
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic and energy level engineering of directly fused porphyrin-conjugated polymers – impact of the central metal cation†
Drialys
Cardenas-Morcoso
 *,
Eloa
Vey
,
Max
Heiderscheid
,
Gilles
Frache
and
Nicolas D.
Boscher
*
*,
Eloa
Vey
,
Max
Heiderscheid
,
Gilles
Frache
and
Nicolas D.
Boscher
*
Materials Research and Technology Department, Luxembourg Institute of Science and Technology, 28 avenue des Hauts-Fourneaux, Esch-sur-Alzette, Luxembourg. E-mail: drialys.cardenas@list.lu; nicolas.boscher@list.lu
First published on 24th January 2022
Abstract
The integration of porphyrins and their derivatives in functional devices for solar-assisted fuel production is both highly attractive and challenging due to the difficulties in processing them. This limitation is overcome in the gas-phase approach, particularly by oxidative chemical vapor deposition (oCVD), leading to the simultaneous synthesis and deposition of conjugated porphyrin coatings. We have investigated the impact of the metal cation of 5,15-diphenyl metalloporphyrins (MDPP; M = Co, Cu, Mg, Zn, Pd, Pt, Ag, Ru, Ag, and FeCl) on the dehydrogenative coupling reaction leading to fused-metalloporphyrin thin films via oCVD and on the optoelectronic properties of the resulting thin films. We found that the nature of the chelated cation strongly affects the intermolecular coupling efficiency, as well as the occurrence of side reactions such as chlorination, intramolecular cyclization, demetallation/re-metalation, and oxidation of the porphyrin core. Moreover, we discussed the influence of the above-mentioned reactions on the optoelectronic properties of the fused metalloporphyrin coatings, in view of their potential application in photo-electrocatalytic systems. This study paves the way toward the engineering and future implementation of porphyrin-based systems for clean and efficient solar fuel production.
Introduction
Porphyrins and their derivatives play a fundamental role in essential natural processes – as chlorophyll in the photosynthesis driven by green plants – due to their outstanding light absorption and charge transport properties. Therefore, porphyrin-based materials are highly attractive in artificial light-harvesting systems,1,2 including photocatalytic and photoelectrochemical approaches towards hydrogen production from solar-assisted water splitting.3–5 The features of porphyrins (e.g. optoelectronic and catalytic properties) can be further enhanced by assembling them into large π-conjugated systems, e.g. porphyrin-based metal–organic frameworks (MOFs),6,7 bridged porphyrin conjugated polymers (CPs)8 and directly fused porphyrin CPs.9,10 In contrast to inorganic semiconductors, even a small modification of the chemical structure of conducting and semiconducting coordination and covalent polymers can lead to a significant change of their optoelectronic properties and hence, the extension of their functional applications.2,6,11 In particular, the modification of the central metal ion and/or meso-substituents can alter the band gap and band edge positions of porphyrin-based MOFs6 and porphyrin-based CPs.8In addition, the metal cation chelated at the center of the metalloporphyrin macrocycle – magnesium(II) in chlorophyll – can act as a single-atom catalytic site, boosting the efficiency of a targeted chemical reaction. For example, cobalt(II)-bridged porphyrin CPs have been demonstrated as excellent earth-abundant element heterogeneous catalysts for the oxygen evolution reaction (OER)12 from water oxidation and the oxygen reduction reaction (ORR).13 When combining with zinc(II) porphyrins in a bridged porphyrin conjugated polymer, cobalt(II) porphyrins are also active for the hydrogen evolution reaction (HER) from water splitting.8 In this latter example, the periodically distributed type II-like heterojunction between adjacent zinc(II) and cobalt(II) porphyrins ensures the charge separation and transfer from zinc(II) porphyrins to cobalt(II) porphyrins.8 Directly fused porphyrin assemblies are also active catalysts.14,15 In particular, bimetallic copper(II)-fused porphyrin dimers were shown to be a highly active HER catalyst, demonstrating the direct fusion of the porphyrin macrocycles to be a promising strategy for enhanced electrocatalyst design.14 More recently, electrochemical and spectroscopic investigations on fused iron(III) porphyrin complexes revealed their ability to store up to six electrons, added to the capability of the π-extended ligand to delocalize electrons across the multimetallic scaffold.16 These properties are particularly promising towards multielectron catalytic reactions driven by fused-metalloporphyrins. Despite these promising results,8,13,14 the difficult processability of porphyrins and particularly of any large π-conjugated porphyrin-based systems, owing to their non-solubility and non-meltability, jeopardizes their successful integration into functional devices.
Unlike wet chemistry methods, oxidative chemical vapor deposition (oCVD) enables the simultaneous synthesis and deposition of directly fused porphyrin CPs coatings in one step, without the need for solvents or post-treatment methods.17 In this process, a suitable volatile oxidant18 – usually iron(III) chloride – is used to promote the dehydrogenative coupling of porphyrins possessing free meso-positions, as represented in Scheme 1, yielding conductive thin films on a wide variety of substrates.17 As a suitable approach for the chemical engineering and molecular design of conjugated polymers,19 oCVD was used to improve the conductivity and functional properties of directly fused porphyrin-based CPs. Indeed, the meso-substituents of porphyrins were reported to have a reduced impact on the polymerization of directly fused porphyrin CPs via oCVD,20 in contrast to solution-based methods.21 However, aryl substituents bearing free ortho-positions were shown to undergo an intramolecular cyclization between the ortho-position of the aryl ring and the beta-position of the porphyrin macrocycle. Intramolecular cyclization is responsible for the molecular flattening of the directly fused porphyrin-conjugated polymer chains, which drastically affects their π–π stacking facility and enhances the electronic properties of the films.20 Preliminary studies carried out by our group on the electro- and photocatalytic activity of directly fused copper(II) porphyrin conjugated polymer thin films for the heterogeneous HER showed that the phenyl substituent, enabling intramolecular cyclization, promotes superior catalytic performance over the mesityl substituent.15
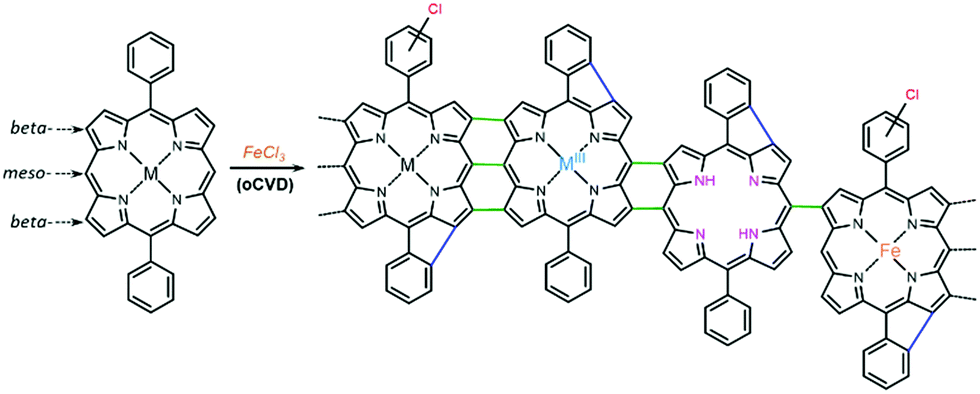 | ||
| Scheme 1 oCVD reaction of a 5,15-diphenyl metalloporphyrin with iron(III) chloride as the oxidant, leading to triple, double, and single intermolecular coupling (green). Other possible side reactions are also represented: intramolecular coupling (blue), chlorination (red), a change in the oxidation state of the central metal cation (light blue), demetallation of the porphyrin core (violet), and further re-metalation with iron originating from the oxidant (orange). | ||
Besides the dehydrogenative intermolecular coupling (polymerization) forming triple, double or single linkages between the porphyrin units and the dehydrogenative intramolecular coupling (cyclization), other side reactions can take place during the oCVD reaction of porphyrins. Namely, the chlorination of the macrocycle and the substituents, the demetallation and potential re-metalation of the porphyrin core, and oxidation of the central metal cation can take place (Scheme 1).22 The occurrence of several of these reactions, e.g. demetallation, re-metalation and oxidation, is of course mainly influenced by the nature of the central metal cation. Additionally, it was shown that the central metal cation in 5,15-dimesityl porphyrins (MDMP; M = Ni(II), Cu(II), Zn(II), Co(II), Pd(II), and Fe(III) Cl) strongly affects the initial C–C coupling step and consequently the formation of triply or doubly linked directly fused porphyrin tapes.23 Therefore, the nature of the central metal cation is expected to have a double influence on the optoelectronic properties of the directly fused porphyrin CPs synthesized by oCVD.
