- T. Senden, E. J. van Harten and A. Meijerink, J. Lumin., 2018, 194, 131–138 CrossRef CAS.
- Z. Yang, Q. Wei, M. Rong, Z. Yang, Z. Wang, Q. Zhou and Q. Wang, Dalton Trans., 2017, 46, 9451–9456 RSC.
- T. Taniguchi, N. Sakamoto, T. Watanabe, N. Matsushita and M. Yoshimura, J. Phys. Chem. C, 2008, 112, 4884–4891 CrossRef CAS.
- J. Qi and X. Zhou, Colloids Surf., A, 2015, 487, 26–34 CrossRef CAS.
- J. Zhang, L. Liu, S. He, J. Peng, F. Du, F. Yang and X. Ye, Inorg. Chem., 2019, 58, 15207–15215 CrossRef CAS PubMed.
- H. Zhu, C. C. Lin, W. Luo, S. Shu, Z. Liu, Y. Liu, J. Kong, E. Ma, Y. Cao, R. S. Liu and X. Chen, Nat. Commun., 2014, 5, 4321 CrossRef PubMed.
- S. Maniv, J. Appl. Crystallogr., 1976, 9, 245 CrossRef.
- S. Chen and P. Gao, J. Appl. Phys., 2020, 128, 010901 CrossRef CAS.
- Z. Dang, J. Shamsi, F. Palazon, M. Imran, Q. A. Akkerman, S. Park, G. Bertoni, M. Prato, R. Brescia and L. Manna, ACS Nano, 2017, 11, 2124–2132 CrossRef CAS PubMed.
- A. P. Lane and D. W. A. Sharp, J. Chem. Soc. A, 1969, 2942–2945 RSC.
- I. W. Forrest and A. P. Lane, Inorg. Chem., 1976, 15, 265–269 CrossRef CAS.
- P. Roonasi and A. Holmgren, Appl. Surf. Sci., 2009, 255, 5891–5895 CrossRef CAS.
- T. Muthukumaran and J. Philip, J. Alloys Compd., 2016, 689, 959–968 CrossRef CAS.
- D. A. Asvestas, P. Pint and S. N. Flengas, Can. J. Chem., 1977, 55, 1154–1166 CrossRef CAS.
- G. J. Kipouros and S. N. Flengas, Can. J. Chem., 1983, 61, 2183–2188 CrossRef CAS.
- R. Král, P. Zemenová, V. Vaněček, A. Bystřický, M. Kohoutková, V. Jarý, S. Kodama, S. Kurosawa, Y. Yokota, A. Yoshikawa and M. Nikl, J. Therm. Anal. Calorim., 2020, 141, 1101–1107 CrossRef.
- G. D. Robbins, R. E. Thoma and H. Insley, J. Inorg. Nucl. Chem., 1965, 27, 559–568 CrossRef CAS.
- L. Gruzdiewa, J. Rak and J. Blazejowski, J. Alloys Compd., 1994, 210, 63–70 CrossRef CAS.
- J. Zachara and W. Wišniewski, J. Therm. Anal., 1995, 44, 929–935 CrossRef CAS.
- K. Mitsuda, H. Kimura and T. Murahashi, J. Mater. Sci., 1989, 24, 413–419 CrossRef CAS.
- Q. Zhou, L. Dolgov, A. M. Srivastava, L. Zhou, Z. Wang, J. Shi, M. D. Dramićanin, M. G. Brik and M. Wu, J. Mater. Chem. C, 2018, 6, 2652–2671 RSC.
- X. Yuan, S. Ji, M. C. De Siena, L. Fei, Z. Zhao, Y. Wang, H. Li, J. Zhao and D. R. Gamelin, Chem. Mater., 2017, 29, 8003–8011 CrossRef CAS.
- E. Song, S. Ye, T. Liu, P. Du, R. Si, X. Jing, S. Ding, M. Peng, Q. Zhang and L. Wondraczek, Adv. Sci., 2015, 2, 1500089 CrossRef PubMed.
- A. N. Novikov, V. S. Kalinov, E. I. Pavalanski, A. P. Stupak, I. E. Svitsiankou and A. P. Voitovich, J. Lumin., 2019, 209, 244–250 CrossRef CAS.
- C. Li, Z. Xu, D. Yang, Z. Cheng, Z. Hou, P. Ma, H. Lian and J. Lin, CrystEngComm, 2012, 14, 670–678 RSC.
- X. Zhang, Z. Quan, J. Yang, P. Yang, H. Lian and J. Lin, Nanotechnology, 2008, 19, 075603 CrossRef PubMed.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- B. Kang and K. Biswas, J. Phys. Chem. C, 2016, 120, 12187–12195 CrossRef CAS.
- H. Fu, G. Yang, S. Gai, N. Niu, F. He, J. Xu and P. Yang, Dalton Trans., 2013, 42, 7863–7870 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc05734k |
| This journal is © The Royal Society of Chemistry 2022 |