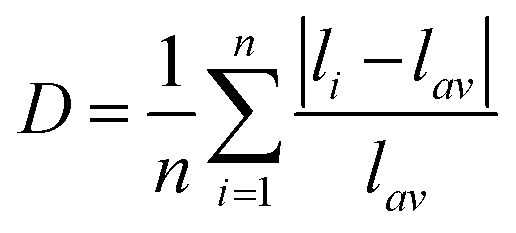DOI:
10.1039/D1TC05680H
(Paper)
J. Mater. Chem. C, 2022,
10, 2095-2102
Coordination units of Mn2+ modulation toward tunable emission in zero-dimensional bromides for white light-emitting diodes†
Received
25th November 2021
, Accepted 6th January 2022
First published on 7th January 2022
Abstract
Organic–inorganic metal halides have become a multifunctional platform for manipulating photoluminescence due to highly efficient and tunable emissions, especially for lead-free Mn2+-based halides. Herein, the zero-dimensional (0D) bromides of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 with different coordination environments were designed and synthesized by a solvent evaporation method. They exhibit green and red broadband emission peaks at 528 nm and 627 nm with high photoluminescence quantum yields of 86.83% and 61.91%, respectively, which are attributed to the d–d transition (4T1(G) → 6A1(S)) of [MnBr4]2− tetrahedral and [Mn3Br12]6− octahedral units. The cases emphasize the effect of organic ligands on the intrinsic emissions of Mn2+ ions, thereby revealing the luminescence mechanism of Mn2+ ions in 0D isolated structures through the Tanabe–Sugano (TS) energy diagram. Thanks to their bright and stable emissions, the fabricated white light-emitting diode (LED) based on (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 provides an outstanding color rendering index (Ra) of 90.8 at a correlated color temperature (CCT) of 3709 K, along with the CIE chromaticity coordinates of (0.3985, 0.3979) and a luminous efficacy of 51.2 lm W−1. This work aims at clarifying the relationship between the coordination units of Mn2+ and tunable emissions, and in particular, proposes a new strategy to explore phosphors excited by blue light for white LEDs.
1. Introduction
Organic–inorganic metal halides (OIMHs) formed by self-assembly of organic cations and inorganic anions have received extensive interest in many fields, such as light-emitting diodes (LEDs), photodetectors, ferroelectrics, solar cells, and so on.1–6 Compared to all-inorganic metal halides, OIMHs have a high degree of design flexibility owing to the various structural dimensions and abundant chemical components.7–9 Among them, as a kind of hybrid material composed of organic cations and spatially isolated anions, zero-dimensional (0D) OIMHs provide an excellent platform for the development of high-efficiency emitting materials;10–12 in particular, 0D Mn2+-based OIMHs have attracted much attention.13–16
Although most of the reported OIMHs possess excellent luminescence properties, the toxicity of Pb2+ and instability limit their practical applications, which lead to the development of lead-free OIMHs becoming imminent.17–20 In comparison, the Mn2+-based OIMHs present the advantages of environmental friendliness, good stability and tunable emission. As is well known the transition metal ion Mn2+ has a 3d5 electronic configuration and generally exhibits visible (vis) photoluminescence (PL) originating from the 4T1 → 6A1 transition, which strongly depends on the crystal field surroundings of Mn2+ in the host lattice.21,22 Octahedrally coordinated Mn2+ shows an orange to red emission with a large full width at half maximum (FWHM > 60 nm), while tetrahedrally coordinated Mn2+ exhibits a green–yellow emission (500–550 nm) with a small FWHM of 25–60 nm.23–27 So far, Mn2+-doped metal halides with orange–red emissions have been reported the most,28–34 but unfortunately, they cannot be well applied in the field of solid-state lighting due to the shortcoming that almost all of them cannot be excited by blue light, resulting in a low efficiency for white LED devices. Most of the Mn2+-based OIMHs exhibit a narrow-band green emission.13–15,35–39 Even if these 0D analogues show a high-efficiency Mn2+ emission with near-unity photoluminescence quantum yields (PLQYs), the relatively small FWHM (20–60 nm) of the PL peak makes it impossible to endow high-performance white LED devices with a high color rendering index (Ra) for the application of solid-state lighting.40,41 In addition, the 0D Mn2+-based OIMHs with a highly efficient red emission have also been reported and popularized recently. However, only a small amount of 0D Mn2+-based OIMHs that can emit red light was investigated so far,3,24,42,43 and there is no comparative analysis of their luminescence mechanism by binding to that of 0D Mn2+-based OIMHs with green light emission.
Organic ligands, as a key component to modify Mn2+ coordination units in OIMHs, play an irreplaceable role in the construction of their electronic band structures and crystal structures. In this work, we employed organic cations including guanidine hydrobromide and 1,1,3,3-tetramethylguanidine hydrobromide, and synthesized two kinds of 0D Mn2+-based bromides with broadband emission by reacting them with MnBr2 respectively. Efficient room-temperature phosphorescence originating from the d−d transition (4T1–6A1) of Mn2+ is demonstrated in these compounds and shows a strong dependence on the crystal field surroundings of Mn2+. Furthermore, we elaborated the electronic features and optical absorption according to the Tanabe–Sugano (TS) diagram of the 3d5 electron orbital of Mn2+ and extended the principle by employing organic ligands to tune the Mn2+ emission in 0D bromides. The as-fabricated white LED device with a high Ra of 90.8 at a correlated color temperature (CCT) of 3709 K demonstrates that (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 can be promising as rare-earth-free and lead-free phosphors in the field of solid-state lighting. Our realizable cases provide a deep understanding about the coordination units of Mn2+ toward tunable emission and propose a new way to explore high-performance OIMHs pumped by blue light for white LEDs.
2. Experimental section
2.1 Materials and preparation
Guanidine hydrobromide (CH6N3Br, 98%, Sigma-Aldrich), 1,1,3,3-tetramethylguanidine (C5H13N3, 98%, Energy chemical), manganese bromide tetrahydrate (MnBr2·4H2O, 98%, Aladdin), hydrobromic acid (HBr, 40%, Aladdin), acetonitrile (CH3CN, 99.5%, Kermel) and ethanol (C2H5OH, 99.7%, Guangfu) were all used as received.
The single crystals of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 were synthesized by a solvent volatilization method. For (C5H14N3)2MnBr4, 1.1518 g (10 mmol) of C5H13N3 were dissolved in 3 mL of ethanol, then 2 mL of hydrobromic acid (40%) were dropped slowly into it. Again, 1.434 g (5 mmol) of MnBr2·4H2O were added into the solution, followed by placing the solution on the magnetic stirrer and stirring it until a clear solution was formed; then the solution was slowly evaporated at room temperature and the single crystals could be obtained after about one week. The obtained crystals were washed repeatedly three times with C2H5OH, and then filtered with a Büchner funnel. Finally, the crystals were dried at 50 °C in an oven for 24 hours. For (CH6N3)2MnBr4, 0.70 g (5 mmol) of CH6N3Br and 0.717 g (2.5 mmol) of MnBr2·4H2O were thoroughly dissolved in 5 mL of acetonitrile. The solution was then slowly evaporated at room temperature and the single crystals of (CH6N3)2MnBr4 can be obtained after about one week. The obtained crystals were washed repeatedly three times with CH3CN, and then filtered by using the Büchner funnel. Finally, the crystals were dried at 50 °C in an oven for 24 hours.
2.2 Characterization
The single-crystal X-ray diffraction data of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 were collected at room temperature using the XtaLAB AFC12 X-ray four-circle single crystal diffractometer (Rigaku) equipped with a CCD-detector, a graphite monochromator and a Mo Kα radiation source. The powder X-ray diffraction data of (CH6N3)2MnBr4 and (C5H14N3)2MnBr4 were collected at room temperature with an X-ray diffractometer (Cu-Kα radiation). The photoluminescence (PL) spectra, photoluminescence excitation (PLEX) spectra, photoluminescence emission (PLEM) spectra, fluorescence decay curve and PLQYs were measured using a FLS-920 fluorescence spectrometer (Edinburgh Instruments Ltd. UK). Temperature-dependent emission spectra were measured on the same spectrophotometer installed with a heating apparatus as the heating source. Thermogravimetric analysis (TGA) was performed on a SETARAM 131 LABSYS device under an argon stream at a heating rate of 10 °C min−1 from RT to 800 °C. The emission spectra, correlated color temperature (CCT), luminous efficacy, and CIE coordinates of the LED devices were measured using an integrating sphere spectroradiometer system (ATA-100, Everfine).
