
Asymmetric sky-blue thermally-activated delayed fluorescence emitters bearing tris(triazolo)triazine moiety for solution-processable organic light-emitting diodes†
Zhou
Fang
a,
Shengyue
Wang
a,
Junxu
Liao
b,
Xinrui
Chen
a,
Yuanyuan
Zhu
a,
Weiguo
Zhu
 *a and
Yafei
Wang
*a
*a and
Yafei
Wang
*a
aJiangsu Collaborative Innovation Center of Photovoltaic Science and Engineering, Jiangsu Engineering Laboratory of Light-Electricity-Heat Energy-Converting Materials and Applications, School of Materials Science & Engineering, Changzhou University, Changzhou 213164, China. E-mail: zhuwg18@126.com; qiji830404@hotmail.com
bSchool of Chemical Engineering and Energy Technology, Dongguan University of Technology, Dongguan, 523808, China
First published on 4th January 2022
Abstract
An efficient blue thermally-activated delayed fluorescence (TADF) emitter is still a challenge in solution-processed organic light-emitting diodes (OLEDs). In this contribution, two asymmetric molecules, TTT-Ac and TTT-2Ac were synthesized and characterized, in which the tris(triazolo)triazine (TTT) fragment is used as the acceptor core and different numbers of acridine (Ac, one or two) units are considered as the donor unit. Both compounds display clear TADF character, concomitant with the emission between 420 and 450 nm in a doped PMMA film. Compared to TTT-2Ac, TTT-Ac shows blue-shifted emission due to its weaker intramolecular charge transfer effect. The solution-processed OLEDs achieve a maximum external quantum efficiency (EQEmax) of 9.26% and 8.10% with the emission peak at about 470 nm for TTT-Ac and TTT-2Ac, respectively. Furthermore, the TTT-Ac-based solution-processed OLED exhibits an EQEmax of 10.01% after optimization of the emitter thickness. This research focuses on the molecular structure–property relationship of TTT-based molecules, which enriches the TTT-based solution-processable sky-blue TADF emitter.
Introduction
Since the first example of an organic thermally-activated delayed fluorescent (TADF) material was utilized in organic light-emitting diodes (OLEDs) by Adachi's research group,1 organic TADF materials have attracted considerable interest from both academe and industry due to their high efficiency and metal-free atoms.2,3 Compared with traditional organic fluorescent materials, TADF materials can harvest both singlet and triplet excitons through the reverse intersystem crossing process, leading to theoretical 100% internal quantum efficiency.4–7 In addition, metal atoms are absent from organic TADF materials, making them definitely low-cost and eco-friendly relative to phosphorescent materials.8–10 According to previous reports,11–15 the prerequisite for constructing a TADF molecule is to achieve a small energy splitting (ΔEST) between singlet (S1) and triplet (T1) excited states, which usually is realized via minimizing the spatial overlap between the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO).To this end, the most effective strategy for TADF materials is to design a twisted molecule with a donor–acceptor/donor–π–acceptor (D–A/D–π–A) skeleton.16–21 This molecular structure can effectively separate the HOMO and LUMO, thus reducing the energy gap between S1 and T1 states. Therefore, many high-efficiency TADF emitters have been developed based on D–A/D–π–A systems, and their external quantum efficiencies (EQEs) in OLEDs exceed 20%.22–29 Unfortunately, blue TADF emitters still lag far behind green and red ones because of the bathochromic shift in the D–A/D–π–A molecules caused by strong intramolecular charge transfer (ICT). Thus, how to balance ICT and blue emission is very important for blue TADF emitters. Recently, some effective methods to achieve blue TADF emitters have been reported, such as decreasing the conjugation degree, utilization of a weak acceptor and realizing multiresonant-TADF.30–33 Although certain blue TADF emitters have obtained excellent efficiency in devices,34–37 the vast majority of them are prepared via full vapor deposition, involving complicated fabrication procedures and high cost. In addition, the reported blue TADF emitters usually contain an acceptor with low-rigidity structure, such as triazine, triphenyl boron, diphenyl sulphone fragments,38–43 which is not beneficial to suppress the molecular deformation in the excited state. Therefore, there is much space to develop novel blue TADF emitters for solution-processable OLEDs.
The tristriazolotriazine (TTT) moiety is a polyazin, which is a popular candidate in liquid crystal studies due to its rigid and planar structure.44,45 Impressively, this fragment was employed as an acceptor group in blue TADF emitters by Yang, Eli and our groups at the same time (Fig. S1, ESI†).46–48 These researches demonstrated that the TTT unit possesses moderate electron-withdrawing ability and high molecular rigidity, which can be employed as an acceptor in blue TADF emitters. After optimizing the device, we recently reported the sky-blue TADF emitter of TTT-Ph-Ac with a highest EQE of 22% in a solution-processed OLED.49 However, the electroluminescent (EL) emission is at about 490 nm, not the real blue region. Very recently, Hojo et al. also reported a green TADF emitter based on a TTT core (Fig. S1, ESI†).50 Unfortunately, the device performance of these emitters was not reported. It is worth noting that all these TTT-based emitters are symmetric molecules with three donor groups, probably a simple synthetic method. As is known, decreasing the number of donor units can effectively reduce the ICT effect, and then lead to a hypochromic shift. With this in mind, the proposal to prepare asymmetric emitters based on TTT acceptor with one or two donor units was made to realize blue emission. Herein, two TTT-based emitters, named TTT-Ac and TTT-2Ac, with one and two acridine (Ac) moieties, respectively, were synthesized and characterized. As expected, both emitters showed blue-shifted emission in solution and film compared to the TTT-based TADF material with three Ac fragments. A solution-processable device employing TTT-Ac as the emitter achieved a maximum EQE of 10.01% with the EL emission peak at 474 nm.