With the aim of elucidating the direct and indirect (regioselectivity of the polymerization) influence of the central metal cation on the band gap and band edge positions of directly fused porphyrin-conjugated polymers, we studied the oCVD reaction of 5,15-diphenyl metalloporphyrins (MDPP; M = Co(II), Cu(II), Mg(II), Zn(II), Pd(II), Pt(II), Ag(II), Ru(II), Ag(II), and Fe(III) Cl). Phenyl was selected as the meso-substituent due to the superior catalytic15 and sensing properties24 of poly(5,15-diphenyl metalloporphyrin) thin films over poly(5,15-dimesityl metalloporphyrin) ones. The occurrence of intermolecular coupling and other side reactions during oCVD was evidenced by ultraviolet-visible-near infrared (UV-Vis-NIR) spectroscopy, laser desorption ionization high-resolution mass spectrometry (LDI-HRMS), and X-ray photoelectron spectroscopy (XPS) analysis. The influence of the reactivity of 5,15-diphenyl metalloporphyrins on the electronic properties of the resulting coatings was evaluated using conductivity measurements by atomic force microscopy (c-AFM). Finally, from the combination of the optical and electronic characterization, we obtained the band alignment of the directly fused porphyrin CP thin films formed by oCVD, allowing their viability in solar-assisted hydrogen production approaches to be assessed.
Experimental
Reactants
5,15-Diphenyl-metalloporphyrins chelated with different central metal cations (Co2+, Cu2+, Mg2+, Zn2+, Pd2+, Pt2+, Ag2+, Ru2+, Ag2+, and Fe3+–Cl−) and the free-base (H2) 5,15 diphenyl-porphyrin were obtained from PorphyChem (98%) and were used without further purification. Based on previous reports,17,18 iron(III) chloride (97%, Sigma-Aldrich) was chosen as the oxidising agent. Dichloromethane (HPLC grade >99.8%, SupraSolv®) was used as the solvent for electrochemical and optical characterization. Tetrabutylammonium hexafluorophosphate (TBAPF6, for electrochemical analysis, ≥99.0%, Sigma-Aldrich) was used as the supporting electrolyte for the electrochemical measurements.Porphyrin monomer characterization
Thermogravimetric analyses (TGAs) of the porphyrin monomers were performed under an inert atmosphere (Ar) with a ramp of 10 K per minute. Cyclic voltammetry measurements were performed to estimate the redox potentials (E1/2) of the porphyrin monomers, in a three-electrode-cell and using an AUTOLAB potentiostat/galvanostat. A platinum working electrode, a platinum wire counter electrode, and a 0.01 M Ag/AgNO3 in acetonitrile reference electrode were used for the experiments. For each metalloporphyrin, cyclic voltammetry was performed using 1 mM of the monomer in dichloromethane, containing 0.1 M TBAPF6 as the supporting electrolyte. The cyclic voltammograms were recorded at a 100 mV s−1 scan rate. Ferrocene was employed as the internal reference.Oxidative chemical vapor deposition (oCVD)
The oCVD reaction was performed in a custom-built oCVD reactor (Scheme S1, ESI†). Two crucibles were used to sublime the porphyrin and the oxidant, respectively. As previously reported, the different metal cations in the porphyrin core affect the sublimation temperature during the oCVD proces.23 Therefore, preliminary experiments were performed to determine the adequate sublimation temperature (from 230 °C to 290 °C) of each porphyrin monomer in order to sublime similar amounts of reactants, while keeping the porphyrin/oxidant ratio as constant as possible. Table S1 in the ESI† summarizes the deposition conditions used for each porphyrin investigated. On the other hand, the temperature of the oxidant's crucible was always kept at 170 °C. Glass microscope slides (Menzel-Gläser Superfrost®), bare and platinum-coated silicon wafers (Siegert Wafer®) and interdigitated chips (OFET Gen4, Fraunhofer) were used as substrates. Prior to deposition, all the substrates were cleaned with absolute ethanol (99.98%, VWR chemicals®) and dried with nitrogen gas. The substrate holder was kept at 150 °C for all the depositions. The pressure inside the oCVD reactor was kept at 10−3 mbar, under an argon (99.999%, Air Liquide) atmosphere. The deposition time was set to 30 minutes for all the experiments. Additionally, reference porphyrin monomers coatings were obtained under the same conditions, without supplying the oxidant.Film characterization
The ultraviolet-visible-near infrared (UV-Vis-NIR) spectra of the thin films deposited on glass slides were recorded with a PerkinElmer Lambda 1050 spectrometer, in the transmission (T) mode, and at a wavelength interval of 300–2500 nm. The absorbance (A) was calculated as: A = −log(T). From the absorbance spectra, the direct optical band gap of the fused-metalloporphyrins was estimated through the Tauc plot as: (αhν)1/n = A(hν − Eg), where α is the absorbance coefficient, n = 1/2 for direct transitions, h is the Planck's constant and ν is the wavelength number. The absorbance coefficient was calculated as: α = ln(10)A/l, where l is the film's thickness. The film's thickness was determined with an alpha-SE spectroscopic ellipsometer. The spectroscopic ellipsometry measurements were acquired at three angles (65°, 70° and 75°), in the wavelength range of 500–900 nm. The data were fitted to a Cauchy model. The films were rinsed with dichloromethane to record the absorbance spectra of the DCM-soluble phase in transparent quartz cuvettes of 3.5 mL capacity and 1 cm of light path. The rinsed glass slides were also analyzed for comparison with the as-deposited films.Scanning electron microscopy (SEM) images were recorded using a FEI Quanta 200F system. X-ray photoelectron spectroscopy (XPS) measurements were performed with a Kratos Axis Ultra DLD, instrument using a monochromatic Al Kα X-ray source of energy of 1486.6 eV, at a power of 105 W. Charge calibration was accomplished by fixing the binding energy of carbon (C 1s) to 285.0 eV.