2.3 Computational methods
The electronic band structure and density of states (DOS) were calculated by CASTEP based on plane-wave pseudopotential density functional theory (DFT).44 Perdew–Burke–Ernzerhof (PBE) functionals in the generalized gradient approximation (GGA) form were used for electronic structure calculations.45 The optimized norm-conserving pseudopotential in Kleinman–Bylander allows us to use a small plane basis set without compromising the required accuracy of the calculation. A kinetic energy cut off value of 900 eV and a Monkhorst–Pack k-point mesh spanning less than 0.03 Å−1 in the Brillouin zone were chosen. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization scheme was employed and the convergence criteria for the structure optimization were set to 5.0 × 10−5 eV per atom, 0.1 eV per Å, 0.2 GPa and 5.0 × 10−3 GPa which is energy, maximum force, maximum stress and maximum displacement, respectively.
3. Results and discussion
3.1 Synthesis and structure evolution
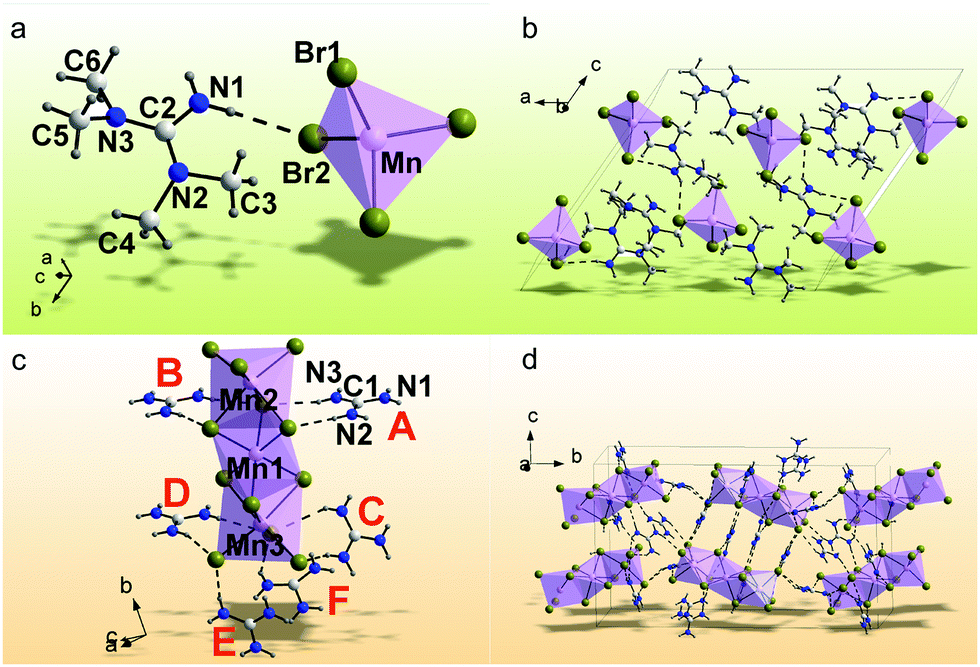
The Mn2+-based bromides of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 were synthesized by a solvent volatilization method, and their single crystal data are collected at room temperature (RT). The orientation matrix and cell parameters of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 were defined and refined for the set of 5077 and 23264 reflections, respectively. The unit cell corresponds to a monoclinic symmetry in both cases. Space groups C2/c and P21/c were determined by utilizing the statistical analysis of the intensities of all the reflections. The absorption correction using spherical harmonics was applied using the ABSPAK program. The crystal structure was solved by using package SHELXS and refined via the anisotropic approach for non-hydrogen atoms using the SHELXL program.46 All the hydrogen atoms of the ligands were positioned geometrically as riding on their parent atoms with d(C–H) = 0.97 Å for the C–H bonds and d(N–H) = 0.86 Å for all other N–H bonds and Uiso(H) = 1.2Ueq (C, N). The main crystal data are shown in Table 1. The crystallographic data are deposited in the Cambridge Crystallographic Data Centre (CCDC # 2094640–2094641). The coordinates of atoms are in Table S1 (ESI†), and the main bond lengths are shown in Table S2 (ESI†). The asymmetric part of the (C5H14N3)2MnBr4 unit cell includes one organic cation of [C5H14N3]2+, one half of Mn2+ and two Br− ions. The Mn2+ ion is coordinated with four Br− ions to form the [MnBr4]2− tetrahedron (Fig. 1a). The tetrahedra are isolated from each other to form a 0D structure (Fig. 1b). There are two N–H⋯Br hydrogen bonds (Table S3, ESI†), which join the organic cations [C5H14N3]2+ with [MnBr4]2− tetrahedra and form a three-dimensional (3D) net. The asymmetric part of the (CH6N3)2MnBr4 unit cell contains six organic cations of [CH6N3]2+, three Mn2+ and twelve Br− ions. All Mn2+ ions are coordinated by six Br− ions to form MnBr6 octahedra (Fig. 1d). These octahedra are linked with each other by sharing faces to form an isolated 0D block (Fig. 1c). There are 33 N–H⋯Br hydrogen bonds (Table S3, ESI†) between the organic cations [CH6N3]2+ and [Mn3Br12]6− polyhedra, all of which stack together to form a 3D net. Fig. S1 (ESI†) shows the PXRD patterns of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4, indicating that the obtained powders are pure phases and are consistent with their single crystal phases, respectively. In addition, the organic functional groups were characterized by FTIR spectra in Fig. S2 (ESI†).47,48 It is observed that (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 both exhibit the absorption peaks at ∼3300 cm−1, ∼1650 cm−1 and ∼1000 cm−1, which are ascribed to the stretching vibration, blending vibration of N–H and the C–N stretching vibration, respectively. Compared with (CH6N3)2MnBr4, (C5H14N3)2MnBr4 possess additional absorption peaks at ∼2950 cm−1, ∼2850−1 and ∼1370 cm−1, which are assigned to the –CH3 asymmetric, symmetric stretching vibrations and deformation vibrations, respectively.