Results and discussion
Materials and thermal properties
As shown in Scheme 1, starting from the commercial materials of 2,4,6-trichloro-1,3,5-triazine, 5-(4-bromobenzene)-1H-tetrazole and 5-phenyltetrazole, the key precursors of TTT-Br and TTT-2Br were prepared by a one-pot Huisgen reaction in a yield of 25% and 38%, respectively. Then, a Buchwald–Hartwig coupling reaction between TTT-Br/TTT-2Br and Ac provided the target TADF emitters (TTT-Ac and TTT-2Ac) in the presence of Pd catalyst, which was evidenced by 1H NMR, 13C NMR, TOF-MS and HRMS (Fig. S2–S11, ESI†). Both compounds exhibited good solubility in ordinary organic solvents, such as toluene, CH2Cl2, CHCl3 and THF. Impressively, excellent thermal stability was found, with the decomposition temperature (Td, at 5 wt% weight loss) determined to be 415 and 417 °C for TTT-Ac and TTT-2Ac, respectively (Fig. S12, ESI†).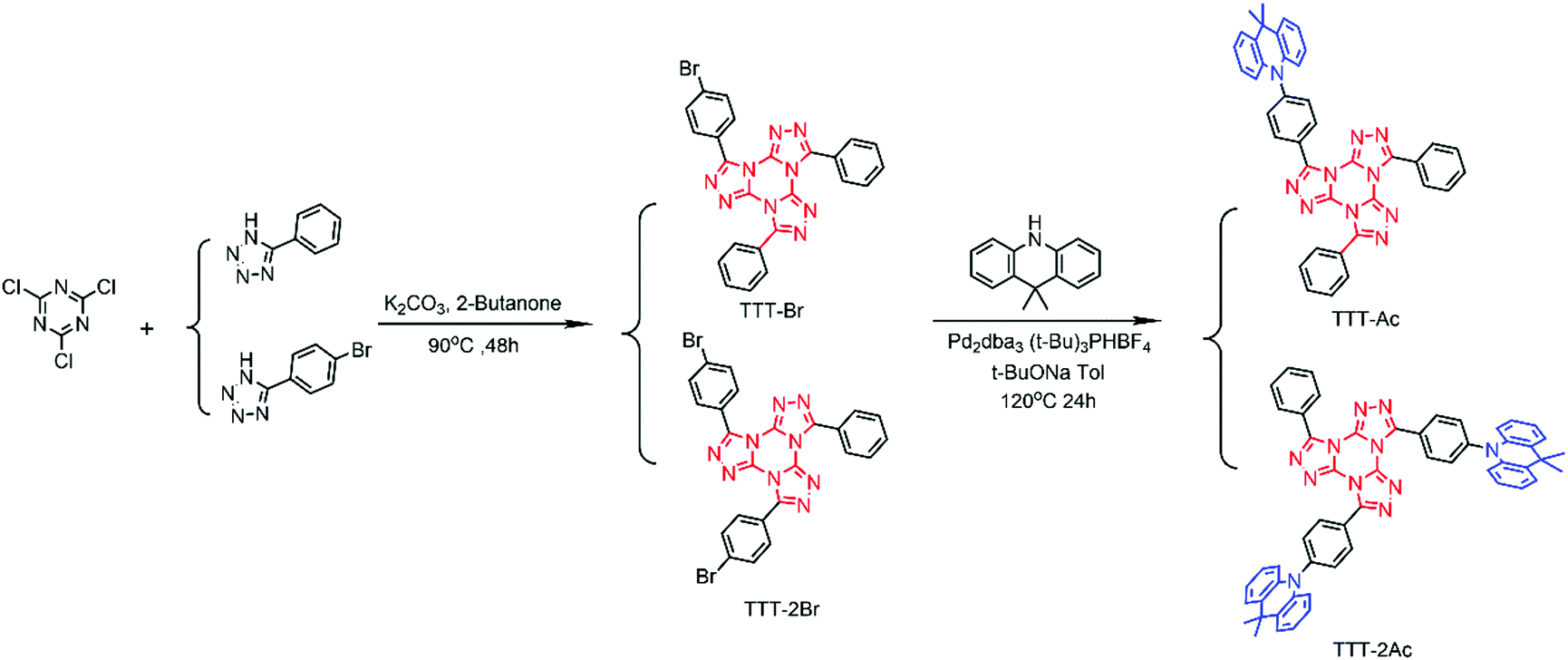 | ||
| Scheme 1 Synthetic route of the TADF emitters. | ||
Theoretical calculations
To better explore the geometric configurations and frontier molecular orbital energy levels of both emitters, density functional theory simulations were conducted based on Gaussian 09 program (b3lyp/6-31g(d)). As shown in Fig. 1, both emitters show a twisted molecular configuration, in which large spatial separations between HOMO and LUMO were observed from orbital pictures. The LUMOs of both compounds are mainly distributed on the TTT acceptor, while the HOMO distributions are mainly governed by the donor units. It is notable that HOMOs and LUMOs of both molecules are weakly overlapped at the bridged phenyl ring, indicating the possibility of high emission efficiency. The theoretical ΔEST values (energy gap between singlet and triplet energies) are evaluated to be 0.0071 for TTT-Ac and 0.0068 for TTT-2Ac. Such small ΔEST values are very conducive to reverse intersystem crossing from T1 to S1. The small ΔEST can be assigned to the vertical twist between the donor and bridged phenyl ring caused by strong electron-donating ability of the Ac unit according to our previous work. However, the amount of donor moieties seems to have less influence in the distribution of HOMO and LUMO. In conclusion, both molecules have separated distribution of HOMO and LUMO while being slightly overlapped at bridged phenyl ring, which may demonstrate great TADF characteristics.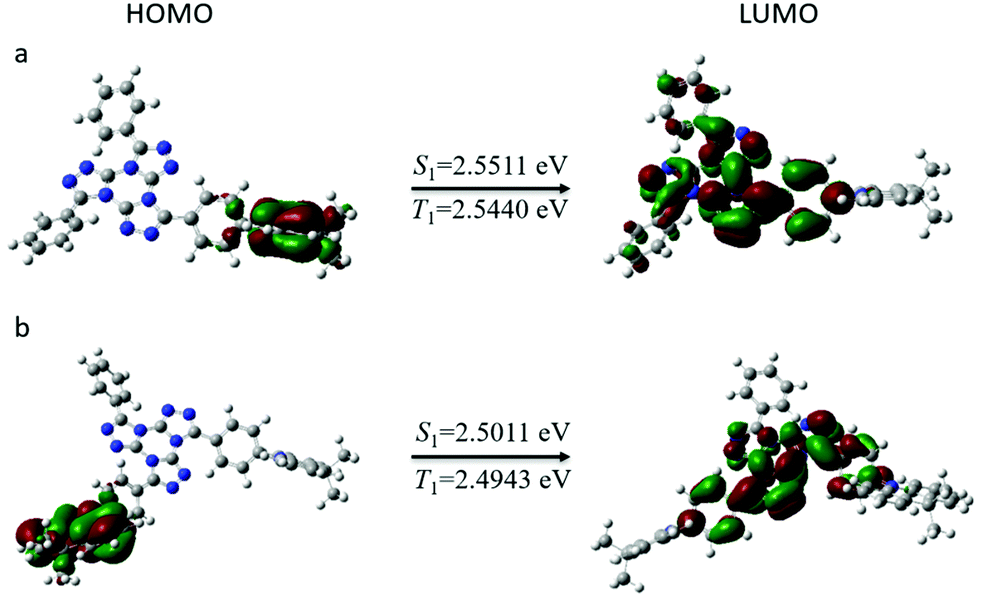 | ||
| Fig. 1 The calculated HOMO and LUMO distributions, and singlet and triplet energies of the compounds: (a) TTT-Ac and (b) TTT-2Ac. | ||
Photophysical properties
The ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra in both toluene solution and doped PMMA films at room temperature are depicted in Fig. 2, and the relevant data are listed in Table 1. Both emitters exhibit similar absorption spectra with two clear absorption bands from 250 nm to 400 nm (Fig. 2a). The intense absorption bands at 283 nm are assigned to the π–π* transition of the aromatic backbone, while the weak absorption bands at about 355 nm are ascribed to the ICT from the donor to the acceptor units. It is worth noting that there is a slightly red-shifted absorption band for TTT-2Ac compared to that for TTT-Ac due to the stronger ICT effect caused by one more donor unit.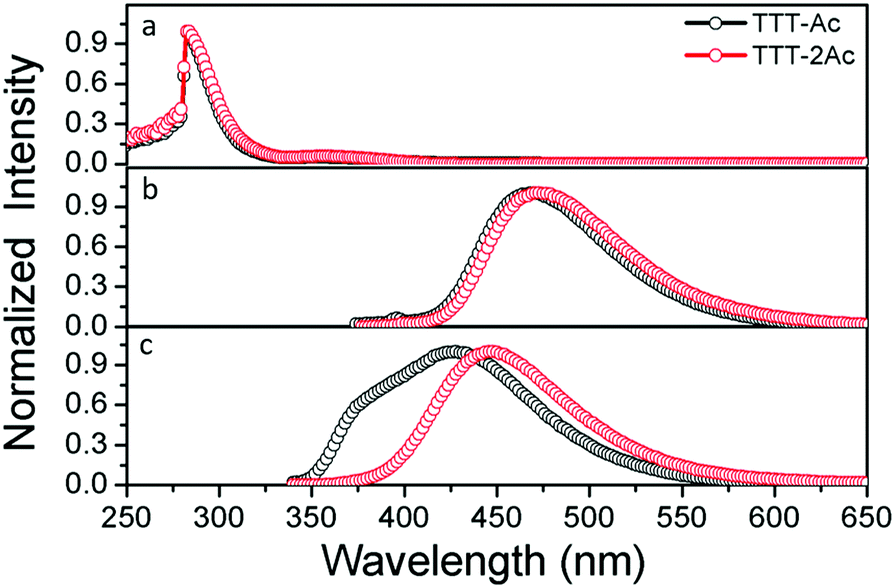 | ||
| Fig. 2 UV-Vis spectra (a) and PL spectra measured in toluene solution (b) and PL spectra in doped PMMA film (c) at room temperature. | ||
