
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAll-dry, one-step synthesis, doping and film formation of conductive polypyrrole†
Afshin
Dianatdar
 a,
Matteo
Miola
a,
Oreste
De Luca
b,
Petra
Rudolf
b,
Francesco
Picchioni
a and
Ranjita K.
Bose
*a
a,
Matteo
Miola
a,
Oreste
De Luca
b,
Petra
Rudolf
b,
Francesco
Picchioni
a and
Ranjita K.
Bose
*a
aDepartment of Chemical Engineering, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, 9747AG Groningen, The Netherlands. E-mail: r.k.bose@rug.nl; Tel: +31 50 3634486
bZernike Institute for Advanced Materials, University of Groningen, 9747 AG Groningen, The Netherlands
First published on 6th December 2021
Abstract
Oxidative chemical vapor deposition (oCVD) is an extremely effective method for solvent-free deposition of highly conductive polypyrrole films, where polymer synthesis, doping, and film formation are combined in a single step. Here we show that by carefully tuning the reaction parameters, namely the deposition temperature, the reactor pressure and the oxidant to monomer flow rate ratio, homogeneous polypyrrole films with a record conductivity of 180 S cm−1 for a solvent-free method were produced. Fourier transform infrared spectroscopy, UV-vis spectrophotometry, X-ray photoelectron spectroscopy, scanning electron microscopy, and four-probe surface resistivity measurements were performed to gain insights into the relationship between different reaction conditions and the structure of oCVD-deposited polypyrrole, the development of defects, the film morphology and its physical properties.
Introduction
Since the 1970s, conjugated polymers or intrinsically conductive polymers (ICPs) have been the subject of a large research effort. They have shown potential for a wide range of applications in the microelectronics and semiconductor industries, energy generation/storage, biomedical devices and catalysis, as thermoelectric materials, and for corrosion protection.1–10 Their popularity derives from their ability to combine the electronic properties of traditional inorganic materials with the mechanical flexibility of polymers. They are relatively cheap and lightweight, making them especially suitable for modern electronic devices intended for skin contact and wearable applications.11–13Among ICPs, polypyrrole (PPy) has been favored as a test material for a variety of applications due to its good electrical conductivity, relative stability and biocompatibility.14–18 Unfortunately, the extended double-bond conjugation that ensures electrical conductivity is also responsible for the poor solubility of PPy, which results in major challenges in its synthesis and processing.19
Considering that in the majority of cases, PPy is used as a thin film on a surface, electrochemical synthesis of PPy on an electrode has been a widely used method, since it eliminates solubility issues and provides better control over the film uniformity and thickness than solvent processing methods like spin-coating.20,21 However, the major drawback of the electrochemical approach is that the electrode material has to be conductive and therefore, this synthesis route cannot be universally applied.22
An alternative method for circumventing the problems with solubility is vapor phase polymerization (VPP).23–28 In the VPP of PPy, the substrate is pretreated with an oxidant before being exposed to pyrrole vapor, and this results in PPy growth on the pre-treated substrate.29–43 VPP simultaneously eliminates the need for conductive substrate and for the solvent. However, the pre-treatment of the substrate with an oxidant involves wet chemistry and therefore presents potential drawbacks related to the substrate sensitivity to a solvent and/or shortcomings arising from coating of 3D substrates.36,44 This is especially important for a large number of applications in which fragile or 3D substrates are used, for example e-textiles, printed electronics on paper, conductive layers on a stretchable substrate, porous substrates for energy storage and biomedical hydrogels.45–49
In 2006, a modified VPP method for deposition of conjugated polymers was first implemented, where both the oxidant and the conjugated monomer are delivered to the substrate in the vapor phase.50,51 The technique is called oxidative chemical vapor deposition (oCVD) and provides more structural control over the synthesis of ICPs. The kinetics of polymerization are controlled by precise reactant feeds along with adjustment of deposition temperature and reactor pressure. The merits of oCVD over solution-processed coating and/or electrochemical techniques have been well established in literature.52,53 In addition to the substrate-independent applicability of oCVD, an important advantage is that this method also allows for the one-step synthesis of ICPs in their doped form.51
Despite the large “library” of the polymers synthesized by oCVD, the synthesis of PPy has not yet been fully explored.51 There are only a few reports of oCVD that use pyrrole as a comonomer (together with thiophene) for deposition.54–56 The aim of the present work is therefore to comprehensively study PPy synthesized via oCVD. We chose antimony pentachloride (SbCl5) as the oxidant as it has recently showed outstanding results in oCVD synthesis.57,58 Different reactants ratios, and operational parameters were systematically investigated to optimize the reaction condition for obtaining highly conductive films.
Experimental
Materials
Pyrrole (>99%) was purchased from TCI Europe, while antimony pentachloride (SbCl5, > 98%) was acquired from Sigma Aldrich. All other solvents and reagents were bought from Acros; all chemicals were used as received. Single-side polished silicon wafers (University Wafer, Inc. USA, 〈100〉 P-Boron), microscopy glass slides, and optical quartz plates (Präzisions Glas & Optik GmbH, Germany) were used as substrates for deposition.Polymer synthesis via oCVD
A custom-made CVD setup was used for PPy deposition (Chart 1). Inside the reactor, the substrates (silicon wafer, glass slide and quartz plate) were fixed onto the bottom wall with an adhesive tape. The reactor bottom temperature was controlled by electrical heating elements. To avoid condensation of the reactants, all feedthrough lines as well as the reactor body were heated to 110 °C, while the reactor body was kept at 40 °C. The reactor pressure was monitored by a pressure transducer (MKS, model 622C11TBE) and controlled by throttle valve (MKS, model 653B-1-40-1).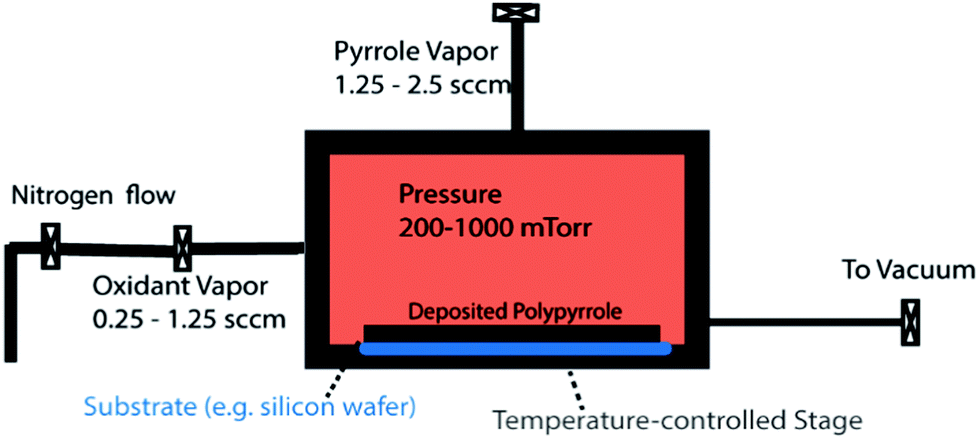 | ||
| Chart 1 A representation of the oCVD reactor. | ||
In a typical reaction, the monomer and the oxidant heated to 60 °C were controllably delivered into the reactor via needle valves (Swagelok, SS-MGVR4-MH and SS-SVR4) from two different inlets set at a right angle from each other on two adjacent reactor walls. In all depositions, N2 was used as the diluent gas as well as the carrier gas for the oxidant. After 15 min, the reaction was stopped by closing the valves on the feed lines and pumping down the reactor for 5 min. The samples were taken out and rinsed with acetone to remove any unreacted monomer and excess oxidant (for resistivity and UV-vis measurements) before being dried in an oven at 140 °C for 15 minutes and then stored in a vacuum desiccator to avoid oxidation.