Table 1 Main crystal structure parameters of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4
| Chemical formula |
(C5H14N3)2MnBr4 |
(CH6N3)2MnBr4 |
| Weight, g mol−1 |
606.96 |
494.76 |
| Temperature (K) |
293 |
293 |
| Space group, Z |
C2/c, 4 |
P21/c, 12 |
|
a (Å) |
19.215 (3) |
9.0171 (3) |
|
b (Å) |
9.2604 (7) |
27.9628 (10) |
|
c (Å) |
15.366 (2) |
16.1020 (5) |
|
β (°) |
125.51 (2) |
93.016 (3) |
|
V (Å3) |
2225.6 (6) |
4054.4 (2) |
|
ρ
calc, g cm−3 |
1.811 |
2.432 |
|
R
int
|
0.0496 |
0.0546 |
| 2θmax, ° |
58.34 |
59.18 |
|
R1 [Fo > 4σ(Fo)] |
0.0697 |
0.0508 |
|
wR2 |
0.1913 |
0.1363 |
| Goof |
0.828 |
0.898 |
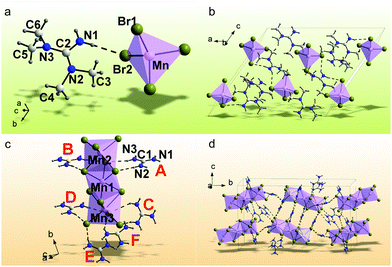 |
| | Fig. 1 Crystal asymmetric structure units of (C5H14N3)2MnBr4 (a) and (CH6N3)2MnBr4 (c). The atoms in the asymmetric part of the unit cell (C5H14N3)2MnBr4 (b) and (CH6N3)2MnBr4 (d) are labeled, the hydrogen bonds in which are presented as dashed lines. The independent [CH6N3]+ cations in the Figure (c) are marked by bold labels A–E. | |
3.2 Photoluminescence tuning and mechanism
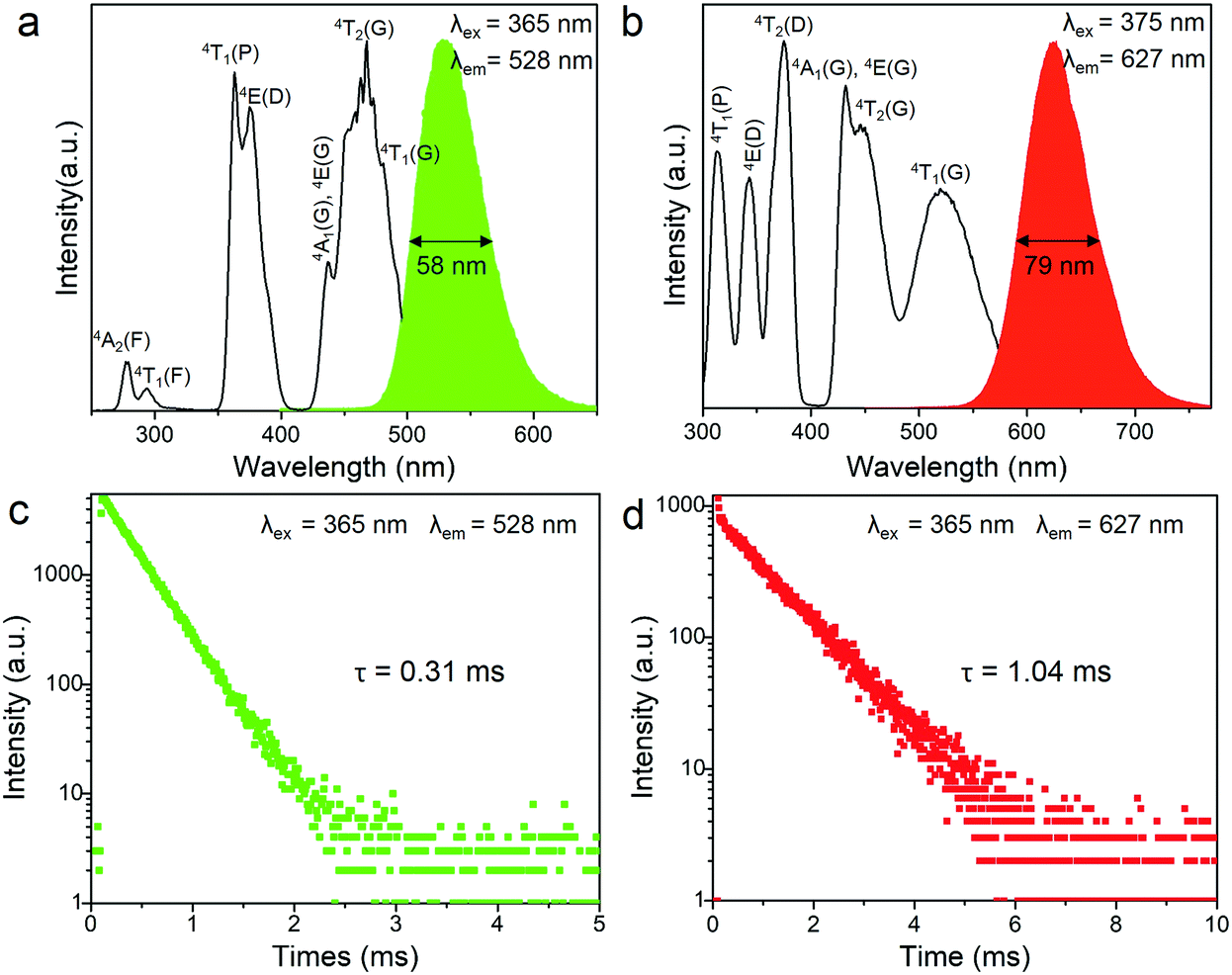
The transition metal ion Mn2+ has a 3d5 electronic configuration. Since the 3d orbital electrons of Mn2+ are exposed in the outer layer, the wavelength position of the emission band of Mn2+ is strongly affected by the crystal field environment. In view of this, the coordination units of Mn2+ can be modified by employing different organic ligands toward tunable emission in 0D bromides. The photoluminescence (PL) and photoluminescence excitation (PLE) spectra of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are shown in Fig. 2a and b. They exhibit a green emission at 528 nm and a red emission at 627 nm under excitation at 365 and 375 nm, respectively, which are originated from the d–d transition (4T1(G) → 6A1(S)) of four coordinated [MnBr4]2− units and six coordinated [Mn3Br12]6− units, respectively. In particular, (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 presents absorption peaks at the blue region, corresponding to the transitions of 6A1 → 4A1(G), 4E(G) and 6A1 → 4T2(G), which enables them to be applied to white LEDs pumped by blue light. The UV-visible absorption spectra of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are shown in Fig. S3 (ESI†). The decay curves monitored at λem = 528 nm and λem = 627 nm were collected under 365 nm excitation, as shown in Fig. 2c and d. They can be fitted by utilizing the following decay equation:49,50| | | I(t) = I0 + A exp(−t/τ) | (1) |
where I(t) and I0 are the luminescence intensity at time t and t » τ, respectively. A is a constant, and τ is the decay time, which is an exponential component. The lifetimes of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are determined to be 0.31 and 1.04 ms, respectively, which possess relatively short lifetimes. Among them, the lifetime value of (C5H14N3)2MnBr4 is consistent with that reported in the literature,14,15 which indicates that the green-light emission originates from the d–d transition (4T1(G) → 6A1(S)) of four coordinated [MnBr4]2− units. The lifetime value of (CH6N3)2MnBr4 has not been proven, but compared with the similar A2BX4 type Mn2+-based chlorides,42,43 its lifetime value is smaller, which is in line with the law of the influence of halogen ions on the lifetime in green-emitting Mn2+-based halides.15 Accordingly, the red emission peaked at 627 nm is attributed to the d–d transition (4T1(G) → 6A1(S)) of six coordinated [Mn3Br12]6− units.