Laser desorption/ionization high-resolution mass spectrometry (LDI-HRMS) measurements were performed on an AP-MALDI UHR ion source (MassTech, Inc.) coupled to an LTQ/Orbitrap Elite (ThermoScientific) system. In-source fragmentation (E = 70 V) was used to prevent the formation of clusters. The measurements were performed on a Si wafer coated with the directly fused porphyrin CP films, placed directly on the sample holder. LDI-HRMS measurements and analysis of metalloporphyrin coatings have been described in detail elsewhere.18,22,23
The conductivity of the oCVD coatings was acquired using the c-AFM mode of an Innova AFM (Bruker) system. Conductive AFM tips, ElectriMulti75-G from BudgetSensors coated with a layer of 5 nm chromium and 25 nm of platinum with nominal spring constants of 3 N m−1 and nominal radius < 25 nm were used. The samples were deposited on monocrystalline silicon wafers coated with platinum and connected to the conductive stage via a silver colloidal ink. A bias of +4 V was applied to the back electrode of the samples, while the grounded conductive tip collected electrons for the current measured using an amplifier (DLPCA-200, Femto). A 109 V A−1 amplification was used for the pA current range, while a 107 V A−1 was used for those ones in the nA range.
Results and discussion
Reactivity of diphenyl-substituted metalloporphyrins in oCVD
Prior to the oCVD processing, the redox potential of the metalloporphyrin monomers was determined through cyclic voltammetry measurements. Fig. S1 of the ESI† show the cyclic voltammograms of the metalloporphyrin monomers. The redox potentials (E1/2) of ZnDPP, CuDPP, CoDPP, MgDPP, PtDPP, AgDPP and RuDPP monomers were estimated at the midpoint potentials for the reversible and quasi-reversible waves, as the average of the anodic and cathodic peak potentials. The cyclic voltammetry of NiDPP, PdDPP, FeDPP, and H2DPP show irreversible redox couples. For these samples, the E1/2 was estimated as the half-potential peak of the voltammograms, corresponding to the potential at half the maximum current in the cyclic voltammograms.25 All the values are summarized in Table 1. The obtained values are consistent with those previously reported for di-mesityl substituted metalloporphyrins.23 All the di-phenyl substituted metalloporphyrins have E1/2 values lower than the oxidation potential of the Fe3+/Fe2+ couple (0.77 V), supporting the selection of an iron(III)-based oxidant for the dehydrogenative coupling of the selected metalloporphyrins.| Porphyrin | E 1/2 (V vs. Fc/Fc+) | Porphyrin | E 1/2 (V vs. Fc/Fc+) |
|---|---|---|---|
| MgDPP | 0.24 | AgDPP | 0.55 |
| CoDPP | 0.27 | H2DPP | 0.56 |
| ZnDPP | 0.41 | PdDPP | 0.60 |
| RuDPP | 0.42 | PtDPP | 0.64 |
| CuDPP | 0.54 | FeClDPP | 0.69 |
| NiDPP | 0.54 |
It is worth mentioning that after the oCVD reaction, the redox potential of the resulting fused-metalloporphyrins can significantly vary. As previously reported by Moore and co-workers for copper and iron-based bi-nuclear fused porphyrins, such a difference of redox events between the monomer and the fused porphyrins is attributed to the enhanced electronic delocalization resulting from the extended aromaticity of the fused architecture.14,16
To set an adequate sublimation temperature of the selected metalloporphyrins during the oCVD reaction, their thermal stability was assessed by thermogravimetry (Fig. S2 in the ESI†). All the di-phenyl substituted metalloporphyrins investigated in this work are thermally stable up to at least 300 °C confirming their suitability for oCVD processing. Two series of thin films were prepared from (i) the oCVD reaction of the metalloporphyrins with iron(III) chloride (denoted as pMDPP) and (ii) from the sublimation of the metalloporphyrins (denoted as sMDPP). The sublimation temperatures used for the oCVD reaction of each metalloporphyrin investigated are summarized in the Table S1 of the ESI,† including the amount of porphyrins and oxidant consumed during the thin film deposition experiments.
The SEM analysis of the films prepared by oCVD shows homogeneous coverage and a relatively smooth and dense surface, as depicted in Fig. S3 of the ESI,† in agreement with previous reports.20,26 The particles and islands disseminated across the surfaces of the oCVD films are attributed to the inclusions of both the unreacted oxidant and oxidant by-products.22
Most of the oCVD coatings exhibit a notable color difference compared to the reference coatings obtained from the sublimation of the metalloporphyrin monomer without any oxidant, as shown in Fig. 1a. Such color changes are indicative of the occurrence of some of the reactions depicted in Scheme 1, including the formation of directly fused porphyrin tapes.17,18 In particular, the oCVD coatings obtained from Zn, Cu, Co, Mg, Ni and Pt di-phenyl substituted porphyrins (pZnDPP, pCuDPP, pCoDPP, pMgDPP, pNiDPP and PtDPP, respectively) exhibit an intense green coloration, different from the orange of the sublimed reference thin films. This is consistent with previous studies on the oCVD reactivity of porphyrins hinting at the retention of the porphyrin macrocycle upon sublimation, and their oxidative dehydrogenation and polymerization in the presence of a suitable oxidant.18 Conversely, thin films from the oCVD reaction of the Pd, Ag, Ru, Fe and free-base porphyrins (pPdDPP, pAgDPP, pRuDPP, pFeDPP, and pH2DPP, respectively) exhibit yellow and reddish colorations, that in some cases do not strongly differ from their respective reference thin films (e.g. sRuDPP and pRuDPP), as depicted in Fig. 1a, suggesting less efficient coupling reaction.
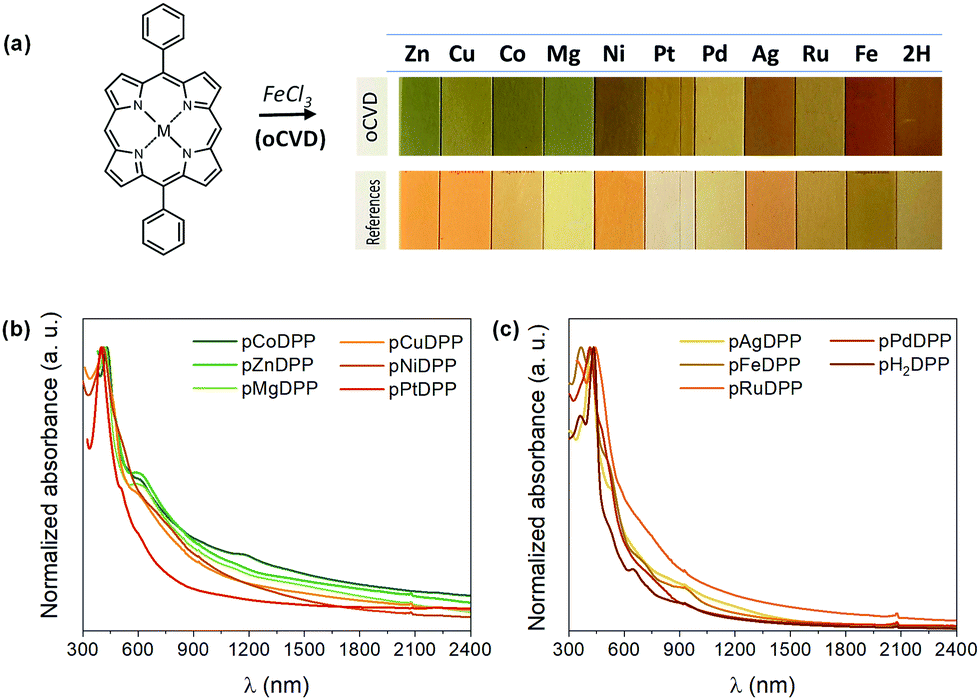 | ||
| Fig. 1 (a) Digital pictures of the as-deposited oCVD coatings from 5,15-diphenyl metalloporphyrins with different central metal cations, in the presence of iron(III) chloride (top row). The films from the sublimed monomers without an oxidant are shown as references (bottom row). (b) and (c) UV-Vis-NIR spectra of the as-deposited oCVD coatings. For better comparison, the absorbance spectra are normalized to their Soret bands. | ||
Further insights into the oCVD reaction efficiency toward directly fused porphyrin CPs are revealed from the UV-Vis-NIR spectra of the as-prepared thin films. Broadened Soret and Q bands, and absorption in the NIR spectral region were observed for pZnDPP, pCuDPP, pCoDPP, pMgDPP, pNiDPP, pPtDPP, and pRuDPP thin films, as depicted in Fig. 1b and c. This enhanced absorption is indicative of the formation of multiply fused porphyrin tapes, due to the intensified and red-shifted Soret and Q-bands deep in the NIR region.