To investigate the influence of the operational parameters on the characteristics of the films, we produced several series of samples: starting out with a pressure (P) and a specific ratio of oxidant to monomer flow rates in sccm (RR), in the range reported in the literature for PPy growth, we varied the substrate temperature (T).55 We then checked how different pressures affect PPy properties. Finally, after having determined the optimal pressure, we varied the RR to arrive at the optimized result. Table 1 lists all the samples produced and specifies the operational parameters in each case. More information about the actual flow rates of the precursors, their vapor pressures, along with the surface concentrations for each experiment has been calculated and included in Table S1 (ESI†).

Characterization
Fourier-transform infrared Spectroscopy (FTIR) spectra in transmission mode were collected on a Shimadzu IRTracer instrument equipped with a deuterated triglycine sulphate (DTGS) detector, in the region of 600–4000 cm−1. Each spectrum was the average of 128 scans and the resolution was set to 4 cm−1. The measurement was done on the PPy-coated silicon wafers.UV-vis absorption spectroscopy was performed with a Jasco V730 double beam spectrophotometer in the range 250–900 nm on PPy-coated quartz. To calculate the conductivity of the films, the sheet resistance (R in eqn (1)) was first measured with a four-point probe (Ossila Ltd, UK.) on PPy-coated microscopy slides. The results were averaged over 10 measurements at different locations on the same sample. The PPy films resistivity was then calculated by:
| ρ = R × t | (1) |
 | (2) |
 is the film conductivity in S cm−1. For thickness measurement, Profilometry (Bruker DektakXT) was used and the thickness of each sample was calculated as the average of three measurements.
is the film conductivity in S cm−1. For thickness measurement, Profilometry (Bruker DektakXT) was used and the thickness of each sample was calculated as the average of three measurements.
Results and discussions
PPy thin film deposition
Typical polymerization of pyrrole proceeds according to Scheme 1.59,60 First, SbCl5 and pyrrole monomer vapors are adsorbed onto the surface of the substrate, and pyrrole is oxidized to form a radical cation (1). Then two of the radical cations undergo C–C coupling forming a radical dication (2). This coupling can take place in three ways namely α–α, α–β, β–β as illustrated in Scheme 2. In this polymerization phase, the α–α linkage is not only the dominant one, but also the desired one to ensure PPy conjugation and charge transport.59,61 The radical dication then undergoes deprotonation resulting in a pyrrole dimer (3). This is followed by further oxidation of the dimer (4), bonding between the dimer with another monomer radical (5) and further deprotonation (6), leading to the oligomer. | ||
| Scheme 1 Mechanistic pathway for oxidative chemical vapor deposition of PPy (1–7); doping of PPy from benzoid form to quinoid form (8). | ||
 | ||
| Scheme 2 Different types of C–C coupling for two pyrrole units during oxidation polymerization. | ||
Subsequent repetitions of the series of steps result in the formation of PPy (7). It is noteworthy that as the reaction continues and the pyrrole chains grow, the number of C–C couplings involving the β position increases.61 Finally, the neutral (benzoid) form of PPy can be oxidized and this electron loss modifies the PPy electronic structure. This transformation from the benzoid to quinoid form is called doping of PPy (8).62,63
The structure of a typical PPy grown by oCVD was characterized by FTIR and XRD, the results presented in Fig. 1. Fig. 1(a) presents the IR spectrum for PPy-T60 with its corresponding assignments of the bands presented in Table 2. The pyrrole vibrations due to intra/inter ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C and C–N stretching modes are evident at 1545 cm−1 cm−1, ring deformation at 964 cm−1 and ring breathing at 1168 cm−1.64–67 The IR spectra of the other samples are provided in Fig. S1–S3 (ESI†) and 1454 cm−1, respectively.65,68 The other bands include C–H in-plane bending at 1040 cm−1 and 1300.
C/C–C and C–N stretching modes are evident at 1545 cm−1 cm−1, ring deformation at 964 cm−1 and ring breathing at 1168 cm−1.64–67 The IR spectra of the other samples are provided in Fig. S1–S3 (ESI†) and 1454 cm−1, respectively.65,68 The other bands include C–H in-plane bending at 1040 cm−1 and 1300.