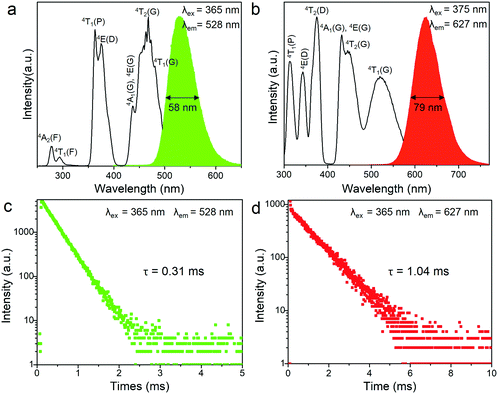 |
| | Fig. 2 The photoluminescence (PL) and photoluminescence excitation (PLE) spectra of (C5H14N3)2MnBr4 (a) and (CH6N3)2MnBr4 (b) at room temperature (RT). Photoluminescence decay curves of (C5H14N3)2MnBr4 (c) and (CH6N3)2MnBr4 (d) at RT under excitation at 365 nm, emission at 528/627 nm. | |
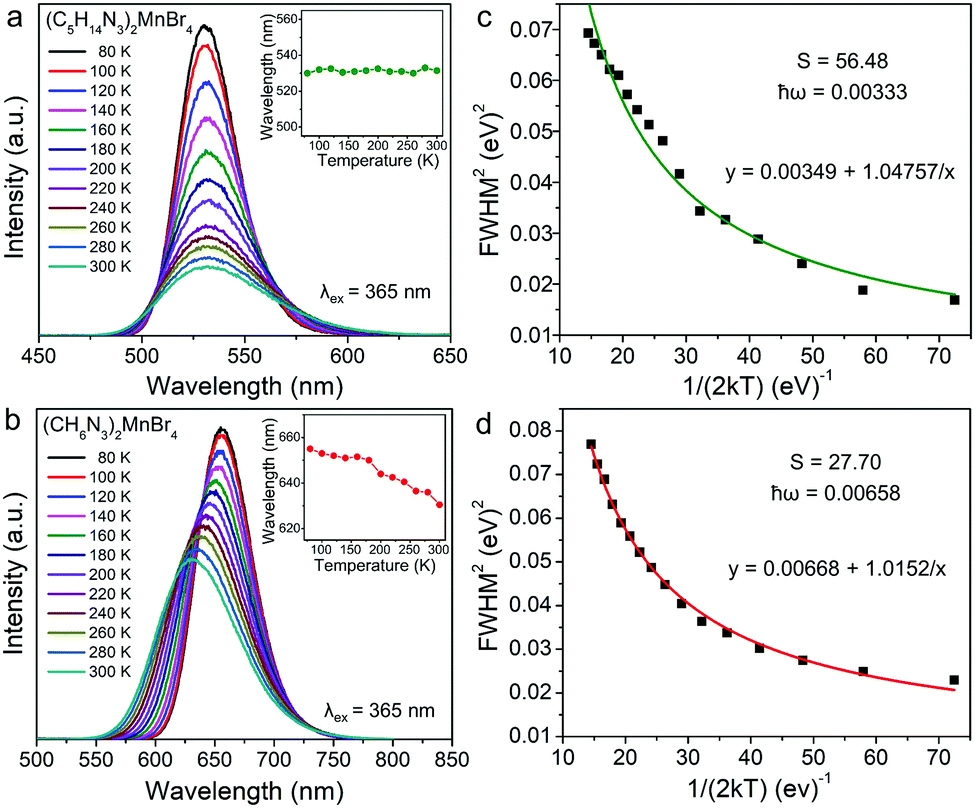
In addition, (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 both exhibit the broadband emissions, with a relatively large FWHM of 58 nm and 79 nm at RT, respectively, which is conducive to the application of such materials in solid-state lighting. Fig. 3a and b show the temperature-dependent PL spectra in the range of 80–300 K. The emission peak position of (C5H14N3)2MnBr4 hardly changed, while the emission peak position of (CH6N3)2MnBr4 showed a blue-shift from 655 nm to 627 nm, which is mainly ascribed to the following two reasons. On the one hand, (CH6N3)2MnBr4 has a high crystal field strength, and the thermally induced host lattice expansion has a significant effect on the crystal field strength, resulting in the value of that reduce significantly.
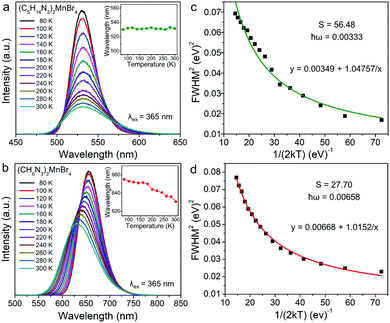 |
| | Fig. 3 Temperature-dependent PL spectra of (C5H14N3)2MnBr4 (a) and (CH6N3)2MnBr4 (b) under 365 nm excitation in the range from 80 to 300 K. Inset: the temperature-dependent emission peak. The FWHM fitting curve of (C5H14N3)2MnBr4 (c) and (CH6N3)2MnBr4 (d) as a function of temperature via the eqn (4) to obtain the Huang–Phys factor S and maximum phonon energy ħω. | |
On the other hand, the crystal expansion reduced the spin–spin coupling energy (Esc) of locally adjacent Mn2+ ions in the crystal such as Frenkel exciton coupling in molecular aggregates. If the separation between Mn ions increases during the process of crystal expansion, the value of Esc value would decrease clearly. With the increase of temperature, the emission peak of (CH6N3)2MnBr4 shows a blue-shift. In addition, the degree of electron–phonon coupling is defined by the Huang–Rhys factor S,37,51,52 which can be solved by the following equation:
| | 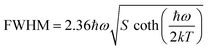 | (2) |
where
ω is the phonon frequency,
ħω is the maximum phonon energy,
k is the Boltzmann constant,
S is Huang–Rhys factor, and
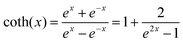
. When
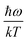
is small enough,
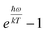
≈
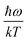
, the following equation can be obtained:
| | 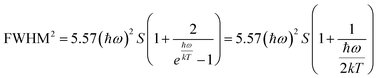 | (3) |
This can be further written as follows:
| | 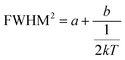 | (4) |
where
a = 5.57 ×
S × (
ħω)
2 and
b = 5.57 ×
S × (
ħω). The Huang–Rhys factor
S reflects the degree of electron–phonon coupling. According to the temperature-dependent PL spectra from 80 to 300 K, the value of
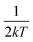
and FWHM
2 can be calculated. Taking
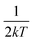
as the horizontal coordinate and FWHM
2 as vertical coordinate, the Huang–Rhys factors
S and
ħω of (C
5H
14N
3)
2MnBr
4 and (CH
6N
3)
2MnBr
4 are obtained by fitting, corresponding to
S = 56.48,
ħω = 0.00333 eV and
S = 27.70,
ħω = 0.00658 eV, and the relatively large Huang–Rhys factor
S indicates a stronger electron–phonon coupling interaction.
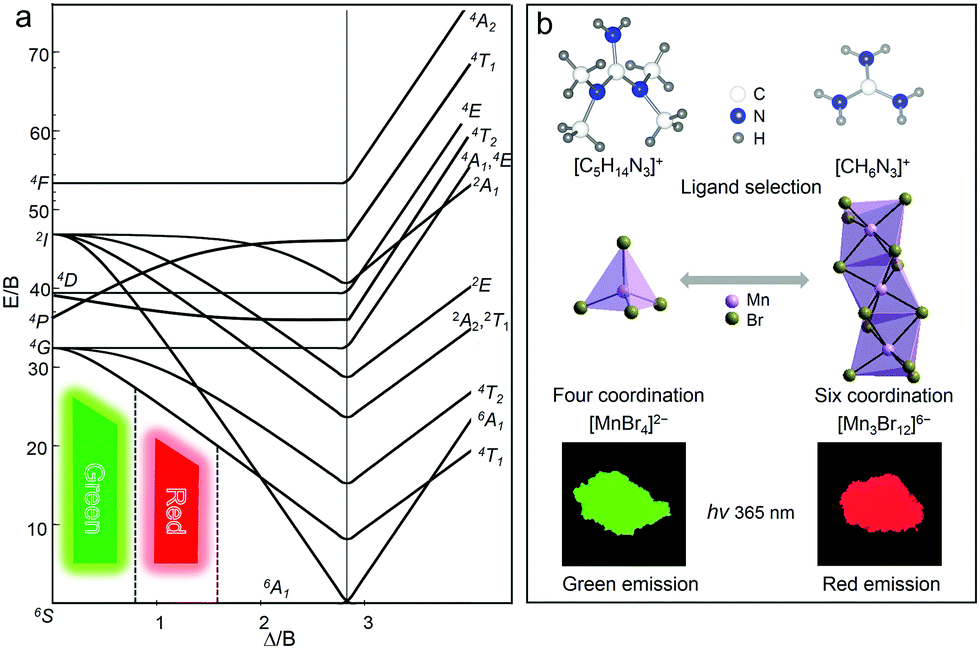
As is well-known, the energy levels of Mn2+ ions have different spin multiplicities, and there is a strong interaction with the crystal field and lattice vibration due to the spatial extension of the d electron wave function, especially under the influence of the two factors including the strength of the crystal field (Δ) and covalency (B) of the Mn–ligand bond. To illustrate the energy level distribution of Mn2+ ions (3d5), a Tanabe–Sugano (TS) diagram is described in Fig. 4a.21,22,53 The electrostatic field generated by the ligand splits the original five degenerate d orbitals of Mn2+ into two or more groups with different energy levels. The ground state electrons transit to different excited states under light irradiation, and then return to the lowest excited state energy level 4T1 through non-radiative relaxation, and finally the electrons return to the ground state with the production of photons. According to the general theory of d-ion emission, the position of the emission peak of Mn2+ can be determined by the crystal field strength.21 In a weaker crystal field, the separation of the 4T1 and 6A1 energy levels is stronger, and hence the green light emission with higher energies is achieved. In contrast, with the increase of the crystal field strength, the emission peak shifts to a lower energy region accompanied by red light emission.
