
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of factors affecting the stability of compounds formed by isovalent substitution in layered oxychalcogenides, leading to identification of Ba3Sc2O5Cu2Se2, Ba3Y2O5Cu2S2, Ba3Sc2O5Ag2Se2 and Ba3In2O5Ag2Se2†
Gregory J.
Limburn
a,
Daniel W.
Davies
 bc,
Neil
Langridge
a,
Zahida
Malik
a,
Benjamin A. D.
Williamson
d,
David O.
Scanlon
b and
Geoffrey
Hyett
*a
bc,
Neil
Langridge
a,
Zahida
Malik
a,
Benjamin A. D.
Williamson
d,
David O.
Scanlon
b and
Geoffrey
Hyett
*a
aSchool of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: g.hyett@soton.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
cResearch Computing Service, Information and Communication Technology, Imperial College London, London, SW7 2AZ, UK
dDepartment of Materials Science and Engineering, Norwegian University of Science and Technology (NTNU), Trondheim 7491, Norway
First published on 8th February 2022
Abstract
Four novel compositions containing chalcogenide layers, adopting the Ba3M2O5M′2Ch2 layered structure have been identified: Ba3Sc2O5Cu2Se2, Ba3Y2O5Cu2S2, Ba3Sc2O5Ag2Se2 and Ba3In2O5Ag2Se2. A comprehensive comparison of experimental and computational results providing the crystallographic and electronic structure of the compounds under investigation has been conducted. Materials were synthesised at 800 °C under vacuum using a conventional ceramic synthesis route with combination of binary oxide and chalcogenide precursors. We report their structures determined by Rietveld refinement of X-ray powder diffraction patterns, and band gaps determined from optical measurements, which range from 1.44 eV to 3.04 eV. Through computational modelling we can also present detailed band structures and propose that, based on their predicted transport properties, Ba3Sc2O5Ag2Se2 has potential as a visible light photocatalyst and Ba3Sc2O5Cu2Se2 is of interest as a p-type transparent conductor. These four novel compounds are part of a larger series of sixteen compounds adopting the Ba3M2O5M′2Ch2 structure that we have considered, of which approximately half are stable and can be synthesized. Analysis of the compounds that cannot be synthesized from this group allows us to identify why compounds containing either M = La, or silver sulfide chalcogenide layers, cannot be formed in this structure type.
Introduction
The mixed-anion layered material Sr3Sc2O5Cu2S2 has been identified as a promising p-type transparent conductor, having an unusually high hole mobility,1,2 which subsequently prompted a computational search for further p-type conductors adopting this structure.3 Isostructural oxysulfides, Ba3Sc2O5Cu2S2 and Ba3In2O5Cu2S2, have been found to be photocatalysts,4 in common with other layered oxysulfides.5,6 The 32522 structure of Sr3Sc2O5Cu2S2 – the designation derived from the ratio of the ions – adopted by these compounds is part of a broader class of oxychalcogenides with isostructural copper or silver chalcogenide anti-litharge layers, separated by oxide layers with varying thickness and co-ordination, often adopting a fragment of the perovskite structure.7–9 The separation of the ‘heavy’ chalcogenide layers by the ‘light’ oxide layers in these materials creates a quantum well leading to hole transport in the chalcogenide layers combined with a large band gap,10,11 resulting in the photocatalysis and p-type conducting functions observed in these materials.Specifically, in the 32522 structure, A3M2O5M′2X2, the ‘heavy’ anion layer [M′2X2]2− adopts the anti-litharge structure, while the oxide layer [A3M2O5]2+ adopts a triple layer fragment of the perovskite structure, but truncated such that the geometry of the M ion is pyramidal rather than octahedral. There is a competing and related structure type which can be formed using ions with the same formal charges, the 42622, with general formula A4M2O6M′2X2, sometimes expressed as the empirical formula AMO3M′X. In comparison to the 32522 structure, the 42622 has an additional rock-salt structured AO layer which shears and displaces the vertex sharing of the apical oxygen of the MO5 polyhedra. Schematic unit cells for the two structures are shown in Fig. 1. It has been previously identified that there is a delicate balance between the stability of the 42622 or 32522 structure for a given combination of ions.12 For example, Sr4Ga2O6Cu2S2 is stable, but the 32522 equivalent is not. Replacing gallium with scandium, it is found that both structures, Sr4Sc2O6Cu2S2 and Sr3Sc2O5Cu2S2, can be formed. With further substitution of strontium by barium it is found that only the 32522 structure is formed, Ba3Sc2O5Cu2S2. The overall trend is that moving from smaller ions (Sr2+, Ga3+) to larger ions (Ba2+, Sc3+) leads to a preference for the 32522 structure over the 42622 structure.