 | ||
| Fig. 1 (a) Fourier transform infrared spectrum of PPy-T60 between 600–3500 cm−1; (b) XRD spectrum of PPy-T60, revealing an amorphous structure. | ||
| Band position (cm−1) | Assignment |
|---|---|
| 1545 | CC/C–C stretching |
| 1454 | C–N stretching |
| 1300 | C–H in-plane bending |
| 1168 | Ring breathing |
| 1040 | C–H in-plane bending |
| 964 | Ring deformation |
The XRD spectrum of PPy-T60 (Fig. 1(b)) shows two very broad peaks at 2θ = 12.3° and 26.3°, indicative of some structural ordering at the nanoscale which is attributed to the interplanar spacing of the PPy chains. This is typical of nanostructured polymers with a low degree of crystallinity, as previously reported.69
Conductivity
The conductivity of the deposited films varied considerably depending on the reaction temperature (T), pressure (P), and reactants ratio at which the deposition had took place (Fig. 2). At a constant pressure of 1000 mTorr and the oxidant to monomer flow rate ratio (RR) of 0.5, no reaction was observed at T = 20 °C during 60 minutes. However, increasing the temperature to 40 °C, resulted in the formation of a PPy thin film with a conductivity of ∼20 S cm−1. Deposition at T = 60 °C boosted the conductivity of the film to 148 S cm−1, but a further increase to T = 80 °C had a negative effect since it produced a film with markedly lower conductivity under 1 S cm−1. In an attempt to understand this behavior, Cox/Cmo was calculated which reflects the relative ratio of surface availability of the oxidant/monomer at each temperature and the values are shown in Fig. 2(a) (detailed information in Table S1, ESI†). It was observed that with an increase of temperature from 40 °C to 60 °C, and subsequently to 80 °C, the surface availability of oxidant to monomer surface concentration is increased steadily. This indicates that even with constant molar ratios of the precursors, their actual concentration at the site of the surface reaction differs when the temperature is changed. This suggests that a higher conductivity at 60 °C compared to 40 °C is a result of higher doping due to a higher presence of dopants. However, an excessive amount of dopant at 80 °C has a detrimental effect due to overoxidation of PPy chains. This is further discussed in the following sections. | ||
| Fig. 2 (a) Conductivity of the deposited PPy thin films, deposited at different T along with calculation of oxidant to monomer surface availability (Cox/Cmo); (b) conductivity of the deposited PPy thin films, deposited at different P, or RR (see Table 1). Referring to Table S1 (ESI†) shows that Cox/Cmo remains constant for pressures series and follows the same trend as RR for oxidant to monomer flow rate ratio series, thus not included in the figure. Note: error bars are included, the maximum SD is less than 5%. | ||
In the second series deposited at a constant temperature of 40 °C and with an RR of 0.5, the pressure was adjusted to 200, 300, 500, and 1000 mTorr. The films produced under these conditions exhibit an increase in conductivity from 46 S cm−1 at a deposition pressure of 200 mTorr to 180 S cm−1 for the one grown at 500 mTorr. A further pressure increase to 1000 mTorr was detrimental since it yielded films with a conductivity of only 20 S cm−1. Finally, RR was changed from 0.1 to 1, and again a change in conductivity was observed; the conductivity increased from 57 S cm−1 to 155 S cm−1 upon increasing RR from 0.1 to 0.2 during deposition. However when the oxidant content of the vapor phase was increased, and RR rose correspondingly to 0.5 and subsequently to 1, the resulting films again exhibited a much lower conductivity.
It should be noted that the film with a conductivity of 180 S cm−1 (obtained for PPy-RR0.2) is more than two times higher than the highest conductivity of PPy reported using VPP26 (83 S cm−1) and comparable with the one produced by the electrochemical method,70 proving the viability of oCVD for synthesizing highly conductive PPy.
The conductivity of PPy is strongly affected by the interrelated parameters of level of doping, conjugation length, and microstructure. Doping results in a reduction of bandgap between highest occupied molecular orbital and lowest unoccupied molecular orbital in conjugated polymers, and yields a higher conjugation length. The conjugation is the length of a sequence of undisturbed conjugated monomeric units, along which charge can travel.71 Previous reports established that the increase in conjugation length is associated with a red-shift of the π–π transition (λmax) in the UV-vis spectrum of the polymer film.72 The full UV-vis spectra of samples are presented in Fig. S4 (ESI†). From the positions of λmax for the deposited PPy films (Fig. 3(a)), it is evident that a higher deposition temperature leads to a lower doping levels (and conjugation length). However, higher pressure and oxidant to monomer flow rate ratio increase the level of doping. For RR, doping reaches a plateau beyond RR = 0.5, most likely because the charge saturation point of the deposited PPy has been reached and the polymer cannot accommodate more charge carriers.73–75
 | ||
| Fig. 3 λ max and ECC as at different (a) temperature, (b) pressures, and (c) oxidant to monomer flow rate ratio. | ||
Achieving higher doping levels when depositing at a higher pressure was reported by Smolin et al. for the oCVD of polyaniline.76 As the relative molar ratio is constant for both samples at different pressures, the mean free path at higher pressure becomes shorter in the gas phase and this results in higher adsorption rate for precursors and hence polymer film formation. Finally, the decrease of conductivity of PPy when oCVD is performed at higher temperatures has been reported before, though in that case the reasons were not investigated.54 Nevertheless, these observations from UV-vis spectroscopy for temperature, pressure and RR are only partly in-line with the values for conductivity, suggesting other aspects affect the films electrical behaviour.
In an early work on PPy, Tian and Zerbi defined a parameter named “effective conjugation coordinate (ECC)” that relates the conductivity to the ratio of CC/C–C to C–N bands calculated from FTIR.67 In this way, the conjugation of PPy is inversely proportional to the ratio of the areas of the bands centered at 1545 to 1454 cm−1 (A1545 cm−1/A1454 cm−1), a concept which has been widely corroborated by other works.77,78 Here, the ECC values were calculated for all the PPy and films and the results are shown in Fig. 3(b). As evident for the temperature series, the film deposited at T = 60 °C showed a lower ECC and therefore a higher conjugation length, followed by the ones grown at T = 40 °C and T = 80 °C, similar to the trend for conductivity values. An analogous behavior is found for the variation of pressure, where ECC value decreases when P at which the film was deposited was increased from 200 mTorr to 300 mTorr and subsequently 500 mTorr. On the other hand, the ECC value showed a sharp rise when pressure during the film growth was increased to 1000 mTorr. Comparing with conductivity, it is clear that dovetails with the ECC value of the samples. For the series of films grown at different RR values, Fig. 3(b) shows that for the thin layers grown with increasing RR from 0.1 to 0.2 and subsequently to 0.5, the ECC value shows a slight decrease. However, the ECC value is significantly higher for the layer deposited with RR = 1, which is in line with the conductivity drop for the film grown at this condition; though for the conductivity, the trend changed at RR = 0.5. These findings suggest that the ECC value is very often but not always, a good indicator of conductivity in these PPy films.
The different reaction conditions also give rise to the changes in the film as visible in the SEM micrographs presented in Fig. S5 (ESI†). The film deposited at 40 °C shows a cluster-like appearance compared to the smooth and homogenous morphology film deposited at T = 60 °C. On the other hand, for the film deposited at RR = 1, the surface morphology shows speckles indicating a higher apparent roughness than that of the film grown at RR = 0.2 (Fig. S5, ESI†).
All of these measurements suggest that there are optimum combinations of T, P, or RR values, at which conductivity is the highest. In order to better understand how these deposition parameters influence the molecular conformation of the film, a few samples were selected for a detailed study by XPS.