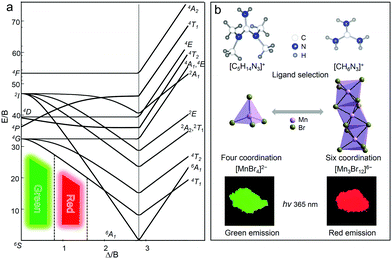 |
| | Fig. 4 (a) Tanabe–Sugano (TS) energy diagram of 3d5 electron orbital of Mn2+. (b) Modifying the coordination units of Mn2+ by selecting different organic ligands to modulate the photoluminescence properties. | |
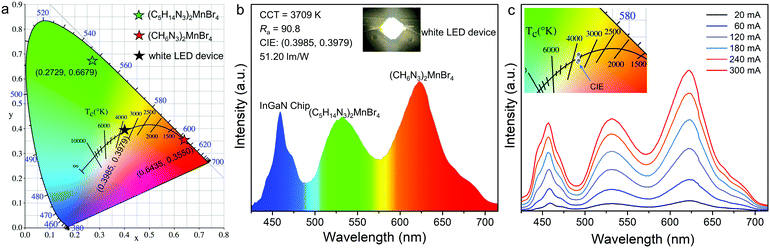
Specifically, the crystal field strength is also affected by the polyhedral distortion index (D) of the polyhedron. With the increase of polyhedral distortion index, the crystal field strength increases, resulting in a large crystal field splitting of the Mn2+ 3d level.54–56 It makes the lowest excited state level 4T1 of Mn2+ closer to the ground state and ultimately results in the red-shift of the emission peak of Mn2+. When the manganese halide anions are changed from the tetrahedron to the octahedron, the polyhedral distortion index will change greatly, which can be defined as57,58
| | 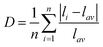 | (5) |
where
li is the distance between the central atom Mn and the coordinated halogen atom, and
lav is the average bond length. For (C
5H
14N
3)
2MnBr
4, the distortion index of [MnBr
4]
2− is 2.16 × 10
−3, while for (CH
6N
3)
2MnBr
4, the distortion index of three MnBr
6 octahedrons in [Mn
3Br
12]
6− are 7.97 × 10
−3, 3.55 × 10
−2 and 3.73 × 10
−2 respectively, with an average value of 2.69 × 10
−2. So the distortion index of [Mn
3Br
12]
6− is much higher that of [MnBr
4]
2−. Therefore, compared with (C
5H
14N
3)
2MnBr
4, the emission peak of (CH
6N
3)
2MnBr
4 have a large redshift, which is coincident with the observed experimental phenomenon. As for the MnX
6 units, they usually show orange emission due to the large polyhedral distortion. If these octahedrons combine together by sharing planes with each other, the orange emission shifts to the red emission, which means the increase in distortion index and crystal field strength. As shown in
Fig. 4b, the coordination units of Mn
2+ can be modified by employing different organic cations toward tunable emission in 0D bromides. The Mn
2+ in (C
5H
14N
3)
2MnBr
4 is located in the tetrahedral weak field, so it exhibits a green light emission. While the Mn
2+ in (CH
6N
3)
2MnBr
4 is located in the octahedral strong field and these MnBr
6 octahedral units form a trimetric [Mn
3Br
12]
6− unit by sharing planes with each other, so it exhibits a red light emission.
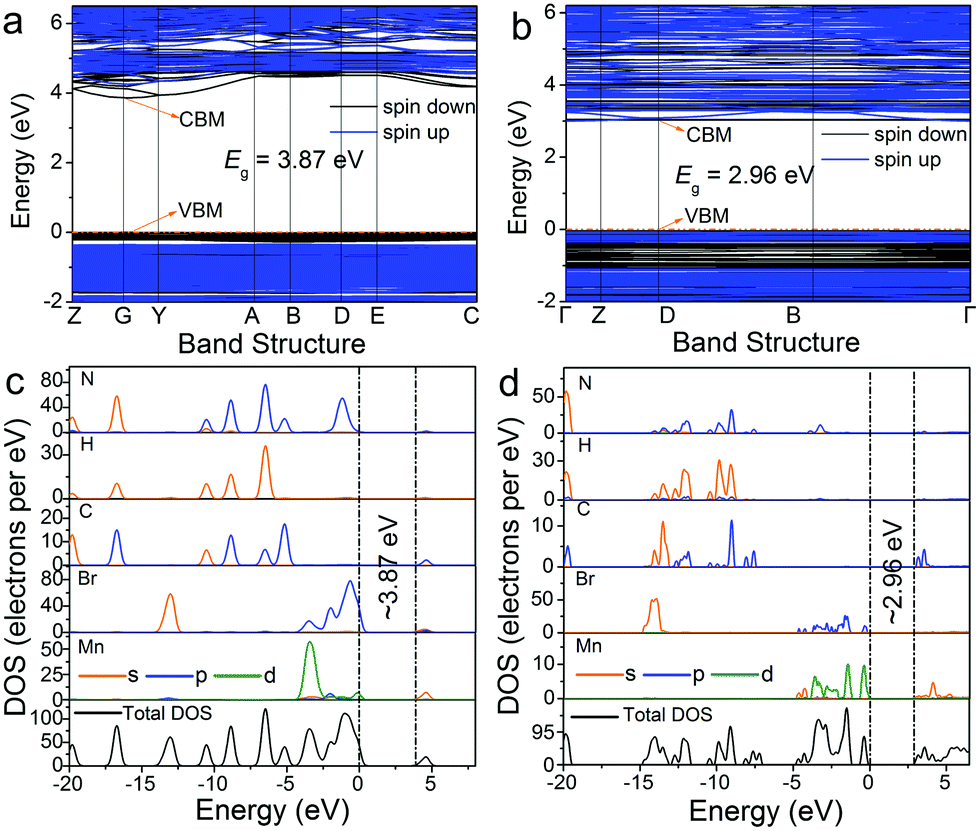
In addition, the electronic energy band structures of 0D bromides with different coordination units of Mn2+ are calculated based on the density functional theory (DFT) approach. As illustrated in Fig. 5a and b, the calculated energy gaps for (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are estimated to be around 3.87 eV at the G point of the Brillouin zone and 2.96 eV at the D point of the Brillouin zone, respectively. The total density states (TDOS) and orbital-projected partial density of states (PDOS) diagrams for (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 (Fig. 5c and d) indicate that the electronic states in the valence band maximum (VBM) are mainly attributed to Br-p and Mn-d orbitals, while those in the conduction band minimum (CBM) are said to principally originate from organic groups.