In particular, the pZnDPP, pCuDPP, pCoDPP, pMgDPP, pNiDPP, and pPtDPP films exhibit appreciable absorption above 1500 nm, pointing to the significant formation of triply linked porphyrin tapes. The low redox potentials of ZnDPP, CoDPP, and MgDPP explain the highest degree of fusing derived from these monomers, as suggested by the UV-Vis-NIR spectra. However, the higher redox potentials of PtDPP and NiDPP, coupled with the high degree of fusing of pPtDPP and pNiDPP indicated by the UV-Vis-NIR spectra, suggest more complex relationships in the successful dehydrogenative coupling of metalloporphyrins in oCVD.
On the other hand, the pPdDPP and pAgDPP films are essentially transparent above 1500 nm, as depicted in Fig. 1c, suggesting the formation of mainly doubly linked porphyrin tapes. Moreover, the absorbance spectra obtained for pFeDPP and pH2DPP films are rather similar to those of their respective reference sublimed films, displaying neither significant shift of the Soret and Q-bands, nor absorption in the NIR region, as shown in Fig. S4 of the ESI.† Such observations confirm that these porphyrins failed to yield directly fused porphyrin tapes, or at least with a lower efficiency.
The analysis of both the thin films after rinsing with a nonpolar solvent, i.e. dichloromethane (DCM), and the collected DCM-soluble phase provides significant insights into the formation of directly fused porphyrin chains and other modifications occurring during the oCVD reaction. As shown in the digital picture of the films (Fig. S5, ESI†) the pZnDPP, pCuDPP, pCoDPP, pMgDPP, pNiDPP, and pPtDPP coatings are almost insoluble in dichloromethane, suggesting the formation of highly fused porphyrin oligomers. The UV-Vis-NIR spectra of these films after DCM rinsing remain superimposable with those of the as-deposited films, as depicted in Fig. S4(a)–(f), ESI,† while there is a clear absence of NIR absorption in the DCM-soluble fractions of these films (Fig. S6(a)–(f), ESI†) pointing to the retention of the multiply fused porphyrin tapes in the films. Conversely, the pPdDPP, pAgDPP, pRuDPP, pFeDPP, and pH2DPP oCVD coatings are visibly partially dissolved in dichloromethane (Fig. S5, ESI†), also confirmed from the UV-Vis-NIR spectra of the oCVD films after rinsing, compared to the as-deposited oCVD films as shown in Fig. S4(g)–(k) (ESI†). The rinsing experiment using DCM was also performed in the reference sublimed samples (in the absence of the oxidant), for comparison. In all cases, the sublimed films were fully dissolved, as expected from the solubility of the porphyrin monomers in non-polar solvents such as DCM, confirmed by the UV-VIS-NIR analysis of the DCM-soluble fraction (Fig. S6, ESI†).
To reach a deeper understanding on the reactivity of diphenyl substituted metalloporphyrins in oCVD, laser desorption/ionization high-resolution mass spectrometry (LDI-HRMS) and X-ray photoelectron spectroscopy (XPS) analyses were performed on both the sublimed references and oCVD films. It is worth mentioning that the LDI-HRMS spectra do not provide a complete representation of all the oligomer chains formed, since the instrument only detects m/z values lower than 4000. In addition, long and highly fused porphyrins are difficult to ionize, hindering the detection of larger oligomers. Yet, the LDI-HRMS analysis of the prepared films, in combination with UV-Vis-NIR and XPS analyses, provides insightful information into the reactivity and conformations of directly fused metalloporphyrins.
Irrespective of the central metal cation, the distribution of peaks corresponding to the presence of monomers (n = 1) and oligomers (n > 1) is seen in the LDI-HRMS spectra of all the oCVD films, as depicted in Fig. S7(a)–(k) of the ESI.† Structures up to pentamers are notably detected, as representatively shown in Fig. 2a corresponding to the pCuDPP oCVD films, indicating the successful fusing of the porphyrin units by dehydrogenative C–C coupling. Furthermore, the chlorination side reaction is clearly present in all the oCVD coatings giving rise to peak distributions that shifted from 35 m/z corresponding to the exchange of hydrogen atoms by chlorine atoms (Fig. 2a). We should note that, unlike previously studied dimesityl substituted porphyrins, where meso-mesityl substituents with blocked ortho positions were used, diphenyl substituted porphyrins are highly prone to intermolecular cyclization. This effect is shown in Fig. 2b where both intermolecular (polymerization) and intramolecular (cyclization) dehydrogenative coupling (Scheme 1) occur to form [(CuDPP)2–(H2)n]+ dimers in which more than three hydrogen pairs (up to three intermolecular C–C bonds between two porphyrins) are eliminated. Indeed, significant contributions of the [(CuDPP)2–8H]+ and [(CuDPP)2–10H]+ species to the experimental signal were observed, pointing to the formation of a minimum of one and two intramolecular C–C coupling of triply fused dimers, respectively.