XPS
Four samples were selected to verify the correlation between film composition, doping level, and conjugation length. PPy-P300 and P1000 were chosen to study the effects resulting from higher deposition rate as a result of higher reactor pressure. PPy-RR0.2 and RR1 were investigated to gain insight in the stoichiometric variation when RR is changed from 0.2 to 0.5 (= PPy-RR1000) and finally to 1. This latter series combines the effect of higher deposition rate and higher oxidation level.The atomic percentages of C, N, Cl, O, and Sb as obtained from the intensities of XPS spectra are shown in Table 3. The XPS analysis gives an insight on the integrity of the pyrrole ring via the C/N ratio, the number of counter ions per pyrrole ring via N/Cl ratio, the amount of impurities present via the Sb content (and its oxides) as well as the presence of possible structural defects via the oxygen content (Fig. 4). Comparing the samples of PPy-P300 and P1000, where the pressure during the deposition was varied but the temperature of the substrate (T = 40 °C) and the oxidant to monomer flow rate ratio (RR = 0.5) were constant, it is evident that the higher pressure resulted in a very high C/N ratio of around 9, much higher than that of a pyrrole monomer (C/N = 4); while the C/N is 3.5 for the film deposited at 300 mTorr. The increased carbon content in PPy-P1000 can be due to defects such as ring cleavage, PPy chain cross-linking and aliphatic chain formation during polymerization, as suggested in the literature.79 In comparison to PPy-P300, the PPy film deposited at the same conditions (T = 40 °C, P = 300 mTorr) but with lower RR = 0.2, has a comparable C/N ratio of 3.4, close to that of a pyrrole monomer. The C/N ratio close to 4 correlates with the high conductivity of 155 S cm−1 obtained for this sample. When RR is increased to 1, the C/N ratio rose to over 20, strongly suggesting that extra carbon is formed in the film as a result of undesired reactions.
| Sample | Composition (%) | |||||
|---|---|---|---|---|---|---|
| C | N | Cl | Sb | O | Sna | |
| a The “Sn” content in PPy-RR0.2 is most likely the result of contamination during sample handling. | ||||||
| PPy-P300 | 54.2 (± 1.4) | 15.3 (± 0.4) | 8.1 (± 0.2) | 10.9 (± 0.2) | 11.5 (± 0.1) | — |
| PPy-P1000 | 42 (± 1.8) | 4.7 (± 0.2) | 12 (± 0.3) | 24.8 (± 0.9) | 16.6 (± 0.4) | — |
| PPy-RR0.2 | 60.6 (± 2) | 17.6 (± 0.3) | 12.2 (± 0.2) | 2.6 (± 0.1) | 5.7 (± 0.2) | 1.3 (± 0.1) |
| PPy-RR1 | 60 (± 0.7) | 2.9 (± 0.7) | 8.6 (± 0.1) | 14.4 (± 0.1) | 14.1 (± 0.3) | |
 | ||
| Fig. 4 Quantification of the N/Cl and C/N ratios, as well as “O+ Sb” atomic contents for PPy-P300, PPy-P1000, PPy-RR0.2, and PPy-RR1. The theoretical C/N atomic ratio for PPy is the same as for the monomer and is shown for comparison. | ||
The number of Cl− counter ion per pyrrole ring is an indication of the doping level and can be quantified via the Cl/N ratio. Since PPy-RR0.2 showed the highest conductivity among these four samples, two counter ions per three pyrrole rings (N/Cl = 1.4) is inferred to be the best doping. When the amount of oxidant present during PPy growth is increased by choosing RR = 1, the N/Cl ratio in the resulting film decreased to ∼0.35. In other words, an average of three Cl− per pyrrole ring testifies to overdoping/overoxidation, in line with Ge et al. who observed that the nitrogen to counter ion ratio dropped from 3 to 0.6 when PPy was electrochemically overoxidized.80 A similar overoxidation was observed when the deposition pressure was increased (PPy-P1000). Excessively-doped PPy was shown to be less stable over time as one pyrrole ring can effectively interact with only one counter ion.81
Concerning impurities and defects, we note that contamination by oxygen up to around 16 at% is present in the films. A similar oxygen contamination has been reported before in PPy synthesis and attributed to physical and chemical interaction of PPy when exposed to air or water in the precursors. In particular, it was suggested that the oxygen is a result of the hydroxyl free carrier (OH−) and/or defect formation (e.g. carbonyl).82–84 Here, PPy-RR0.2 sample shows just below 15 at% of Sb and O; while for samples produced at higher P and RR, this percentage increases reaching over 40 at% for PPy-P1000.
C 1s XPS spectra
The detailed XPS spectra of the C 1s core level region suggest the presence of five chemical environments as shown in Fig. 5(a)–(d). The spectral contributions at binding energies (B.E.s) of 284.2 eV and 285.2 eV are assigned to carbon at β and α position of a pyrrole ring, respectively (Scheme 2).82 The percentage with which these species contribute to the total C 1s intensity is listed in Table 4. The photoemission intensity is directly proportional to the atoms in that environment present in the probed volume. A single pyrrole ring has the ratio of α/β = 1. However, it is reported that the XPS α-carbon signal is often more intense than β-carbon.79 Different interpretations have been suggested: for example, Zeller et al. claimed that chain linking such as α–β and β–β may affect the electron density around the β position to a greater extent than α position.82 Other researchers have proposed a different structural arrangement of doped PPy compared to a pyrrole ring.79 In addition, it has been observed that this ratio (independent of the initial value) tends to decrease after overoxidation in presence of excess dopants.79,82 | ||
| Fig. 5 High resolution XPS data of C 1s region for (a) PPy-P300, (b) PPy-P1000 (= PPy-RR0.5), (c) PPy-RR0.2, and (d) PPy-RR1; the fit is also shown and for each component the relative contribution to the total spectral intensity of the respective line is reported in Table 4. | ||
| Chemical environments | Relative contribution to the total spectral intensity of the C 1s core level line (%) | Binding energy (eV) | |||
|---|---|---|---|---|---|
| PPy-P300 | PPy-P1000 | PPy-RR0.2 | PPy-RR1 | ||
| β-carbon | 21.9 | 29.8 | 22.8 | 38.2 | 284.2 |
| α-carbon | 44.9 | 44.7 | 44.9 | 40.1 | 285.2 |
| C–N/C–O | 21.4 | 18.8 | 21.0 | 14.6 | 286.2 |
| CN/CO/C–N+ |
8.0 | 4.0 | 7.9 | 4.4 | 287.4 |
| COO/CON/CN+ |
3.8 | 2.7 | 3.5 | 2.9 | 288.8 |
With this in mind, we note that this ratio equals to around 2.0 for PPy-P300 and it retains its value when the deposition pressure was increased from 300 mTorr to 1000 mTorr. However, α/β carbon was reduced by about 50% when oxidant to monomer flow rate ratio was increased from 0.2 to 1 (Fig. 5(c)vs.Fig. 6(d)).