 |
| | Fig. 5 Electronic energy band structures of (C5H14N3)2MnBr4 (a) and (CH6N3)2MnBr4 (b). The total and orbital projection of partial density of states of (C5H14N3)2MnBr4 (c) and (CH6N3)2MnBr4 (d). | |
3.3 Stability and application for LEDs
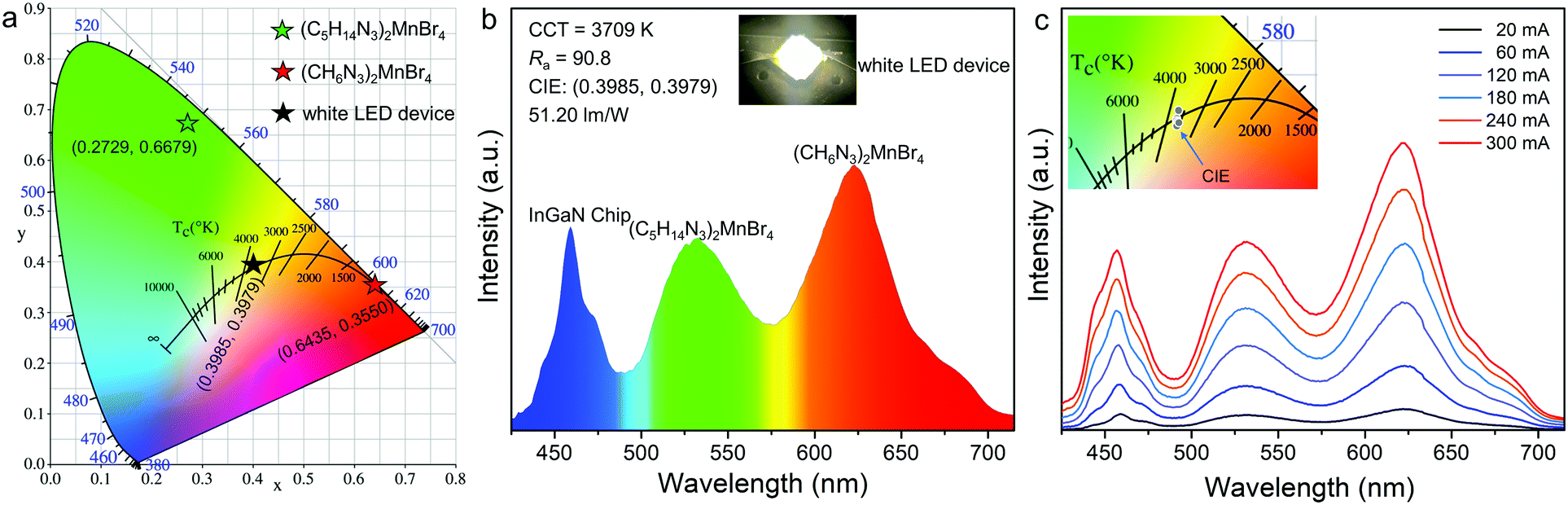
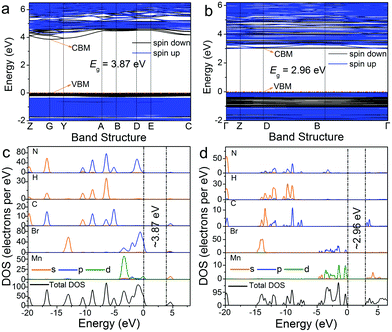
The 0D bromides of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 can be effectively excited by a 450 nm blue light and exhibit relatively high photoluminescence quantum yields (PLQYs) of 86.83% and 61.91%, which enables the corresponding white light LED devices encapsulated by them to achieve a high luminous efficacy in the practical application of solid-state lighting. Meanwhile, their chemical stabilities and temperature quenching (TQ) behaviors were evaluated. The thermogravimetric (TG) results (Fig. S4, ESI†) show that (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are chemically stable up to 281 °C and 289 °C, respectively. The temperature dependent PL spectra (Fig. S5, ESI†) indicate that their thermal quenching behaviors are obvious, especially after reaching 100 °C. To further evaluate their practical application prospects in solid-state lighting, the white LED device was fabricated by utilizing the green-emitting (C5H14N3)2MnBr4 and red-emitting (CH6N3)2MnBr4 (Fig. 6a and b). The results present a correlated color temperature of 3709 K, along with CIE chromaticity coordinates of (0.3985, 0.3979), which means that there is no need to add commercial phosphors containing rare earth as a strategic resource to produce white light emission. The white LED device exhibits a luminous efficacy (LE) of 51.20 lm W−1 and a high color rendering index (Ra) of 90.8, indicating the potential of the as-synthesized Mn2+-based 0D bromides in solid-state lighting. Moreover, the emission spectra and enlarged view of CIE chromaticity coordinates present no remarkable change under different driving currents from 20 to 300 mA (Fig. 6c), revealing a good electroluminescence stability of the fabricated white LEDs. It is found that the 0D bromides of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are suitable to act as the high-performance green and red phosphors for white LED applications.
 |
| | Fig. 6 (a) CIE chromaticity diagram of (C5H14N3)2MnBr4, (CH6N3)2MnBr4, and the fabricated white LED device. (b) Emission spectrum and photograph of the white LED device fabricated with the green emitting (C5H14N3)2MnBr4 and the red emitting (CH6N3)2MnBr4 on a blue LED chip (460 nm) under a current of 20 mA. (c) Emission spectra of the white LED device under the driving currents ranges from 20 to 300 mA, and the inset showing the CIE chromaticity coordinates changes. | |
4. Conclusions
In summary, the 0D bromides of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 with variable coordination units of Mn2+ were designed and synthesized by a facile method of solvent evaporation. They exhibit a green emission at 528 nm and a red emission at 627 nm with a relatively large FWHM of 59 nm and 79 nm under excitation under UV/blue light, which are attributed to the d–d transition (4T1(G) → 6A1(S)) of four coordinated [MnBr4]2− units and six coordinated [Mn3Br12]6− units, respectively. According to the temperature-dependent PL spectra from 80 K to 300 K and Tanabe–Sugano (TS) energy diagram, the 0D bromides of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 show relatively large Huang–Rhys factor S (S = 56.48; S = 27.70), and the distortion index of [Mn3Br12]6− (D = 2.69 × 10−2) is much higher than that of [MnBr4]2− (D = 2.16 × 10−3), resulting in the emission position of (CH6N3)2MnBr4 having a large redshift compared to that of (C5H14N3)2MnBr4. The cases emphasize the influence of organic ligands on the intrinsic emissions of Mn2+ ions, and it is found that the 0D bromides of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are suitable to act as the high-performance green and red phosphors for white LED applications. The fabricated white LED device shows an excellent Ra of 90.8 at a correlated color temperature of 3709 K, along with CIE chromaticity coordinates of (0.3985, 0.3979). This work aims at clarifying the relationship between the coordination units of Mn2+ and emission tuning, and in particular proposes a new way to explore phosphors excited by blue light for white LEDs.
Author contributions
G. Z. and M. X. Z. conceived the project, wrote and revised the paper, and was primarily responsible for the experiment. J. D. synthesized the materials, performed the experimental characterization, and wrote a part of the paper. X. J. and J. Z. performed the DFT calculations. M. M performed the structure analysis. Q. R., J. Z., and S. L. contributed the luminescence mechanism analysis. All authors contributed to the general discussion.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The present work was supported by the Natural Science Foundation of China (21871167), the 1331 Project of Shanxi Province and the Postgraduate Innovation Project of Shanxi Normal University (2019XBY018), the Beijing Natural Science Foundation (No. 2214068) and funded by RFBR according to the research project no. 19-52-80003.
Notes and references
- L. N. Quan, B. P. Rand, R. H. Friend, S. G. Mhaisalkar, T. W. Lee and E. H. Sargent, Chem. Rev., 2019, 119, 7444–7477 CrossRef CAS PubMed.
- Y. H. Dong, Y. S. Zou, J. Z. Song, X. F. Song and H. B. Zeng, J. Mater. Chem. C, 2017, 5, 11369–11394 RSC.
- Y. Zhang, W. Q. Liao, D. W. Fu, H. Y. Ye, Z. N. Chen and R. G. Xiong, J. Am. Chem. Soc., 2015, 137, 4928–4931 CrossRef CAS PubMed.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- Q. Q. He, C. K. Zhou, L. J. Xu, S. Lee, X. S. Lin, J. Neu, M. Worku, M. Chaaban and B. W. Ma, ACS Mater. Lett., 2020, 2, 633–638 CrossRef CAS.
- P. P. Du, L. Gao and J. Tang, Front. Optoelectron., 2020, 13, 235–245 CrossRef.
- G. J. Zhou, B. B. Su, J. L. Huang, Q. Y. Zhang and Z. G. Xia, Mater. Sci. Eng., R, 2020, 141, 100548 CrossRef.