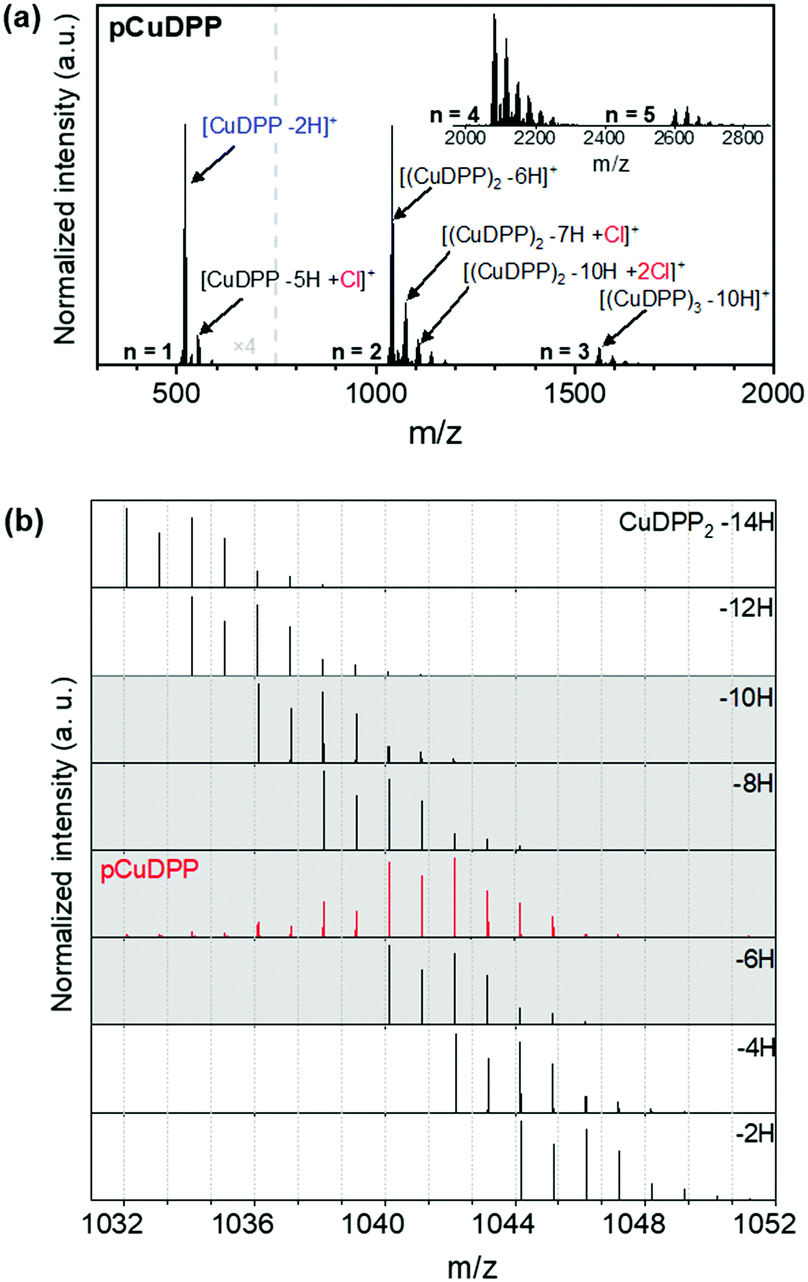 | ||
| Fig. 2 (a) LDI-HRMS spectrum of the oCVD film prepared from CuDPP, in the 400–3000 m/z range, showing the oligomer formation due to dehydrogenative C–C coupling (n = degree of oligomerization) and the occurrence of chlorination side reaction. (b) LDI-HRMS spectrum in the dimer region of pCuDPP (red) in comparison with the simulated spectra of the dimers [(CuDPP)2–(H2)n]+ (n comprising between 1 and 5). | ||
According to their respective UV-Vis-NIR spectra that suggest the formation of highly fused porphyrin tapes (Fig. 1b), the LDI-HRMS spectra of the pCoDPP, pNiDPP, and pPtDPP oCVD films are dominated by peaks related to altered monomers and oligomers [(MDPP)n–(H2)m]+ (m ≥ n) in which multiple hydrogen pairs are eliminated. These observations are in good agreement with the formation of double and triple linkages between the porphyrin units chelated with Cu(II), Co(II), and Ni(II) cations, as previously reported for the oCVD reaction of 5,15-dimesityl-metalloporpyrins (MDMPs),20 being ascribed to the mixed a1u/a2u molecular symmetry of their radical cations. Furthermore, besides the intramolecular cyclization enabled by the free-ortho positions of the phenyl substituent and the chlorination induced using iron(III) chloride, no other side reactions were detected for pCuDPP, pNiDPP, and pPtDPP. In the case of pCoDPP, a change in the oxidation state of the central metal cation Co2+ was detected from XPS analysis (Fig. S8a, ESI†). Precisely, the XPS Co 2p core level peak is broadened and shifted 1.1 eV towards higher binding energies compared to the sublimed porphyrin (sCoDPP) where only Co2+ is present. This is indicative of the presence of both Co2+ and Co3+ oxidation states in the directly fused porphyrin CPs formed from the oCVD reaction of CoDPP. This observation is supported by a similar shift observed in the XPS N 1s core level region, depicted in Fig. S8b (ESI†). These phenomena of cobalt-based porphyrins have already been evidenced in the literature.23,27,28 It is worth mentioning that the analysis of the XPS spectra of Pd 3d, Ni 2p, Ru 3d, Cu 2p, and Pt 4f core levels of the reference and oCVD thin films (Fig. S9a–f, ESI†) suggests no alterations of these central metal cations.
UV-Vis-NIR analysis also suggested an efficient intermolecular dehydrogenative coupling reaction for the pZnDPP and pMgDPP coatings, fostered by the low redox potentials of the respective monomers (Table 1). However, although the signals corresponding to the formation of doubly and triply fused oligomers were detected and further confirmed by comparison with the simulated isotopic patterns (Fig. S10 of ESI†), a pronounced demetallation of the porphyrin cores is observed in the LDI-HRMS spectra (Fig. 3a and c). A more remarkable effect is observed for pMgDPP whose LDI-HRMS spectrum is dominated by the peaks related to fused free-base porphyrin dimers [(H2DPP)2–6H]+. Demetallation is also evidenced by the XPS analysis of the oCVD films. The N 1s region of a free-base porphyrin is characterized by the presence of two signals at 400.0 eV and 397.9 eV binding energies, characteristics of amino- and imine–pyrrole nitrogen environments. As shown in Fig. 3b and d, the pZnDPP and pMgDPP films displayed signals at 398.5 eV and 398.4 eV, respectively, corresponding to nitrogen in its initial Zn2+ and Mg2+ chemical environments. As shown in Fig. 3b and d, the pZnDPP and pMgDPP films displayed signals at 398.5 eV and 398.4 eV, respectively, corresponding to nitrogen in its initial Zn2+ and Mg2+ chemical environments. However, higher energy signals at 400.2 eV and 400.3 eV in pZnDPP and pMgDPP, respectively, confirm the presence of protonated nitrogen, and hence the presence of free-base porphyrin units in both films. Moreover, XPS analysis of the metal core level of pMgDPP films (Fig. S11a in the ESI†) showed a significant shift of the Mg 2p-related signal, supporting the modification of the chemical environment of the porphyrin center. Such a shift is not observed in the pZnDPP films (Fig. S11b in the ESI†) due to a less remarkable demetallation effect, consistent with the LDI-HRMS analysis. Demetallation in pZnDPP and pMgDPP is not surprising since Zn2+ and Mg2+ are known labile cations29 and the demetallation of the zinc(II) porphyrin was previously reported in the oCVD reaction of ZnDMP.20
 | ||
| Fig. 3 LDI-HRMS spectra in the dimer regions (n = 2) of the oCVD films prepared from: (a) ZnDPP, (c) MgDPP and (e) AgDPP. XPS spectra of the N 1s regions of (b) sublimed reference and oCVD films prepared from ZnDPP. The sublimed free-base porphyrin spectrum is shown for comparison. (d) Sublimed reference and oCVD films prepared from MgDPP and sublimed free-base porphyrins for comparison. (f) Sublimed reference and oCVD films prepared from AgDPP, and oCVD films from FeClDPP for comparison. | ||
Evidence of demetallation followed by significant re-metalation of the porphyrin core was observed in the LDI-HRMS spectra of the pAgDPP film that is dominated by the peaks related to fused iron porphyrin dimers [(FeDPP)2–7H–Cl]+ (Fig. 3e). In consistency with this, the XPS analysis in the N 1s region of the signal corresponding to the pAgDPP oCVD film (Fig. 3f) shows a slight shift toward higher energies, i.e. to a value closer to the binding energy characteristic of the nitrogen environment in the pFeDPP film (399.1 eV) confirming that demetallation and re-metalation occur at a significant degree during the oCVD reaction of AgDPP. In addition, the substantial incorporation of iron into the resulting film was evidenced in the elemental composition, from the XPS analysis of the Fe 2p core level (Table S2 of the ESI†).