For the C 1s components at higher B.E.s, different assignments have been reported in the literature, accompanied with proposed mechanistic pathways (Scheme 3(a)).79–82,85–87 The first component at 286.2 eV, has been suggested to stem from C–O, C–N, and C–Cl bonds.81 Yet, these bonds point to very different chemical environments: C–O is a structural defect in PPy, C–N exists in neutral or doped PPy, and C–Cl can only be present in an over-doped sample where the covalent bond between carbon and chlorine breaks the conjugation.87,88 The debate on this goes back decades, when Zeller et al.82 argued that the main component at 286.2 eV arises from non α–α carbon linkage rather than C–O (without any discussion about C–N bonds). For the contribution at a B.E. around 287.4 eV, CO (overoxidation, Scheme 3a), CN (deprotonation, Scheme 3b), or C–N+ in the quinoid structure of PPy (eqn (8), Scheme 1) should be considered.86 In this case, the first two environments negatively impact the electron transfer along the polymer chains; while C–N+ (suggesting a polaron) can positively affect charge transport.63 Finally, the peak at 288.8 eV which could be due to oxygen contamination due to COO/CON bonds, or again it could be CN+ as an alternative chemical environment in the quinoid structure of PPy. Since no CO fingerprint at 1700–1750 cm−1 was observed in FTIR (see Fig. S1–S3, ESI†), the presence of carbonyl group (CO) is excluded. However, Tabaciarova et al.81 reported an oxidation study of PPy prepared with different dopants where they assigned weak and broad peaks at 1700 cm−1 and 1745 cm−1 to N–CO, and C–CO moieties. Such an assignment has been adopted for electrochemically grown PPy as well.89 Nevertheless, we attribute the deconvoluted XPS peaks to carbon–nitrogen environments, as this is more in line with the rest of the data. Calculating from Table 4, the cumulative spectral intensity of high B.E. components of the selected PPy films (∑(%) at 286.2 eV, 287.4 eV, and 288.8 eV) decreases at higher RR and P. However, as synthesis at higher RR and P are expected to be associated with overoxidation (thus higher oxygen content) of the produced film, the observed decrease should instead relate to carbon–nitrogen environments (including polaron/bipolaron).
 | ||
| Scheme 3 (a) a proposed mechanism for carbonyl defect formation in PPy;80,85 (b) A proposed PPy deprotonation mechanism.64,82 | ||
N 1s XPS spectra
The detailed XPS of the N 1s core level region are shown in Fig. 6 and the percentages with which the various components contribute to the total N 1s spectral intensity are presented in Table 5. The N 1s line bears the signature of four chemical species: imine type nitrogen (N–) at a B.E. of 398 eV, N–H from the pyrrole ring at 400 eV, C–N+ at 401.2 eV and CN+ at 402.6 eV.80,86,90,91 Synthesis at higher P and RR leads to a decrease in pyrrole ring nitrogen (N–H), while imine-like nitrogen (N–), polaron (C–N+) and bipolaron (CN+) increased. Imine-like nitrogen has been attributed to the nucleophilic attack of water (moisture) during or after polymerization (Scheme 3(b)), resulting in the deprotonation of nitrogen.82 Deprotonation increased with overoxidation as well (films produced with RR = 1 and P = 1000 mTorr), as also observed in literature.80 On the other hand, the formation of both polaron (C–N+) and bipolaron (CN+) is favored for PPy produced at higher RR and P.
 | ||
| Fig. 6 XPS spectra of the N 1s core level region for (a) PPy-P300, (b) PPy-P1000 (= PPy-RR0.5), (c) PPy-RR0.2, and (d) PPy-RR1, providing information on the formation of polaron and bipolaron species; the fit is also shown and the relative contribution of each component to the total N 1s spectral intensity is reported in Table 5. | ||
| Chemical environments | Relative contribution to the total spectral intensity of the N 1s core level line (%) | Binding energy (eV) | |||
|---|---|---|---|---|---|
| PPy-P300 | PPy-P1000 | PPy-RR0.2 | PPy-RR1 | ||
| N– |
5.5 | 10.1 | 5.9 | 7.2 | 398.0 |
| N–H | 57 | 48.6 | 58.7 | 19.5 | 400.0 |
| C–N+ | 26.9 | 29.7 | 25.4 | 56.3 | 401.2 |
| CN+ |
10.6 | 11.7 | 10.1 | 17.1 | 402.6 |
Another interesting aspect is the ratio between nitrogen atoms in polaron to bipolaron ([C–N+]/[CN+]) environments as deduced from the N 1s spectral intensities. Zuzana et al. investigated the PPy molecular structure evolution during oxidation polymerization and concluded that the depletion of reactants over time allows a stronger nucleophilic attack and results in a higher amount of defects.92 They concluded that the lower reaction kinetics give rise to higher number of defects which in turn is associated with higher polaron to bipolaron ratios.
In our case, fresh monomer and oxidant are continually fed into the reactor and unreacted precursors are pumped out. This avoids an increased defect formation during reaction, as no reactant depletion occurs and the kinetics of the PPy film formation is solely controlled by pressure and oxidant to monomer flow rate ratio. Under these conditions, except for the sample deposited at RR = 1, the other three samples retained a polaron to bipolaron ratio of 2.5. This validates that the evolution of bipolaron formation could be used as an indicator of the doping level of oCVD-polymerized PPy. In the case of RR = 1, polaron to bipolaron ratio of 3.3 is especially detrimental to the PPy structure as reflected in the conductivity data (Fig. 6(d)).