- P. F. Fu, S. L. Hu, J. Tang and Z. W. Xiao, Front. Optoelectron., 2021, 14, 252–259 CrossRef.
- C. K. Zhou, H. Lin, S. Lee, M. Chaaban and B. W. Ma, Mater. Res. Lett., 2018, 6, 552–569 CrossRef CAS.
- L. J. Xu, S. Lee, X. Lin, L. Ledbetter, M. Worku, H. Lin, C. K. Zhou, H. Liu, A. Plaviak and B. W. Ma, Angew. Chem., Int. Ed., 2020, 59, 14120–14123 CrossRef CAS PubMed.
- M. Z. Li and Z. G. Xia, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- C. K. Zhou, L. J. Xu, S. Lee, H. R. Lin and B. W. Ma, Adv. Opt. Mater., 2020, 9, 2001766 CrossRef.
- L. L. Mao, P. J. Guo, S. X. Wang, A. K. Cheetham and R. Seshadri, J. Am. Chem. Soc., 2020, 142, 13582–13589 CrossRef CAS PubMed.
- G. J. Zhou, Z. Y. Liu, J. L. Huang, M. S. Molokeev, Z. W. Xiao, C. G. Ma and Z. G. Xia, J. Phys. Chem. Lett., 2020, 11, 5956–5962 CrossRef CAS PubMed.
- V. Morad, I. Cherniukh, L. Pöttschacher, Y. Shynkarenko, S. Yakunin and M. V. Kovalenko, Chem. Mater., 2019, 31, 10161–10169 CrossRef CAS PubMed.
- P. F. Fu, Y. L. Sun, Z. G. Xia and Z. W. Xiao, J. Phys. Chem. Lett., 2021, 12, 7394–7399 CrossRef CAS PubMed.
- Z. W. Xiao, Z. N. Song and Y. F. Yan, Adv. Mater., 2019, 31, 1803792 CrossRef CAS.
- K. Dave, M. H. Fang, Z. Bao, H. T. Fu and R. S. Liu, Chem. – Asian J., 2020, 15, 242–252 CrossRef CAS PubMed.
- Q. Q. Fan, G. V. Biesold-McGee, J. Z. Ma, Q. N. Xu, S. Pan, J. Peng and Z. Q. Lin, Angew. Chem., Int. Ed., 2020, 59, 1030–1046 CrossRef CAS PubMed.
- G. Q. Ji, C. Z. Han, S. L. Hu, P. F. Fu, X. Chen, J. G. Guo, J. Tang and Z. W. Xiao, J. Am. Chem. Soc., 2021, 143, 10275–10281 CrossRef CAS PubMed.
- B. B. Su, G. J. Zhou, J. L. Huang, E. H. Song, A. Nag and Z. G. Xia, Laser Photonics Rev., 2020, 15, 2000334 CrossRef.
- Q. Zhou, L. Dolgov, A. M. Srivastava, L. Zhou, Z. L. Wang, J. Shi, M. D. Dramićanin, M. G. Brik and M. M. Wu, J. Mater. Chem. C, 2018, 6, 2652–2671 RSC.
- Q. K. Ba, A. Jana, L. H. Wang and K. S. Kim, Adv. Funct. Mater., 2019, 29, 1904768 CrossRef CAS.
- H. Y. Ye, Q. Zhou, X. Niu, W. Q. Liao, D. W. Fu, Y. Zhang, Y. M. You, J. Wang, Z. N. Chen and R. G. Xiong, J. Am. Chem. Soc., 2015, 137, 13148–13154 CrossRef CAS PubMed.
- A. K. Guria, S. K. Dutta, S. Das Adhikari and N. Pradhan, ACS Energy Lett., 2017, 2, 1014–1021 CrossRef CAS.
- G. C. Hu, B. Xu, A. F. Wang, Y. Guo, J. J. Wu, F. Muhammad, W. Meng, C. Y. Wang, S. Q. Sui, Y. Liu, Y. C. Li, Y. Zhang, Y. G. Zhou and Z. T. Deng, Adv. Funct. Mater., 2021, 31, 2011191 CrossRef CAS.
- T. M. Jiang, W. B. Ma, H. Zhang, Y. Tian, G. Lin, W. G. Xiao, X. Yu, J. B. Qiu, X. H. Xu, Y. Yang and D. X. Ju, Adv. Funct. Mater., 2021, 31, 2009973 CrossRef CAS.
- F. Locardi, M. Cirignano, D. Baranov, Z. Dang, M. Prato, F. Drago, M. Ferretti, V. Pinchetti, M. Fanciulli, S. Brovelli, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 12989–12995 CrossRef CAS PubMed.
- W. Y. Liu, Q. L. Lin, H. B. Li, K. F. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS PubMed.
- J. D. Majher, M. B. Gray, T. A. Strom and P. M. Woodward, Chem. Mater., 2019, 31, 1738–1744 CrossRef CAS.
- A. Biswas, R. Bakthavatsalam and J. Kundu, Chem. Mater., 2017, 29, 7816–7825 CrossRef CAS.
- C. K. Zhou, Y. Tian, O. Khabou, M. Worku, Y. Zhou, J. Hurley, H. R. Lin and B. W. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 40446–40451 CrossRef CAS PubMed.
- S. K. Dutta, A. Dutta, S. Das Adhikari and N. Pradhan, ACS Energy Lett., 2018, 4, 343–351 CrossRef.
- G. J. Zhou, X. X. Jiang, M. Molokeev, Z. S. Lin, J. Zhao, J. Wang and Z. G. Xia, Chem. Mater., 2019, 31, 5788–5795 CrossRef CAS.
- L. K. Gong, Q. Q. Hu, F. Q. Huang, Z. Z. Zhang, N. N. Shen, B. Hu, Y. Song, Z. P. Wang, K. Z. Du and X. Y. Huang, Chem. Commun., 2019, 55, 7303–7306 RSC.
- C. L. Jiang, N. Zhong, C. H. Luo, H. C. Lin, Y. Y. Zhang, H. Peng and C. G. Duan, Chem. Commun., 2017, 53, 5954–5957 RSC.
- S. Zhang, Y. F. Zhao, J. D. Zhou, H. Ming, C. H. Wang, X. P. Jing, S. Ye and Q. Y. Zhang, Chem. Eng. J., 2021, 421, 129886 CrossRef CAS.
- M. Z. Li, J. Zhou, M. S. Molokeev, X. X. Jiang, Z. S. Lin, J. Zhao and Z. G. Xia, Inorg. Chem., 2019, 58, 13464–13470 CrossRef CAS PubMed.
- G. J. Zhou, Z. Y. Liu, M. S. Molokeev, Z. W. Xiao, Z. G. Xia and X.-M. Zhang, J. Mater. Chem. C, 2021, 9, 2047–2053 RSC.
- M. Zhao, H. X. Liao, M. S. Molokeev, Y. Y. Zhou, Q. Y. Zhang, Q. L. Liu and Z. G. Xia, Light Sci. Appl., 2019, 8, 38 CrossRef PubMed.
- L. Y. Lian, P. Zhang, G. Liang, S. Wang, X. Wang, Y. Wang, X. W. Zhang, J. B. Gao, D. L. Zhang, L. Gao, H. S. Song, R. Chen, X. Z. Lan, W. X. Liang, G. D. Niu, J. Tang and J. B. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 22749–22756 CrossRef CAS PubMed.
- S. Y. Wang, X. X. Han, T. T. Kou, Y. Y. Zhou, Y. Liang, Z. X. Wu, J. L. Huang, T. Chang, C. Y. Peng, Q. L. Wei and B. S. Zou, J. Mater. Chem. C, 2021, 9, 4895–4902 RSC.
- A. Sen, D. Swain, T. N. Guru Row and A. Sundaresan, J. Mater. Chem. C, 2019, 7, 4838–4845 RSC.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- Y. F. Zhao, R. R. Yang, W. Wan, X. P. Jing, T. Wen and S. Ye, Chem. Eng. J., 2020, 389, 124453 CrossRef CAS.