Both LDI-HRMS and XPS analyses indicate different reactivities of ZnDPP, MgDPP and AgDPP in oCVD, according to different C–C coupling (k1), demetallation (k2) and re-metalation (k3) rates. As shown in Scheme 2a, the oCVD reaction of ZnDPP is characterized by a slow demetallation rate that is balanced by the low redox potential of ZnDPP allowing an efficient intermolecular oxidative C–C coupling. As a result, it yields highly fused porphyrins with partially demetallated oligomers (Fig. 3a and b). Porphyrin units resulting from re-metalation were detected by LDI-HRMS, yet with a slighter contribution compared to the formation of demetallated oligomers (Fig. S7a, ESI†). Analogously, the lowest redox potential of the MgDPP monomer allows a fast reactivity toward intermolecular oxidative dehydrogenative coupling. However the high lability of Mg2+ ions in porphyrins26 further leads to yielding highly fused porphyrin oligomers containing free-base, magnesium and iron porphyrin units (Scheme 2b), e.g. [(MgDPP)(H2DPP)–9H–Cl]+ and [(MgDPP)(FeDPP)–6H–Cl]+ (Fig. S7d, ESI†). The re-metalation of porphyrin cores during the oCVD reaction with iron(III) from the oxidant and its by-products is likely promoted by the faster demetallation of MgDPP compared to ZnDPP. It is important to mention that the formation of both free-base (demetallation) and iron(III) porphyrins (re-metalation) with higher redox potentials than MgDPP (Table 1) jeopardizes the formation of longer-fused porphyrin chains.
 | ||
| Scheme 2 Representation of the competing mechanisms during the oCVD reaction of (a) ZnDPP, (b) MgDPP, and (c) AgDPP porphyrins and classification of their intermolecular C–C coupling (k1), demetallation (k2, represented by grey arrows) and re-metalation (k3, represented by orange arrows) rates. Intramolecular cyclization and chlorination side reactions are shown representatively. | ||
Conversely, a lower extension of the intermolecular coupling was evidenced by UV-Vis-NIR for the pPdDPP, pRuDPP, pFeDPP, pH2DPP, and pAgDPP films (Fig. 1c). In the case of pPdDPP, pRuDPP, and pFeDPP, LDI-HRMS does not provide any insight that could explain such behavior. Nevertheless, both PdDPP and FeClDPP possess high redox potentials (E1/2) that likely contribute to less efficient dehydrogenative coupling rates. In addition, the SOMO of palladium(II) diphenyl porphyrin radical cations is known to possess an a1u symmetry that induces regioselectivity towards meso–beta linkage.30–32 Therefore, predominately doubly linked fused porphyrin tapes are formed and absorption remains moderate in the NIR region.
On the other hand, ruthenium can exhibit multiple oxidation states when chelated in a porphyrin macrocycle.33 The oxidation of the central metal cation may therefore compete with the oxidative coupling of the porphyrins. Additionally, ruthenium porphyrins easily form oxo complexes,33 which may also interfere with the formation of highly conjugated and long-fused porphyrin chains. Di-nuclear ruthenium porphyrin oxo complexes, i.e. [(RuDPP)2 +O]+ and [(RuDPP)3 +2O]+, are notably observed in the LDI-HRMS spectra of the sublimed reference film sRuDPP (Fig. S7i, ESI†).
In the case of pH2DPP, none of the oligomers detected by LDI-HRMS contained free-based porphyrin units (Fig. S7k, ESI†). Specifically, all the porphyrin units constituting the oligomers seem chelated with iron originating from the oxidant. The importance of the metalation reaction is confirmed by the observation of the XPS N 1s core level spectrum where a single contribution is observed at 399.1 eV, the same as that corresponding to the nitrogen environment in pFeDPP films (Fig. S12, ESI†). This indicates that the metalation of the porphyrin macrocycle is essential to form fused porphyrin oligomers in oCVD, even though the high redox potential (E1/2) of FeClDPP yields weak dehydrogenative coupling rates.
The reactivity of AgDPP in oCVD points to a fast rate of demetallation (forming H2DPP) followed by iron(III) incorporation into the porphyrin core (Scheme 2c). Indeed, mostly iron-based oligomers are detected in the LDI-HRMS spectrum of pAgDPP (Fig. 3e and Fig. S7h, ESI†), while a single contribution, corresponding to a metal–nitrogen environment shifted to higher binding energy compared to the sublimed AgDPP monomer, is observed in the XPS N 1s core level spectrum (Fig. 3f). In addition, AgDPP has a relatively high redox potential (compared to ZnDPP and MgDPP), and it is even higher for FeClDPP, making the oxidative C–C coupling reaction once again difficult. As a result, most likely shorter oligomers will be formed during the oCVD of AgDPP, with a lower degree of fusing, in good agreement with the previous analysis of the UV-Vis-NIR spectra of both the as-deposited and dichloromethane-rinsed oCVD coatings.
oCVD reactivity versus optoelectronic properties of the fused-metalloporphyrin coatings
The expansion of the π-conjugated system is known to increase the energy of the highest occupied molecular orbital (HOMO) and leads to a low excitation gap in directly fused porphyrin tapes.9 Consistently, the dehydrogenative coupling of metalloporphyrins by oCVD leads to a broadened and shifted valence band edge towards lower binding energies compared to the sublimed reference films, as shown in Fig. S13 of the ESI.† From the valence band region of the XPS spectra, the valence band maxima (VBM) of the metalloporphyrin-based films were estimated (Fig. S14 of the ESI†). The values are summarized in Table S3 (ESI†) and presented in Fig. 4a. | ||
| Fig. 4 (a) Valence band maxima (VBM) estimated from the XPS analysis of sublimed and oCVD processed films. (b) Histograms of the currents obtained from the c-AFM measurements of the oCVD films. | ||
Interestingly, pCuDPP and pNiDPP thin films show the most significant reduction of their VBMs, with a decrease of 1.28 eV. This is consistent with the previous UV-Vis-NIR analysis evidencing the prevalence of highly conjugated fused porphyrin chains (likely containing a significant proportion of triply linked units). Although highly conjugated structures are suggested by the optical characterization, pCoDPP and pZnDPP thin films show a lower reduction of their VBMs. The shifts of the VBMs were even less prominent for pPtDPP, pPdDPP, pAgDPP, and pRuDPP thin films, as shown in Fig. 4a. This behavior suggests a lower efficiency of the intermolecular dehydrogenative coupling for these cations, in agreement with the UV-Vis-NIR analysis. A negligible VBM shift is observed for pMgDPP, pFeDPP, and pH2DPP thin films in respect of their reference sublimed films. Multiple behaviors can be responsible for these low VBM shifts upon oCVD processing: i.e. high redox potentials (E1/2) for FeClDPP, PdDPP and PtDPP, a pronounced ability to form meso–beta linkages for PdDPP and RuDPP, oxidation of the central metal cation for CoDPP, demetallation of the porphyrin macrocycle for ZnDPP, AgDPP and MgDPP, and re-metalation with iron for H2DPP, AgDPP, MgDPP and ZnDPP. Particularly, the valence band edge XPS spectra of pH2DPP, pMgDPP and pFeDPP coatings almost all overlap, confirming the demetallation and re-metalation with iron originating from the oxidant.