Cl 2p XPS spectra
To understand how SbCl5 interacts with PPy, we collected the XPS spectra of the Cl 2p core level region as shown in Fig. 7. We observe that chlorine is present in three different environments; the percentages with which the various components contribute to the total Cl 2p spectral intensity are reported in Table 6. The Cl− anion gives rise to the peak at a B.E. of 197.4 eV, Cl–N+ to the one at 198.4 eV and covalently bonded Cl with carbon is responsible for the XPS signal at 199.8 eV.93 Synthesis at higher P had a minimal effect on the amount of Cl− present, though it increased more than two times when RR was increased from 0.2 to 1. It is known that Cl− may outgas from PPy over time and/or could possibly attack the PPy chains and disrupt the conjugation.81 Cl–N+ species is responsible for charge transfer and did not show any significant variation when P and/or RR were changed. For Cl–C bonding, changing the pressure during synthesis had a negligible effect. However, PPy-RR0.2 showed a high content (>32%) of C–Cl bonds than PPy-RR1 (∼8%). This interestingly suggests that at higher RR, chlorine tends to stay in free ionic state compared to lower RR which favors C–Cl bonding. | ||
| Fig. 7 XPS spectra of Cl 2p core level region for (a) PPy-P300, (b) PPy-P1000 (= PPy-RR0.5), (c) PPy-RR0.2, and (d) PPy-RR1; the fit is also shown and the relative contribution for each component to the total Cl 2p spectral intensity is reported in Table 6. | ||
| Chemical environments | Relative contribution to the total spectral intensity of the Cl 2p core level line (%) | Binding energy (eV) | |||
|---|---|---|---|---|---|
| PPy-P300 | PPy-P1000 | PPy-RR0.2 | PPy-RR1 | ||
| Cl− | 38.5 | 34.7 | 19.3 | 46.4 | 197.4 |
| Cl–N+ | 46.4 | 51.6 | 48.4 | 46 | 198.4 |
| Cl–C | 15.1 | 13.8 | 32.2 | 7.6 | 199.8 |
O 1s and Sb 3d XPS spectra
Further information on the bonding of the SbCl5 dopant comes from the detailed XPS spectra of the region comprising the O 1s and Sb 3d core level lines, which are shown in Fig. S6 (ESI†) together with the corresponding fits. For each component, the relative contribution to the total spectral intensity of the respective line is reported in Table S2 in the ESI.† Antimony remains as an impurity in the PPy structure and exists in two forms: Sb(III), which gives rise to a contribution at 530.7 eV in the 3d5/2 line and Sb(0), which is at the origin of the contribution at 527.8 eV in the 3d5/2 line. Synthesis at higher pressure slightly increased the net Sb content of the films. In the RR series, increasing RR from 0.2 to 1 significantly increased the Sb content. The O 1s component at a B.E. of 530.89 eV is assigned to oxygen bonding with a metal (here Sb–O), while the contribution O 1s at 532.4 eV stems from CO.94
In summary, the PPy charge transport properties can only be understood through extensive spectroscopic studies. For a clear picture, at least five parameters extracted from the XPS spectra should be taken into account: (i) the C/N atomic ratio and type of carbon–carbon bonds (Cα/Cβ), informing about the conjugation of PPy; the N/Cl atomic ratio, the C–N+/CN+ ratio as these bonds inform on polaron/bipolaron formation, and the “O + Sb” content, which provides insight on how the dopant (SbCl5) interacts with PPy for a given set of operational parameters.
It is of utmost importance to find the oCVD operational parameters that yield a PPy structure that best retains the integrity of the pyrrole ring. This was achieved by controlling the precursors ratio to obtain a C/N atomic ratio close to 4. In terms of doping, PPy growth at higher RR and pressure both result in higher doping and translate into a lower N/Cl ratio, a lower C–N+/CN+ ratio and a higher impurity content. Notably, the increase in oxidant to monomer flow rate ratio has a stronger effect on the dopant interaction with the polymer backbone as evident from the change of the N/Cl. Increasing the pressure during deposition instead increases the total impurity content of PPy.
Conclusion
In this work oCVD has been demonstrated as a reliable method for one-step synthesis of conductive and homogenous PPy films. Temperature, pressure and oxidant to monomer flow rate ratio during oCVD control the doping and conductivity of the films. FTIR and UV-vis spectroscopy provide only basic information about PPy formation and the general trends in doping levels of the different PPy films. Additional insights about the nature of the interaction between PPy and the dopant were provided by XPS, which is the key to understand the conductivity behavior and the morphology development of these thin films. When deposited with a very low oxidant to monomer flow rate ratio (RR = 0.1), the film is not fully doped resulting in a conductivity of 57 S cm−1; the conductivity increased with increasing RR, reaching a maximum of 155 S cm−1. A higher oxidant to monomer flow rate ratio resulted in a higher C–N+/CN+ ratio, assigned to a higher polaron to bipolaron ratio, suggesting a structural barrier to the coupling of two polarons. Keeping an oxidant to monomer flow rate ratio of 0.2 and 0.5 at pressures of 300 and 500 mTorr, respectively, resulted in PPy films with the highest conductivities. In the case of deposition at pressure of 1000 mTorr however, the reaction kinetics showed a similar effect as overoxidation as seen in the N/Cl ratio, while it did not show an effect on the type of carbon–carbon coupling or the ratio of polaron to bipolaron. Finally, when depositing at higher temperature, the lower probability of reactants adsorption that is indirectly related to difference in RR, resulted in films with different morphology and with a different doping level, which in turn affect the conductivity. Such a conclusion is in line with the amorphous structure of the deposited PPy, since crystallinity cannot influence the properties of films deposited at these moderate temperatures of 40–80 °C.
In summary, this work provides insight into how to obtain conductive polypyrrole films using an all-dry technique. These results could contribute towards further electronic device development using polypyrrole in a range of applications from microelectronics to semiconductors, energy generation/storage, and biomedical devices.
Author contributions
Conceptualization: A. D., R. K. B., F. P.; methodology: A. D.; investigation: A. D.; resources: R. K. B., F. P., P. R.; data curation: A. D., M. M. (XPS), O. D. L. (XPS); writing – original draft preparation: A. D.; writing – review and editing: M. M., R. K. B., F. P., P. R., O. D. L.; visualization: A. D,. M. M. (XPS), O. D. L. (XPS); supervision: R. K. B., F. P., P. R.; project administration: R. K. B., F. P. All authors have read and agreed to the submitted version of the manuscript.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work benefitted from financial support by the Advanced Materials research program of the Zernike National Research Centre under the Bonus Incentive Scheme of the Dutch Ministry for Education, Culture and Science.Notes and references
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, Chem. Rev., 2012, 112, 4687–4735 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- Y. Liao, H. Wang, M. Zhu and A. Thomas, Adv. Mater., 2018, 30, 1705710 CrossRef PubMed.
- M. Ates, J. Adhes. Sci. Technol., 2016, 30, 1510–1536 CrossRef CAS.
- A. Kondyurin, K. Tsoutas, Q. X. Latour, M. J. Higgins, S. E. Moulton, D. R. McKenzie and M. M. M. Bilek, ACS Biomater. Sci. Eng., 2017, 3, 2247–2258 CrossRef CAS PubMed.