| Compound | λ abs /nm | λ em /nm | λ em /nm | τ p /ns | Φ p /% | τ d /μs | Φ d /% | Φ total /% | T d /°C | E g opt |
|---|---|---|---|---|---|---|---|---|---|---|
| a In toluene at room temperature. b 10 wt% in PMMA at room temperature. c In N2 at a heating rate of 20 °C min−1. | ||||||||||
| TTT-Ac | 283, 353 | 468 | 426 | 10.9 | 21 | 27.2 | 42 | 63 | 415 | 2.99 |
| TTT-2Ac | 283, 355 | 471 | 446 | 11.9 | 10 | 20 | 37 | 47 | 413 | 2.96 |
At an excitation wavelength of 350 nm, the PL spectra with emission peaks at 468 and 471 nm are observed in dilute toluene solution for TTT-Ac and TTT-2Ac (Fig. 2b), respectively. Compared to our previous reports,51 this result demonstrates that decreasing the number of donor moieties effectively leads to blue-shifted emission owing to a reduced ICT effect. The broad and structureless shape of the PL profiles implies that the emission is mainly generated from the charge transfer (CT) state. In addition, the solvent-dependent emission spectra (Fig. S13, ESI†) also point to there being a strong CT state in both emitters, evidenced by the clear solvatochromism effect. Compared to solution, the doped PMMA films exhibit a blue shift with emission peaks at 426 and 446 nm for TTT-Ac and TTT-2Ac, respectively (Fig. 2c). Two factors could be responsible for this phenomenon: (i) the suppressed molecular motions in the solid state; and (ii) and the polarity of the PMMA film.51 Owing to the stronger ICT effect, compound TTT-2Ac shows distinct red-shifted emission spectra compared with TTT-Ac in both solution and film. Impressively, a shoulder at about 380 nm is detected for TTT-Ac, which can be attributed to emission from a localized state of the Ac unit.52
The low-temperature (77 K) fluorescence and phosphorescence spectra of both emitters were measured in doped PMMA (10 wt%) film (Fig. 3a and b). According to the onsets of fluorescence and phosphorescence spectra, the S1/T1 energy levels were evaluated to be 3.19/2.84 eV for TTT-Ac and 3.09/2.9 eV for TTT-2Ac. Therefore, the corresponding ΔEST values were calculated to be 0.35 eV (TTT-Ac) and 0.19 eV (TTT-2Ac). The smaller ΔEST of TTT-2Ac was attributed to its stronger ICT effect. To verify the TADF characteristics of the emitters, the transient PL decay profiles were measured in doped PMMA films at room temperature. Both emitters showed double-exponential decay curves consisting of prompt and delayed components. The lifetimes of prompt/delayed components were 1.89 ns/44.79 μs for TTT-Ac and 10.04 ns/34.6 μs for TTT-2Ac. Both the small ΔEST values and the delayed components indicate the participation of the triplet exciton through the reverse intersystem crossing process. The PL quantum yields (PLQYs) of the two emitters in doped PMMA films were measured to be 63% and 47% for TTT-Ac and TTT-2Ac, respectively. Based on the PLQY and lifetime, the photophysical constants, such as the rate constants of fluorescence (kr), nonradiative transition (knr), intersystem crossing (kISC) and reverse intersystem crossing (krISC), were calculated according to a previous method (formulas S(1)–S(6) and Table S1, ESI†). Compared to TTT-Ac, compound TTT-2Ac exhibits a higher krISC of 2.055 × 105 s−1, probably due to the smaller ΔEST and multiple channels. Unfortunately, relatively slow kr and large knr are determined for TTT-2Ac, leading to a small emission efficiency.