- H. Y. Huang, R. T. Yang, D. Chinn and C. L. Munson, Ind. Eng. Chem. Res., 2003, 42, 2427–2433 CrossRef CAS.
- R. Pang, C. Li, L. Shi and Q. Su, J. Phys. Chem. Solids, 2009, 70, 303–306 CrossRef CAS.
- S. P. Lee, C. H. Huang, T. S. Chan and T. M. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 7260–7267 CrossRef CAS PubMed.
- X. F. Zhou, W. Y. Geng, J. Y. Li, Y. C. Wang, J. J. Ding and Y. Y. Wang, Adv. Opt. Mater., 2020, 8, 1902003 CrossRef CAS.
- H. Siddique, Z. L. Xu, X. D. Li, S. Saeed, W. T. Liang, X. Q. Wang, C. Gao, R. C. Dai, Z. P. Wang and Z. M. Zhang, J. Phys. Chem. Lett., 2020, 11, 9572–9578 CrossRef CAS PubMed.
- J. Ruiz-Fuertes, S. López-Moreno, J. López-Solano, D. Errandonea, A. Segura, R. Lacomba-Perales, A. Muñoz, S. Radescu, P. Rodríguez-Hernández, M. Gospodinov, L. L. Nagornaya and C. Y. Tu, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125202 CrossRef.
- Z. H. Leng, R. F. Li, L. P. Li, D. K. Xue, D. Zhang, G. S. Li, X. Y. Chen and Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 33322–33334 CrossRef CAS PubMed.
- K. A. Denault, J. Brgoch, M. W. Gaultois, A. Mikhailovsky, R. Petry, H. Winkler, S. P. DenBaars and R. Seshadri, Chem. Mater., 2014, 26, 2275–2282 CrossRef CAS.
- Z. Q. Ming, J. W. Qiao, M. S. Molokeev, J. Zhao, H. C. Swart and Z. G. Xia, Inorg. Chem., 2020, 59, 1405–1413 CrossRef CAS PubMed.
- L. Zhou, J. F. Liao, Z. G. Huang, J. H. Wei, X. D. Wang, H. Y. Chen and D. B. Kuang, Angew. Chem., Int. Ed., 2019, 58, 15435–15440 CrossRef CAS PubMed.
- W. Baur, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1195–1215 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 showing PXRD patterns, FTIR spectra, UV-visible absorption spectra, thermogravimetric (TG) curves, and temperature-dependent PL spectra of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4; Table S1 listing fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters. Table S2 listing the main bond lengths. Table S3 listing hydrogen-bond geometry in the structures. Crystallographic data of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 (CIF). CCDC 2094640 and 2094641. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc05680h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Jialiang
Ding‡
a,
Xingxing
Jiang
*a,
Jialiang
Ding‡
a,
Xingxing
Jiang
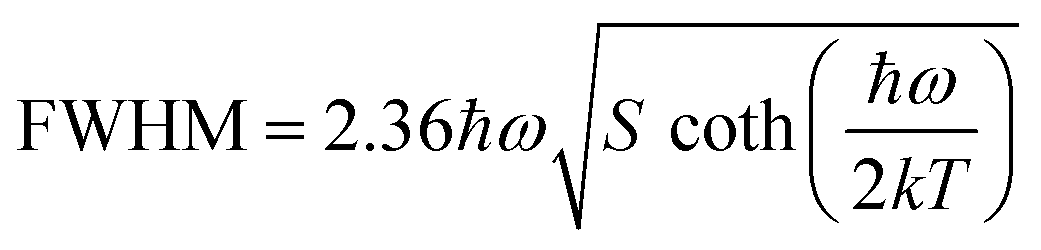
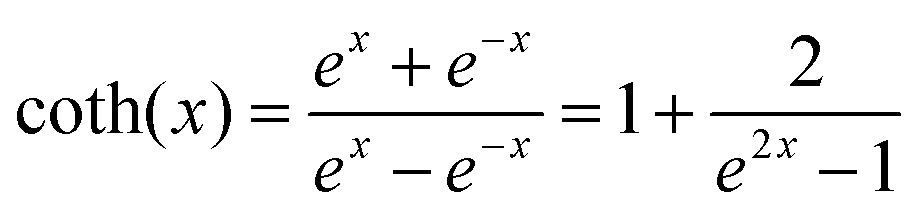 . When
. When 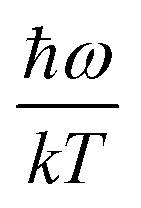 is small enough,
is small enough, 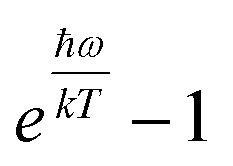 ≈
≈ 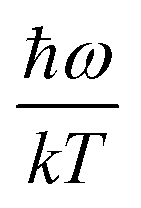 , the following equation can be obtained:
, the following equation can be obtained: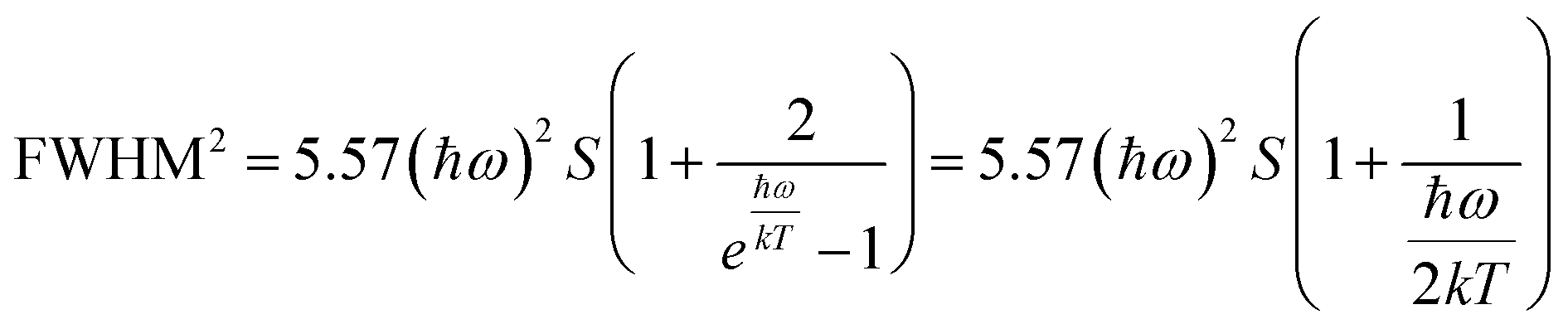
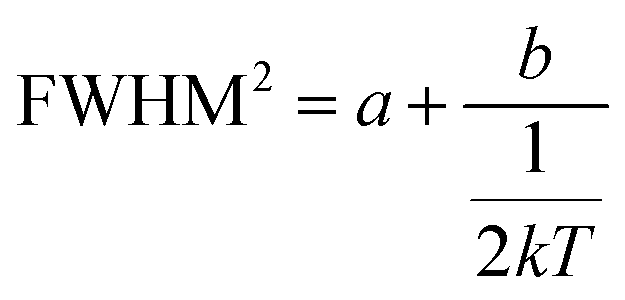
 and FWHM2 can be calculated. Taking
and FWHM2 can be calculated. Taking  as the horizontal coordinate and FWHM2 as vertical coordinate, the Huang–Rhys factors S and ħω of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are obtained by fitting, corresponding to S = 56.48, ħω = 0.00333 eV and S = 27.70, ħω = 0.00658 eV, and the relatively large Huang–Rhys factor S indicates a stronger electron–phonon coupling interaction.
as the horizontal coordinate and FWHM2 as vertical coordinate, the Huang–Rhys factors S and ħω of (C5H14N3)2MnBr4 and (CH6N3)2MnBr4 are obtained by fitting, corresponding to S = 56.48, ħω = 0.00333 eV and S = 27.70, ħω = 0.00658 eV, and the relatively large Huang–Rhys factor S indicates a stronger electron–phonon coupling interaction.