- K. Namsheer and C. S. Rout, RSC Adv., 2021, 11, 5659–5697 RSC.
- Q. Zhou and G. Shi, J. Am. Chem. Soc., 2016, 138, 2868–2876 CrossRef CAS PubMed.
- R. Kumar, J. Travas-Sejdic and L. P. Padhye, Chem. Eng. J. Adv., 2020, 4, 100047 CrossRef.
- P. Müller and M. Schmid, Foods, 2019, 8, 16 CrossRef PubMed.
- E. Hsu, K. Barmak, A. C. West and A. H. A. Park, Green Chem., 2019, 21, 919–936 RSC.
- M. Irimia-Vladu, E. D. Głowacki, G. Voss, S. Bauer and N. S. Sariciftci, Mater. Today, 2012, 15, 340–346 CrossRef CAS.
- R. Jain, N. Jadon and A. Pawaiya, TrAC, Trends Anal. Chem., 2017, 97, 363–373 CrossRef CAS.
- J. G. Ibanez, M. E. Rincón, S. Gutierrez-Granados, M. Chahma, O. A. Jaramillo-Quintero and B. A. Frontana-Uribe, Chem. Rev., 2018, 118, 4731–4816 CrossRef CAS PubMed.
- J. Stejskal and M. Trchová, Conducting polypyrrole nanotubes: a review, Springer International Publishing, 2018, vol. 72 Search PubMed.
- S. A. Alshehri, A. Al-yasari, F. Marken and J. Fielden, Macromolecules, 2020, 53(24), 11120–11129 CrossRef CAS.
- M. Goel and M. Thelakkat, Macromolecules, 2020, 53, 3632–3642 CrossRef CAS.
- T. Q. Nguyen, R. Y. Yee and B. J. Schwartz, J. Photochem. Photobiol., A, 2001, 144, 21–30 CrossRef CAS.
- Y. Yao, H. Dong and W. Hu, Polym. Chem., 2013, 4, 5197–5205 RSC.
- A. Salleo, R. J. Kline, D. M. DeLongchamp and M. L. Chabinyc, Adv. Mater., 2010, 22, 3812–3838 CrossRef CAS PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- L. Dall’Acqua, C. Tonin, A. Varesano, M. Canetti, W. Porzio and M. Catellani, Synth. Met., 2006, 156, 379–386 CrossRef.
- K. F. Babu, R. Senthilkumar, M. Noel and M. A. Kulandainathan, Synth. Met., 2009, 159, 1353–1358 CrossRef CAS.
- S. Shang, X. Yang, X. M. Tao and S. S. Lam, Polym. Int., 2010, 59, 204–211 CAS.
- A. Laforgue and L. Robitaille, Chem. Mater., 2010, 22, 2474–2480 CrossRef CAS.
- H. Wang, Y. Xue and T. Lin, Soft Matter, 2011, 7, 8158–8161 RSC.
- X. Qian, J. Chen and X. An, Appita J., 2010, 63, 102–107 CAS.
- T. Ojio and S. Miyata, Polym. J., 1986, 18, 95–98 CrossRef CAS.
- A. Mohammadi, M. Hasan, B. Liedberg and I. Lundstrom, Synth. Met., 1986, 14, 189–197 CrossRef CAS.
- F. He, M. Omoto, T. Yamamoto and H. Kise, J. Appl. Polym. Sci., 1995, 55, 283–287 CrossRef CAS.
- S. N. Tan and H. Ge, Polymer, 1996, 37, 965–968 CrossRef CAS.
- S. P. Khedkar and S. Radhakrishnan, Thin Solid Films, 1997, 303, 167–172 CrossRef CAS.
- J. Kim, D. Sohn, Y. Sung and E. R. Kim, Synth. Met., 2003, 132, 309–313 CrossRef CAS.
- J. Kim, S. Kwon, S. Han and Y. Min, Jpn. J. Appl. Phys., Part 1, 2004, 43, 5660–5664 CrossRef CAS.
- S. S. Najar, A. Kaynak and R. C. Foitzik, Synth. Met., 2007, 157, 1–4 CrossRef CAS.
- G. Han and G. Shi, Thin Solid Films, 2007, 515, 6986–6991 CrossRef CAS.
- A. Kaynak, S. S. Najar and R. C. Foitzik, Synth. Met., 2008, 158, 1–5 CrossRef CAS.
- P. Subramanian, N. B. Clark, L. Spiccia, D. R. MacFarlane, B. Winther-Jensen and C. Forsyth, Synth. Met., 2008, 158, 704–711 CrossRef CAS.
- N. O. Savage, Sens. Actuators, B, 2009, 143, 6–11 CrossRef.
- D. O. Kim, P. C. Lee, S. J. Kang, K. Jang, J. H. Lee, M. H. Cho and J. Do Nam, Thin Solid Films, 2009, 517, 4156–4160 CrossRef CAS.
- Y. K. Lee, K. J. Lee, D. S. Kim, D. J. Lee and J. Y. Kim, Synth. Met., 2010, 160, 814–818 CrossRef CAS.
- T. Bashir, M. Skrifvars and N. K. Persson, Polym. Adv. Technol., 2011, 22, 2214–2221 CrossRef CAS.
- R. C. Foitzik, A. Kaynak and F. M. Pfeffer, Synth. Met., 2006, 156, 637–642 CrossRef CAS.
- Y. Miao, L. Wan, X. Ling, B. Chen, L. Pan and Y. Gao, ACS Appl. Electron. Mater., 2020, 2, 855–862 CrossRef CAS.
- Y. Guo, M. T. Otley, M. Li, X. Zhang, S. K. Sinha, G. M. Treich and G. A. Sotzing, ACS Appl. Mater. Interfaces, 2016, 8, 26998–27005 CrossRef CAS PubMed.
- S. K. Kang, J. Koo, Y. K. Lee and J. A. Rogers, Acc. Chem. Res., 2018, 51, 988–998 CrossRef CAS PubMed.
- J. Stejskal, I. Sapurina, J. Vilcǎkova, M. Jurciǎ, M. Trchova, Z. Kolska, J. Prokes and I. Krivka, ACS Appl. Polym. Mater., 2021, 3(2), 1107–1115 CrossRef CAS.
- T. G. Yun, B. Il Hwang, D. Kim, S. Hyun and S. M. Han, ACS Appl. Mater. Interfaces, 2015, 7, 9228–9234 CrossRef CAS PubMed.