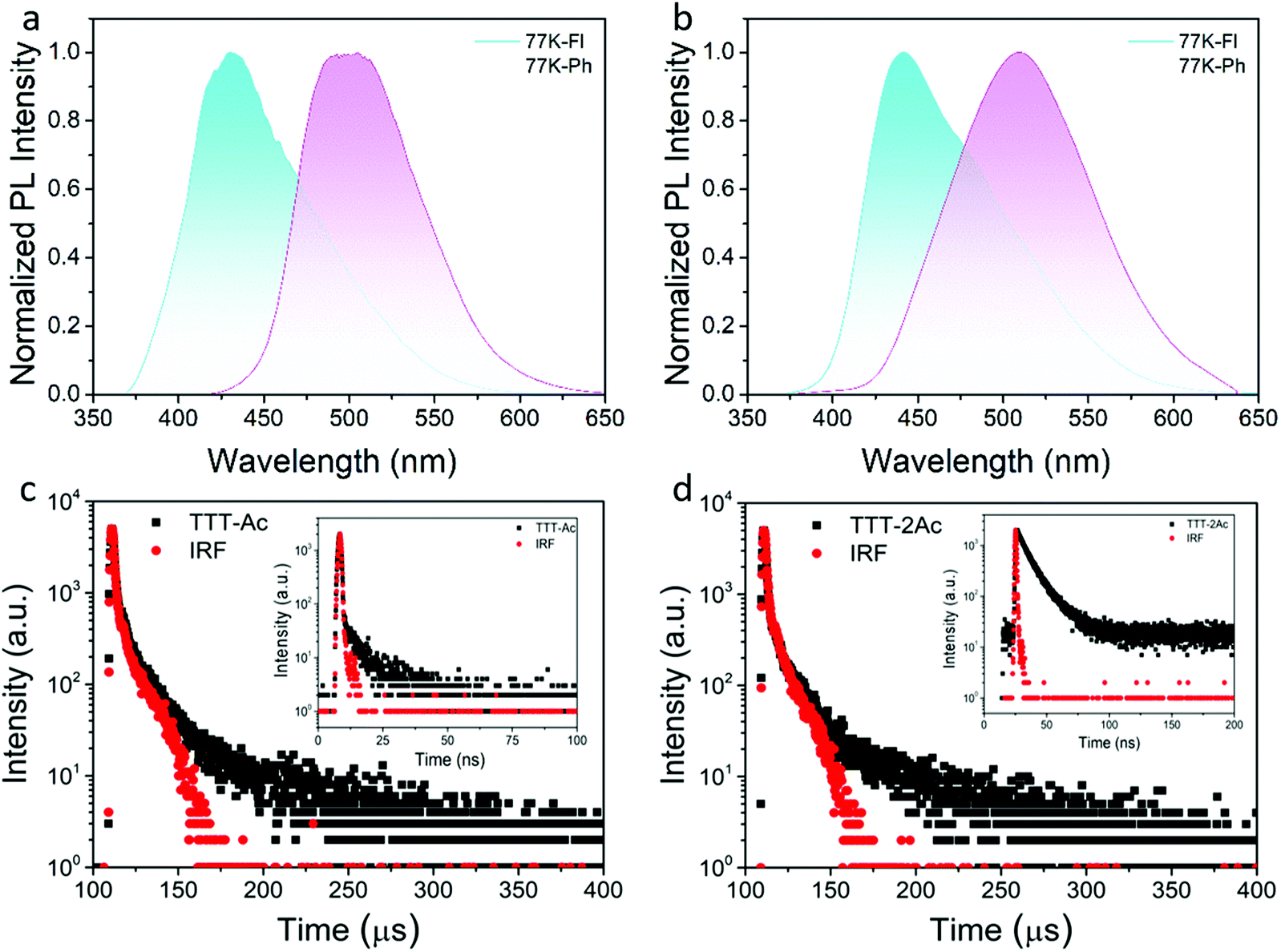 | ||
| Fig. 3 PL spectra of TTT-Ac (a) and TTT-2Ac (b) in 10 wt%-doped PMMA film at 77 K and transient PL prompt (inset) and delayed curves in 10 wt%-doped PMMA films at room temperature (c and d). | ||
Electrochemical properties
To investigate the electrochemical properties, cyclic voltammetry measurements were performed on the neat film in degassed CH3CN solution (0.1 M). As shown in Fig. S14 (ESI†), an obvious oxidation process was observed with reversible oxidation potentials at 0.95 and 0.68 V for TTT-Ac and TTT-2Ac, respectively. The HOMO energy levels were −5.29 and −5.13 eV for TTT-Ac and TTT-2Ac according to the empirical formula of EHOMO = −(VOX − VFc/Fc+ + 4.8) eV. Compared to TTT-Ac, TTT-2Ac shows a shallow HOMO level due to its stronger electron-donating ability. The LUMO energy levels were calculated to be −2.3 and −2.17 eV for TTT-Ac and TTT-2Ac based on their HOMO levels and optical bandgaps.Electroluminescent properties
To further explore the EL properties of both compounds, TTT-Ac and TTT-2Ac were used as dopants in solution-processed OLEDs with a device configuration of ITO/PEDOT:PSS (40 nm)/mCP:EML (X wt%, X = 10, 20, 30) (30 nm)/DPEPO (9 nm)/TmPyPB (45 nm)/LiF (0.5 nm)/Al (120 nm). The emitting layer is a blend of mCP and TTT-Ac or TTT-2Ac with different dopant concentrations of 10, 20 and 30 wt%. Schematic diagrams of the energy levels and chemical structures of materials in the devices are shown in Fig. S15(ESI†), and the relevant data are listed in Table 2.| Material | Dopant/wt% | V ON/V | L max/cd m−2 | CEmax/cd A−1 | EQEmax/% | CIE (x, y) | Peak/nm |
|---|---|---|---|---|---|---|---|
| TTT-Ac | 10 | 4.0 | 734.9 | 14.64 | 8.60 | (0.16, 0.19) | 468 |
| 20 | 3.6 | 1078 | 16.21 | 9.26 | (0.16, 0.21) | 470 | |
| 30 | 3.6 | 1205 | 13.44 | 7.33 | (0.16, 0.23) | 474 | |
| TTT-2Ac | 10 | 4.8 | 1011 | 13.45 | 7.04 | (0.16, 0.24) | 474 |
| 20 | 3.6 | 1384 | 16.61 | 8.10 | (0.17, 0.26) | 474 | |
| 30 | 3.6 | 1395 | 10.10 | 4.79 | (0.18, 0.30) | 482 |
As shown in Fig. 4, both emitters show sky-blue emissions in the devices, and their CIE (Commission International de l’Eclairage) coordinates vary from (0.16, 0.19) to (0.18, 0.30) (Fig. S16, ESI†). Similar to the PL profiles, the TTT-Ac-based devices exhibit blue-shifted emission relative to the TTT-2Ac-based devices. On the other hand, the host emission peaks are absence from the EL spectra, implying the complete energy transfer from mCP to the dopant.