The intrinsic conductivity of the directly fused porphyrin CP thin films arises from the combination of multiple features including the conjugation degree and length of the fused porphyrin tapes,34 the oxidation state of the central metal cation, intramolecular cyclization, and the demetallation/re-metalation of the porphyrin core. Indeed, demetallation and further re-metalation with Fe3+ from the oxidising agent might lead to the presence of axial ligands that hinder the π–π stacking of the chains, and thus the conductivity. Moreover, the presence of Fe3+ as the metal center might also favor the electron trapping, also affecting the conductivity. The c-AFM measurements reveal the notable influence of the metal cation on the intrinsic conductivity of the fused porphyrin films, as shown in Fig. 4b.
The negative impact of the demetallation and re-metalation of the porphyrin macrocycles is reflected on the moderate conductivity of the pZnDPP films supposedly composed of triply fused porphyrin chains but partly composed of FeClDPP units. Such behavior is even more pronounced for pMgDPP that exhibits the lowest conductivity despite the fair dehydrogenative coupling rate, due to the significant demetallation and re-metalation. As result, a similar electronic feature is observed for pH2DPP, pMgDPP, and pFeDPP oCVD coatings, in agreement with the VBM estimated from the XPS analysis shown in Fig. 4a. It is worth noting that the lowest conductivity displayed for the pH2DPP and pFeDPP coatings is expected from the lower yield in the formation of extended fused porphyrin tapes and the presence of Fe3+ at the center of the porphyrin macrocycles. On the other hand, the excellent conductivity of the pCuDPP film is in good agreement with the UV-Vis-NIR and XPS characterization pointing to a high efficiency of the intermolecular coupling, no alteration of the porphyrin core and the highest shift of the VBM.
Surprisingly, the pRuDPP film also displays a high conductivity, despite the lower coupling efficiency previously suggested for this coating. This observation hints at the presence of large oligomers due to a higher conjugation length of single-fused units. Conversely, the pNiDPP thin film was less conductive than expected from the high VBM shift, pointing to a poor ratio of triple:double linkages, and possible axial ligands at the Ni2+ center, as suggested in previous reports with di-mesityl-substituted porphyrins (DMPs).23
The careful assessment and understanding of the optoelectronic properties are key to further photo-electrocatalytic applications – including solar-assisted solar fuel production – with directly fused metalloporphyrin CP photoelectrodes processed by oCVD. Therefore, the optical band gaps (Eg) of the fused metalloporphyrin films were estimated from the absorbance spectra using the Tauc plots, as presented in Fig. S15 of the ESI,† and summarized in Table S4 of the ESI.† All the conjugated metalloporphyrin coatings showed Eg values between 2.25 eV and 2.69 eV, a suitable interval for effective photon absorption, with the possibility of generating a sufficient driving force toward photocatalytic solar fuel production.35 Furthermore, given the VBM previously estimated from the valence band edge region of the XPS (Fig. 4a), and the Eg calculated from the absorbance spectra, the energy band alignment for the fused-metalloporphyrin coatings chelating different metal cations was estimated, as presented in Fig. 5.
 | ||
| Fig. 5 Energy band diagram of the fused metalloporphyrin thin films (pMDPP, M= Zn, Cu, Co, Mg, Ni, Pt, Pd, Ag, Ru, FeCl, and H2). The redox potentials for proton reduction and water oxidation reactions in acid media are presented as references. | ||
All the directly fused porphyrin thin films show adequate conduction band minimum (CBM) values to allow electron injection toward the hydrogen evolution reaction (HER). Moreover, the role of Zn, Cu and Co species as catalytic-active centers boosting the HER efficiency in porphyrin-based systems36–40 has been reported, in addition to the earth-abundance and non-toxicity of these elements. Therefore, the oCVD coatings of fused-metalloporphyrins of Zn(II), Cu(II) and Co(II) are highly promising for the development of photo-electrocatalytic hydrogen production devices.
Indeed, the theoretical maximum photocurrent achievable for each conjugated metalloporphyrin film can be estimated from its corresponding absorbance spectrum as: jabs = (e/hc)∫λ·I(λ)λ·(1 − 10−A)dλ, where I(λ) is the spectral irradiance, e is the elemental charge, h is the Planck constant and c is the speed of light. These values are summarized in Table S4 of the ESI.† The pCoDPP film shows the highest expected photocurrent value, ideally being able to reach up to 5.77 mA cm−2, which corresponds to 5.29% of solar-to-hydrogen efficiency (STH). However, the combination of metalloporphyrins with suitable band alignment into a heterostructured device, providing both enhanced light absorption and charge transfer, can lead to a considerable increase in the performance of photoelectrodes based on these materials. In addition, the coatings from pNiDPP, pPtDPP, and pPdDPP could also be suitable for hole injection toward the oxygen evolution reaction, due to the adequate VBM position, below the water oxidation potential. It is worth noting that Ni species are well-known active catalytic sites for the OER,41,42 which can be beneficial to improve the reaction kinetics at the metalloporphyrin-based photoelectrode surface.
Therefore, owing to their facile processability when operating from the gas-phase and their suitable optoelectronic properties – as well as the ability of the existing metal cations to act as active catalytic sites – fused-metalloporphyrin coatings processed by oCVD can emerge as a new generation of photoelectrode materials for solar-assisted fuel production.
Conclusions
We have investigated the impact of the central metal cation on the dehydrogenative coupling of metalloporphyrins and on the optoelectronic properties of the resulting conjugated metalloporphyrin-based photoelectrodes. It was found out that the nature of the chelated cation has a strong influence on the regioselectivity for the C–C coupling, as well as on the occurrence of side reactions. In particular, the demetallation of the porphyrin core plays a significant role in the constitution of conjugated polymers, following different kinetics according to the nature of the metal cation. The impact of the reactivity during the oCVD was evidenced in the optical band gap, band alignment and conductivity analysis of each metalloporphyrin coating. These results pave the way for the investigation and further implementation of conjugated metalloporphyrin-based photoelectrodes for clean and efficient solar-assisted fuel production.Author contributions
D.C.M: investigation, formal analysis, writing – original draft, and writing – review and editing; E.V., M. H. and G. F: investigation, formal analysis, and writing – review and editing; N. B: conceptualization, methodology, formal analysis, funding acquisition, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the European Research Council (ERC) through the ERC Consolidator Grant project CLEANH2 (grant agreement no. 865985). B. Marcolini, R. Vaudemont, J. Guillot, C. Vergne and P. Grysan of LIST are acknowledged for the TGA, XPS and c-AFM measurements.References
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS PubMed.