- J. P. Lock, S. G. Im and K. K. Gleason, Macromolecules, 2006, 39, 5326–5329 CrossRef CAS.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1–27 CAS.
- K. K. Gleason, CVD Polym., 2015, 1, 1–11 Search PubMed.
- R. Yang, A. Asatekin and K. K. Gleason, Soft Matter, 2012, 8, 31–43 RSC.
- S. Vaddiraju, K. Seneca and K. K. Gleason, Adv. Funct. Mater., 2008, 18, 1929–1938 CrossRef CAS.
- S. Nejati, A. Patel, G. R. Wallowith and K. K. S. Lau, Nanosci. Nanotechnol. Lett., 2015, 7, 50–55 CrossRef.
- A. Castro-Carranza, J. C. Nolasco, S. Bley, M. Rückmann, F. Meierhofer, L. Mädler, T. Voss and J. Gutowski, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1537–1544 CrossRef CAS.
- S. Kaviani, M. Mohammadi Ghaleni, E. Tavakoli and S. Nejati, ACS Appl. Polym. Mater., 2019, 1, 552–560 CrossRef CAS.
- M. Mirabedin, H. Vergnes, N. Caussé, C. Vahlas and B. Caussat, Appl. Surf. Sci., 2021, 554, 149501 CrossRef CAS.
- Y. Tan and K. Ghandi, Synth. Met., 2013, 175, 183–191 CrossRef CAS.
- M. Sravanthi and K. G. Manjunatha, Mater. Today: Proc., 2021, 1–5 Search PubMed.
- S. Sadki, P. Schottland, N. Brodie and G. Sabouraud, Chem. Soc. Rev., 2000, 29, 283–293 RSC.
- S. G. Im and K. K. Gleason, Macromolecules, 2007, 40, 6552–6556 CrossRef CAS.
- P. Camurlu, RSC Adv., 2014, 4, 55832–55845 RSC.
- H. Xie, M. Yan and Z. Jiang, Electrochim. Acta, 1997, 42, 2361–2367 CrossRef CAS.
- R. G. Davidson and T. G. Turner, Synth. Met., 1995, 72, 121–128 CrossRef CAS.
- A. V. Syugaev, N. V. Lyalina, A. N. Maratkanova and A. G. Kurenya, Synth. Met., 2020, 262, 116350 CrossRef CAS.
- B. Tian and G. Zerbi, J. Chem. Phys., 1990, 92, 3886–3891 CrossRef CAS.
- R. Kostić, D. Raković, S. A. Stepanyan, I. E. Davidova and L. A. Gribov, J. Chem. Phys., 1995, 102, 3104–3109 CrossRef.
- E. A. Sanches, S. F. Alves, J. C. Soares, A. M. Da Silva, C. G. Da Silva, S. M. De Souza and H. O. Da Frota, J. Nanomater., 2015, 129678 Search PubMed.
- H. Masuda and D. K. Asano, Synth. Met., 2003, 135–136, 43–44 CrossRef CAS.
- H. Kuzmany and J. Kürti, Synth. Met., 1987, 21, 95–102 CrossRef.
- P. Kar, Doping in Conjugated Polymers, 2013.
- M. Lögdlund, R. Lazzaroni, S. Stafström, W. R. Salaneck and J. L. Brédas, Phys. Rev. Lett., 1989, 63, 1841–1844 CrossRef PubMed.
- T. H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 452 Search PubMed.
- D. S. Kelkar and A. B. Chourasia, Indian J. Phys., 2012, 86, 101–107 CrossRef CAS.
- Y. Y. Smolin, M. Soroush and K. K. S. Lau, Ind. Eng. Chem. Res., 2017, 56, 6221–6228 CrossRef CAS.
- J. Lei, Z. Cai and C. R. Martin, Synth. Met., 1992, 46, 53–69 CrossRef CAS.
- V. P. Menon, J. Lei and C. R. Martin, Chem. Mater., 1996, 8, 2382–2390 CrossRef CAS.
- L. Atanasoska, K. Naoi and W. H. Smyrl, Chem. Mater., 1992, 4, 988–994 CrossRef CAS.
- H. Ge, G. Qi, E. T. Kang and K. G. Neoh, Polymer, 1994, 35, 504–508 CrossRef CAS.
- J. Tabačiarová, M. Mičušík, P. Fedorko and M. Omastová, Polym. Degrad. Stab., 2015, 120, 392–401 CrossRef.
- M. V. Zeller and S. J. Hahn, J. Mol. Electron., 1988, 4, 327–334 Search PubMed.
- P. Pfluger, M. Krounbi, G. B. Street and G. Weiser, J. Chem. Phys., 1983, 78, 3212–3218 CrossRef CAS.
- S. J. Hahn, W. J. Gajda, P. O. Vogelhut and M. V. Zeller, Synth. Met., 1986, 14, 89–96 CrossRef CAS.
- F. Beck, P. Braun and M. Oberst, Ber. Bunsen-Ges., 1987, 91, 967–974 CrossRef CAS.
- C. Malitesta, I. Losito, L. Sabbatini and P. G. Zambonin, J. Electron Spectrosc. Relat. Phenom., 1995, 76, 629–634 CrossRef CAS.
- E. Benseddik, M. Makhlouki, J. C. Bernede, S. Lefrant and A. Proń, Synth. Met., 1995, 72, 237–242 CrossRef CAS.
- P. S. Tóth, C. Janáky, O. Berkesi, T. Tamm and C. Visy, J. Phys. Chem. B, 2012, 116, 5491–5500 CrossRef PubMed.
- L. Qu, G. Shi, F. Chen and J. Zhang, Macromolecules, 2003, 36, 1063–1067 CrossRef CAS.
- H. S. Munro, D. Parker and J. G. Eaves, ESCA Studies of Polyaniline and Polypyrrole, Springer, Berlin, Heidelberg, 1987 Search PubMed.
- E. Pigois-Landureau, Y. F. Nicolau and M. Delamar, Synth. Met., 1995, 72, 111–119 CrossRef CAS.
- Z. Morávková, O. Taboubi, I. M. Minisy and P. Bober, Synth. Met., 2021, 271, 3–8 CrossRef.
- B. Saoudi, N. Jammul, M. M. Chehimi, A. S. Jaubert, C. Arkam and M. Delamar, Spectroscopy, 2004, 18, 519–535 CrossRef CAS.
- O. E. Linarez Pérez, M. D. Sánchez and M. López Teijelo, J. Electroanal. Chem., 2010, 645, 143–148 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc05082f |
| This journal is © The Royal Society of Chemistry 2022 |