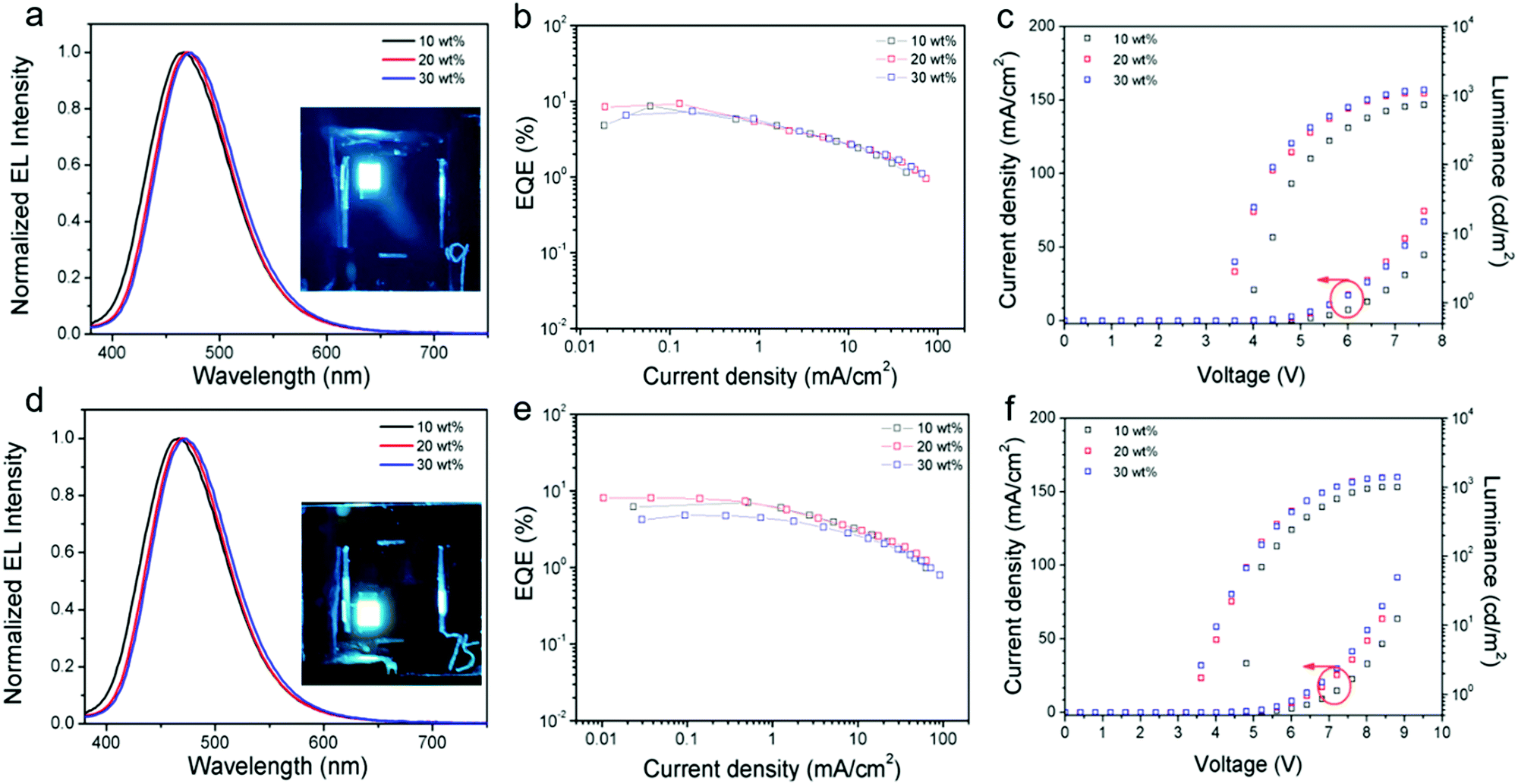 | ||
| Fig. 4 EL spectra (a) (inset: emission picture of the device); EQE–current density curves (b); and current density–voltage–luminance (J–V–L) curves (c) of TTT-Ac. EL spectra (d) (inset: emission picture of the device); EQE–current density curves (e); and current density–voltage–luminance (J–V–L) curves (f) of TTT-2Ac. | ||
As seen from the current density–voltage–luminance (J–V–L) curves, both current density and luminance are increased with increasing dopant concentration, suggesting a charge trapping mechanism in the devices. Both the emitters exhibit the best device performance at 20 wt% dopant concentration. The turn-on voltage (Von at 1 cd m−2) and maximum luminescence were 3.6 V/1205 cd m−2 and 3.6 V/1395 cd m−2 for TTT-Ac- and TTT-2Ac-based devices, respectively. Owing to the higher PLQY, the TTT-Ac-based device achieved a better performance with maximum current efficiency (CE) of 16.21 cd A−1 and EQE of 9.26%, while devices based on TTT-2Ac achieved a maximum CE of 16.61 cd A−1 and EQE of 8.10%.