- I. Obraztsov, W. Kutner and F. D'Souza, Sol. RRL, 2017, 1, 1600002 CrossRef.
- V. Nikolaou, G. Charalambidis, K. Ladomenou, E. Nikoloudakis, C. Drivas, I. Vamvasakis, S. Panagiotakis, G. Landrou, E. Agapaki, C. Stangel, C. Henkel, J. Joseph, G. Armatas, M. Vasilopoulou, S. Kennou, D. M. Guldi and A. G. Coutsolelos, ChemSusChem, 2021, 14, 961–970 CrossRef CAS PubMed.
- D. Khusnutdinova, A. M. Beiler, B. L. Wadsworth, S. I. Jacob and G. F. Moore, Chem. Sci., 2017, 8, 253–259 RSC.
- M. Joseph and S. Haridas, Int. J. Hydrogen Energy, 2020, 45, 11954–11975 CrossRef CAS.
- A. Aziz, A. R. Ruiz-Salvador, N. C. Hernández, S. Calero, S. Hamad and R. Grau-Crespo, J. Mater. Chem. A, 2017, 5, 11894–11904 RSC.
- E. Tavakoli, A. Kakekhani, S. Kaviani, P. Tan, M. M. Ghaleni, M. A. Zaeem, A. M. Rappe and S. Nejati, J. Am. Chem. Soc., 2019, 141, 19560–19564 CrossRef CAS PubMed.
- J. Wang, L. Xu, T. Wang, S. Chen, Z. Jiang, R. Li, Y. Zhang and T. Peng, Adv. Funct. Mater., 2021, 31, 2009819 CrossRef CAS.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed.
- T. Ishizuka, Y. Saegusa, Y. Shiota, K. Ohtake, K. Yoshizawa and T. Kojima, Chem. Commun., 2013, 49, 5939–5941 RSC.
- M. C. Scharber and N. S. Sariciftci, Adv. Mater. Technol., 2021, 2000857 CrossRef CAS.
- H. Jia, Z. Sun, D. Jiang and P. Du, Chem. Mater., 2015, 27, 4586–4593 CrossRef CAS.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- D. Khusnutdinova, B. L. Wadsworth, M. Flores, A. M. Beiler, E. A. Reyes Cruz, Y. Zenkov and G. F. Moore, ACS Catal., 2018, 8, 9888–9898 CrossRef CAS.
- A. M. Huerta-Flores, G. Bengasi, K. Baba and N. D. Boscher, ACS Appl. Energy Mater., 2020, 3, 9848–9855 CrossRef CAS.
- E. A. Reyes Cruz, D. Nishiori, B. L. Wadsworth, D. Khusnutdinova, T. Karcher, G. Landrot, B. Lassalle-Kaiser and G. F. Moore, ChemElectroChem, 2021, 8, 3614–3620 CrossRef CAS.
- G. Bengasi, K. Baba, G. Frache, J. Desport, P. Gratia, K. Heinze and N. D. Boscher, Angew. Chem., Int. Ed., 2019, 58, 2103–2108 CrossRef CAS PubMed.
- K. Baba, G. Bengasi, D. El Assad, P. Grysan, E. Lentzen, K. Heinze, G. Frache and N. D. Boscher, Eur. J. Org. Chem., 2019, 2368–2375 CrossRef CAS.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1801564 CrossRef.
- G. Bengasi, J. S. Desport, K. Baba, J. P. Cosas Fernandes, O. De Castro, K. Heinze and N. D. Boscher, RSC Adv., 2020, 10, 7048–7057 RSC.
- T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- G. Bengasi, K. Baba, O. Back, G. Frache, K. Heinze and N. D. Boscher, Chem. – Eur. J., 2019, 25, 8313–8320 CrossRef CAS PubMed.
- G. Bengasi, L. Quétu, K. Baba, A. Ost, J. P. Cosas Fernandes, P. Grysan, K. Heinze and N. D. Boscher, Eur. J. Inorg. Chem., 2020, 1938–1945 CrossRef CAS.
- G. Bengasi, R. Meunier-Prest, K. Baba, A. Kumar, A. L. Pellegrino, N. D. Boscher and M. Bouvet, Adv. Electron. Mater., 2020, 6, 2000812 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 714–723 CAS.
- K. Baba, G. Bengasi, F. Loyer, J. P. C. Fernandes, D. El Assad, O. De Castro and N. D. Boscher, ACS Appl. Mater. Interfaces, 2020, 12, 37732–37740 CrossRef CAS PubMed.
- X. Ke, R. Kumar, M. Sankar and K. M. Kadish, Inorg. Chem., 2018, 57, 1490–1503 CrossRef CAS PubMed.
- K. Laba, M. Lapkowski, D. L. Officer, P. Wagner and P. Data, Electrochim. Acta, 2020, 330, 135140 CrossRef CAS.
- M. Speckbacher, L. Yu and J. S. Lindsey, Inorg. Chem., 2003, 42, 4322–4337 CrossRef CAS PubMed.
- Y. Nakamura, N. Aratani, A. Tsuda, A. Osuka, K. Furukawa and T. Kato, J. Porphyrins Phthalocyanines, 2003, 07, 264–269 CrossRef CAS.
- C.-M. Feng, Y.-Z. Zhu, S.-C. Zhang, Y. Zang and J.-Y. Zheng, Org. Biomol. Chem., 2015, 13, 2566–2569 RSC.
- M. Kamo, A. Tsuda, Y. Nakamura, N. Aratani, K. Furukawa, T. Kato and A. Osuka, Org. Lett., 2003, 5, 2079–2082 CrossRef CAS PubMed.
- J. P. Collman, C. E. Barnes, P. J. Brothers, T. J. Collins, T. Ozawa, J. C. Gallucci and J. A. Ibers, J. Am. Chem. Soc., 1984, 106, 5151–5163 CrossRef CAS.
- N. Algethami, H. Sadeghi, S. Sangtarash and C. J. Lambert, Nano Lett., 2018, 18, 4482–4486 CrossRef CAS PubMed.
- R. van de Krol, Y. Liang and J. Schoonman, J. Mater. Chem., 2008, 18, 2311–2320 RSC.
- Y. Liu, Y. Han, Z. Zhang, W. Zhang, W. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613–2622 RSC.
- H. Lei, Y. Wang, Q. Zhang and R. Cao, J. Porphyrins Phthalocyanines, 2020, 24, 1361–1371 CrossRef CAS.
- S. Cui, M. Qian, X. Liu, Z. Sun and P. Du, ChemSusChem, 2016, 9, 2365–2373 CrossRef CAS PubMed.
- B. B. Beyene, S. B. Mane and C.-H. Hung, J. Electrochem. Soc., 2018, 165, H481–H487 CrossRef CAS.
- P. T. Smith, B. P. Benke, L. An, Y. Kim, K. Kim and C. J. Chang, ChemElectroChem, 2021, 8, 1653–1657 CrossRef CAS.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W.-G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225 CrossRef CAS.
- I. Elghamry, A. S. Alablan, M. A. Alkhalifah and M. E. Abdelsalam, J. Electrochem. Soc., 2021, 168, 016510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc05452j |
| This journal is © The Royal Society of Chemistry 2022 |