Based on our previous report,49 the emitter layer thickness plays a key role in the device performance. Herein, taking TTT-Ac as an example, the influence of emitter layer thickness on the device performance is explored. The solution-processed devices with the same configuration of ITO/PEDOT:PSS (40 nm)/mCP:EML (X nm, X = 20, 25 and 30 nm)/DPEPO (9 nm)/TmPyPB (45 nm)/LiF (0.5 nm)/Al (120 nm) were fabricated, and the performance data of the devices are summarized in Fig. 5 and Fig. S17, Table S2 (ESI†). As expected, the devices showed the best performance with 25 nm EML layer, concomitant with the maximum CE of 17.37 cd A−1 and EQE of 10.01%. This phenomenon could be ascribed to the balanced hole and electron currents influenced by the thickness of the emitter layer.53 Compared to the reported TTT-based TADF emitters (Fig. S1 and S18, ESI†), TTT-Ac shows the bluest emission with high efficiency in solution-processable OLED.
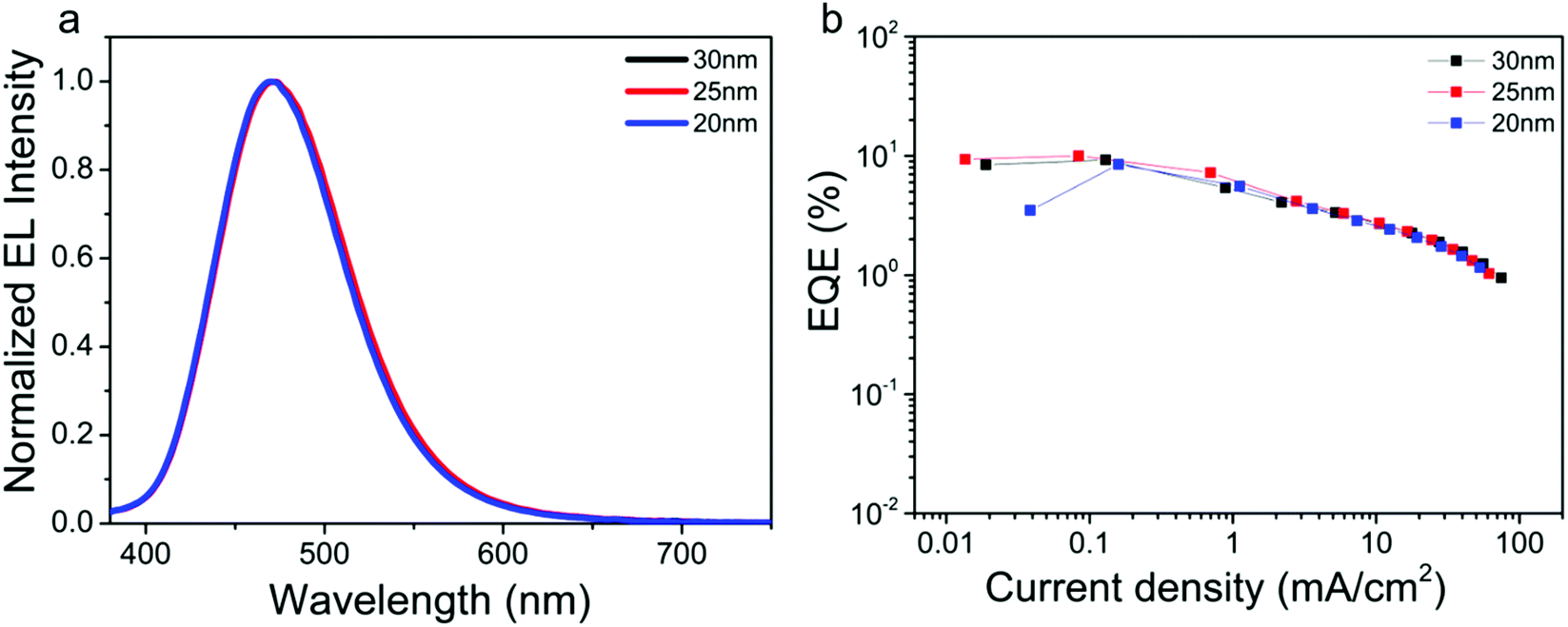 | ||
| Fig. 5 EL spectra (a) and EQE–current density curves (b) of TTT-Ac with different EML thickness of 20 wt% dopants. | ||
Conclusion
In conclusion, two sky-blue TADF emitters, TTT-Ac and TTT-2Ac, were prepared by combining the electron acceptor of TTT and one or two Ac fragments. The influence of the donor number on the photophysical properties was investigated in detail. The HOMO and LUMO of the two compounds were spatially separated, leading to small ΔEST. Intense deep blue emissions in doped films were obtained with PLQYs of 63% (426 nm) for TTT-Ac and 47% (446 nm) for TTT-2Ac. The solution-processed OLEDs based on TTT-Ac and TTT-2Ac achieved maximum EQEs of 9.26% at 470 nm and 8.10% at 474 nm, respectively. Furthermore, by optimizing the emitter layer thickness, the TTT-Ac-based solution-processed OLEDs exhibited a maximum EQE of 10.01%. This work provided a novel method for designing sky-blue emitters for solution-processed OLEDs.Experimental section
General procedure for synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane (1:1) as eluent to afford white solid (60 mg, yield 40%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.46 (d, J = 8.5 Hz, 2H), 8.19–8.15 (m, 4H), 7.71–7.58 (m, 9H), 7.49 (dd, J = 7.6, 1.4 Hz, 2H), 7.07–6.95 (m, 5H), 6.43 (d, J = 7.1 Hz, 2H), 1.71 (s, 6H). 13C NMR (126 MHz, CDCl3) δ (ppm) 151.00 (d, J = 15.1 Hz), 150.42, 143.64, 141.77, 140.67, 138.57, 132.08, 131.78, 130.20, 128.68, 126.07, 123.91, 121.64, 116.36, 109.47, 34.80, 32.00. TOF-MS (ESI) m/z calcd for C39H28N10 [M + H]+: 637.72; found: 637.29; HRMS (ESI) m/z found: 637.256144 C39H28N10 (Fig. S4, S6, S8 and S10, ESI†).
:dichloromethane (1:1) as eluent to afford yellow solid (70 mg, yield 40%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.48 (dd, J = 8.5, 2.7 Hz, 4H), 8.22–8.17 (m, 2H), 7.71–7.60 (m, 7H), 7.50 (d, J = 7.6 Hz, 4H), 7.08–7.02 (m, 4H), 6.99 (t, J = 7.4 Hz, 4H), 6.47–6.42 (m, 4H), 1.71 (s, 12H). 13C NMR (126 MHz, CDCl3) δ (ppm) 151.16, 150.47, 145.00, 140.85, 132.81, 132.19, 131.32, 130.73, 130.22, 128.76, 126.53, 125.31, 123.77, 123.40, 121.17, 114.56, 36.12, 31.04. TOF-MS (ESI) m/z calcd for C54H41N11 [M + H]+: 844.00; found: 844.348. HRMS (ESI) m/z found: 844.360839, C54H42N11 (Fig. S5, S7, S9 and S11, ESI†).
:dichloromethane (1:1) as eluent to afford white solid (60 mg, yield 40%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.46 (d, J = 8.5 Hz, 2H), 8.19–8.15 (m, 4H), 7.71–7.58 (m, 9H), 7.49 (dd, J = 7.6, 1.4 Hz, 2H), 7.07–6.95 (m, 5H), 6.43 (d, J = 7.1 Hz, 2H), 1.71 (s, 6H). 13C NMR (126 MHz, CDCl3) δ (ppm) 151.00 (d, J = 15.1 Hz), 150.42, 143.64, 141.77, 140.67, 138.57, 132.08, 131.78, 130.20, 128.68, 126.07, 123.91, 121.64, 116.36, 109.47, 34.80, 32.00. TOF-MS (ESI) m/z calcd for C39H28N10 [M + H]+: 637.72; found: 637.29; HRMS (ESI) m/z found: 637.256144 C39H28N10 (Fig. S4, S6, S8 and S10, ESI†).
:dichloromethane (1:1) as eluent to afford yellow solid (70 mg, yield 40%). 1H NMR (400 MHz, CDCl3) δ (ppm) 8.48 (dd, J = 8.5, 2.7 Hz, 4H), 8.22–8.17 (m, 2H), 7.71–7.60 (m, 7H), 7.50 (d, J = 7.6 Hz, 4H), 7.08–7.02 (m, 4H), 6.99 (t, J = 7.4 Hz, 4H), 6.47–6.42 (m, 4H), 1.71 (s, 12H). 13C NMR (126 MHz, CDCl3) δ (ppm) 151.16, 150.47, 145.00, 140.85, 132.81, 132.19, 131.32, 130.73, 130.22, 128.76, 126.53, 125.31, 123.77, 123.40, 121.17, 114.56, 36.12, 31.04. TOF-MS (ESI) m/z calcd for C54H41N11 [M + H]+: 844.00; found: 844.348. HRMS (ESI) m/z found: 844.360839, C54H42N11 (Fig. S5, S7, S9 and S11, ESI†).
Fabrication and measurements of OLED device
The substrates were successively cleaned with isopropyl alcohol, acetone, detergent, deionized water, and isopropyl alcohol in an ultrasonic bath and then dried overnight in an oven. The substrates were pre-treated with oxygen plasma to increase the work function of the ITO film. Then, 40 nm-thick PEDOT:PSS was spin-coated onto the ITO substrates at 3200 rpm for 30 s and annealed at 150 °C for 15 min. And then emissive layer was spin-coated and annealed at 60 °C or 110 °C for 30 min using a precursor containing different materials co-dissolved in chlorobenzene or toluene. The films of DPEPO, TmPyPB, LiF and aluminum were prepared by thermal evaporation under a vacuum of 1 × 10−4 Pa. Each sample had an active area of 0.04 cm2.
All the devices were encapsulated before characterization to prevent degradation and emission quenching caused by oxygen and water. The EL spectra and J–V–R curves were obtained with a Photoresearch Spectra Scan PR735 photometer and a Keithley 2400 source meter constant current source at room temperature. The EQE values were calculated by assuming a Lambertian distribution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the NSFC (51773021, 51911530197, U1663229, 51473140), the Six Talent Peaks project in Jiangsu Province (XCL-102), and the Natural Science Fund for Colleges and Universities in Jiangsu Province (no. 19KJA430002). The NMR testing was carried out by the Analysis and Testing Center, NERC Biomass of Changzhou University.References
- K. Goushi and C. Adachi, Appl. Phys. Lett., 2012, 101, 023306 CrossRef.
- Y. Zou, S. Gong, G. Xie and C. Yang, Adv. Opt. Mater., 2018, 6, 1800568 CrossRef.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Brase, Adv. Mater., 2021, 33, 2005630 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- S. K. Jeon, H. L. Lee, K. S. Yook and J. Y. Lee, Adv. Mater., 2019, 31, 1803524 CrossRef PubMed.
- X. Liang, Z. L. Tu and Y. X. Zheng, Chem. – Eur. J., 2019, 25, 5623–5642 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- P. Ganesan, W. Y. Hung, J. Y. Tso, C. L. Ko, T. H. Wang, P. T. Chen, H. F. Hsu, S. H. Liu, G. H. Lee, P. T. Chou, A. K. Y. Jen and Y. Chi, Adv. Funct. Mater., 2019, 29, 1900923 CrossRef.
- S. Tan, X. Wu, Y. Zheng and Y. Wang, Chin. Chem. Lett., 2019, 30, 1951–1954 CrossRef CAS.
- Z. Wei, K. Zhang, C. K. Kim, S. Tan, S. Wang, L. Wang, J. Li and Y. Wang, Chin. Chem. Lett., 2021, 32, 493–496 CrossRef CAS.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2016, 4, 11355–11381 RSC.
- T. Huang, W. Jiang and L. Duan, J. Mater. Chem. C, 2018, 6, 5577–5596 RSC.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 6, 1800255 CrossRef.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- J. M. Teng, Y. F. Wang and C. F. Chen, J. Mater. Chem. C, 2020, 8, 11340–11353 RSC.
- Q. Xue and G. Xie, Adv. Opt. Mater., 2021, 9, 2002204 CrossRef CAS.
- W. Che, Y. Xie and Z. Li, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2019, 7, 2172–2198 RSC.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- D. G. Chen, T. C. Lin, C. L. Chen, Y. T. Chen, Y. A. Chen, G. H. Lee, P. T. Chou, C. W. Liao, P. C. Chiu, C. H. Chang, Y. J. Lien and Y. Chi, ACS Appl. Mater. Interfaces, 2018, 10, 12886–12896 CrossRef CAS.
- T. Yang, Z. Cheng, Z. Li, J. Liang, Y. Xu, C. Li and Y. Wang, Adv. Funct. Mater., 2020, 30, 2002681 CrossRef CAS.
- Z. Li, D. Yang, C. Han, B. Zhao, H. Wang, Y. Man, P. Ma, P. Chang, D. Ma and H. Xu, Angew. Chem., Int. Ed., 2021, 60, 14846–14851 CrossRef CAS PubMed.
- F.-M. Xie, Z.-D. An, M. Xie, Y.-Q. Li, G.-H. Zhang, S.-J. Zou, L. Chen, J.-D. Chen, T. Cheng and J.-X. Tang, J. Mater. Chem. C, 2020, 8, 5769–5776 RSC.
- J. Jin, H. Jiang, Q. Yang, L. Tang, Y. Tao, Y. Li, R. Chen, C. Zheng, Q. Fan, K. Y. Zhang, Q. Zhao and W. Huang, Nat. Commun., 2020, 11, 842 CrossRef CAS PubMed.
- R. Braveenth, H. Lee, J. D. Park, K. J. Yang, S. J. Hwang, K. R. Naveen, R. Lampande and J. H. Kwon, Adv. Funct. Mater., 2021, 2105805 CrossRef CAS.
- Y. K. Chen, J. Jayakumar, C. M. Hsieh, T. L. Wu, C. C. Liao, J. Pandidurai, C. L. Ko, W. Y. Hung and C. H. Cheng, Adv. Mater., 2021, 33, 2008032 CrossRef CAS.
- H. J. Park, H. L. Lee, H. J. Lee, K. H. Lee, J. Y. Lee and W. P. Hong, Chem. Mater., 2019, 31, 10023–10031 CrossRef CAS.
- X. Liang, Z. P. Yan, H. B. Han, Z. G. Wu, Y. X. Zheng, H. Meng, J. L. Zuo and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 CrossRef CAS PubMed.
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- X. Zheng, R. Huang, C. Zhong, G. Xie, W. Ning, M. Huang, F. Ni, F. B. Dias and C. Yang, Adv. Sci., 2020, 7, 1902087 CrossRef CAS PubMed.
- W. Li, W. Li, L. Gan, M. Li, N. Zheng, C. Ning, D. Chen, Y. C. Wu and S. J. Su, ACS Appl. Mater. Interfaces, 2020, 12, 2717–2723 CrossRef CAS PubMed.
- H. Abroshan, Y. Zhang, X. Zhang, C. Fuentes-Hernandez, S. Barlow, V. Coropceanu, S. R. Marder, B. Kippelen and J. L. Brédas, Adv. Funct. Mater., 2020, 30, 2005898 CrossRef CAS.
- J. Zhang, Q. Wei, N. Fei, M. Zhao, L. Xie, L. Cao, X. Zhang, G. Xie, T. Wang and Z. Ge, ACS Appl. Mater. Interfaces, 2021, 13, 12305–12312 CrossRef CAS.
- R. K. Konidena, J. Lim and J. Y. Lee, Chem. Eng. J., 2021, 416, 129097 CrossRef CAS.
- J. H. Yun, K. H. Lee, J. Y. Lee and W. P. Hong, Chem. Eng. J., 2021, 411, 128550 CrossRef CAS.
- H. Yang, Q. Liang, C. Han, J. Zhang and H. Xu, Adv. Mater., 2017, 29, 1700553 CrossRef PubMed.
- H. J. Kim, M. Godumala, S. K. Kim, J. Yoon, C. Y. Kim, H. Park, J. H. Kwon, M. J. Cho and D. H. Choi, Adv. Opt. Mater., 2020, 8, 1902175 CrossRef CAS.
- Y. Wada, S. Kubo and H. Kaji, Adv. Mater., 2018, 30, 1705641 CrossRef PubMed.
- K. Wu, Z. Wang, L. Zhan, C. Zhong, S. Gong, G. Xie and C. Yang, J. Phys. Chem. Lett., 2018, 9, 1547–1553 CrossRef CAS PubMed.
- L. Zhan, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 17651–17655 CrossRef CAS.
- J. Luo, S. Gong, Y. Gu, T. Chen, Y. Li, C. Zhong, G. Xie and C. Yang, J. Mater. Chem. C, 2016, 4, 2442–2446 RSC.
- I. H. Lee, W. Song, J. Y. Lee and S.-H. Hwang, J. Mater. Chem. C, 2015, 3, 8834–8838 RSC.
- T. Rieth, N. Roder, M. Lehmann and H. Detert, Chem. – Eur. J., 2018, 24, 93–96 CrossRef CAS PubMed.
- T. Rieth, T. Marszalek, W. Pisula and H. Detert, Chem. – Eur. J., 2014, 20, 5000–5006 CrossRef CAS PubMed.
- S. K. Pathak, Y. Xiang, M. Huang, T. Huang, X. Cao, H. Liu, G. Xie and C. Yang, RSC Adv., 2020, 10, 15523–15529 RSC.
- F. Hundemer, E. Crovini, Y. Wada, H. Kaji, S. Bräse and E. Zysman-Colman, Mater. Adv., 2020, 1, 2862–2871 RSC.
- S. Wang, X. Wang, K. H. Lee, S. Liu, J. Y. Lee, W. Zhu and Y. Wang, Dyes Pigm., 2020, 182, 108589 CrossRef CAS.
- X. Chen, S. Wang, H. L. Lee, J. Y. Lee, X. Liao, L. Li, W. Zhu and Y. Wang, Adv. Opt. Mater., 2021, 2101518 CrossRef CAS.
- R. Hojo, D. M. Mayder and Z. M. Hudson, J. Mater. Chem. C, 2021, 9, 14342–14350 RSC.
- Y. Liu, Z. Yin, X. Wang, E. Baranoff, D. Zhou, K. Zhang, Z. Ren, S. Wang, W. Zhu and Y. Wang, J. Mater. Chem. C, 2020, 8, 8971–8979 RSC.
- B. Li, Z. Yang, W. Gong, X. Chen, D. W. Bruce, S. Wang, H. Ma, Y. Liu, W. Zhu, Z. Chi and Y. Wang, Adv. Opt. Mater., 2021, 9, 2100180 CrossRef CAS.
- M. R. Fadavieslam, Optik, 2019, 182, 452–457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc05372h |
| This journal is © The Royal Society of Chemistry 2022 |