 | ||
| Fig. 1 Structure map of layered mixed anion phases showing adoption of either 32522 or 42622. Examples with copper sulphide heavy layers are indicated by red circles, copper selenide by blue triangles, iron arsenides by black squares, and the less common layers (iron phosphide, cobalt arsenide, nickel phosphide and nickel arsenide) by green triangles. | ||
A survey of the literature indicates that there are a total of 10 compounds adopting the 32522 structure, where full structural details are available. The 42622 is slightly more numerous with 14 examples known. These mixed anion compounds have been found with a range of trivalent M ions in the [A4M2O6]2+ or [A3M2O5]2+ layers, including ions of chromium, gallium, vanadium, iron, manganese, scandium, and indium. The smaller Cr3+, Ga3+ and V3+ ions are only found with the 42622 structure, the larger Fe3+, Mn3+ and Sc3+ have been found in both the 32522 and 42622 types, while the largest, In3+, has only been found in the 32522 structure. A structure map plotting the average M–O bond length and the average A–O bond length is shown in Fig. 1, this indicates that for a given M–O bond length the 32522 structure has a larger A ion site that the 42622 structure. Therefore, the structure map indicates that novel compositions that might adopt the 32522 structure are more likely to be found in compositions containing larger A ions.
In this work we report on our attempt to identify new materials adopting the 32522 structure with optical and transport properties which will make them viable for p-type conducting or photocatalysis applications. In order to prevent the intra-band trapping states that would be detrimental to both these applications, this requires that the M ion must be a d0 and d10 trivalent cation.13 Our search of this space will include the Sc3+ ion, and the less studied larger ions, In3+, Y3+ and La3+ all combined with the largest practicable alkaline earth ion, Ba2+, which based on the analysis of the structure map should favour the formation of the 32522 structure. In a systematic review we will report our attempts to combine these four oxide layers; [Ba3Sc2O5]2+, [Ba3In2O5]2+, [Ba3Y2O5]2+, and [Ba3La2O5]2+, with the four heavy anion layers; [Cu2S2]2−, [Cu2Se2]2−, [Ag2S2]2− and [Ag2Se2]2−. This provides a matrix of 16 compounds in which to study the structural and property trends, by XRD, spectroscopy and computational modelling. Four of these sixteen are known, although one has not had a detailed structure published previously. We find that no lanthanum containing compound can be formed, nor any compound with a [Ag2S2]2− heavy layer, and we will discuss the factors behind the instability of these compositions. However, we can report Ba3Sc2O5Cu2Se2, Ba3Sc2O5Ag2Se2, Ba3In2O5Ag2Se2, and Ba3Y2O5Cu2Se2 as novel compounds.
Experimental section
Solid state synthesis
Layered intergrowth compounds of the form Ba3M2O5M′2Ch2 were targeted in 0.5 g batches by the conventional ceramic synthesis route – i.e. direct combination of appropriate stoichiometric ratios of binary precursors. As shown in eqn (1), for our target layered compounds we combined BaO, BaCh, M2O3 and M′2O in the ratio, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:1, where Ch = S or Se; M = Sc, In, Y or La; and M′ = Cu or Ag. The powders were mixed and ground together in a nitrogen-filled glove box, before being pressed into pellets using a hydraulic press with a pressure of 0.75 GPa. The pellets were placed in alumina crucibles and sealed inside a quartz tube under vacuum to form an evacuated ampoule. The samples were reacted at 800 °C for 12 hours, followed by regrinding and resealing under vacuum for a further heat treatment at 800 °C for 12 hours. For eight of the compositions the first two cycles of synthesis failed to yield a single-phase product, a third cycle was attempted at a higher temperature of 900 °C for 72 hours in each case, but this still did not produce the target phase.
:2:1:1, where Ch = S or Se; M = Sc, In, Y or La; and M′ = Cu or Ag. The powders were mixed and ground together in a nitrogen-filled glove box, before being pressed into pellets using a hydraulic press with a pressure of 0.75 GPa. The pellets were placed in alumina crucibles and sealed inside a quartz tube under vacuum to form an evacuated ampoule. The samples were reacted at 800 °C for 12 hours, followed by regrinding and resealing under vacuum for a further heat treatment at 800 °C for 12 hours. For eight of the compositions the first two cycles of synthesis failed to yield a single-phase product, a third cycle was attempted at a higher temperature of 900 °C for 72 hours in each case, but this still did not produce the target phase.| BaO + 2BaCh + M2O3 + M′2O → Ba3M2O5M′2Ch2 | (1) |
Characterisation
A Bruker D2 diffractometer equipped with a copper Kα X-ray source and Lynx Eye detector was used to collect powder X-ray diffraction patterns, in the range of 10° < 2θ < 100°, with a 0.02° step size and a scan time of 3 hours. Collected diffraction patterns were analysed using Rietveld refinement carried out with the GSAS-II software package.15 Spectroscopic data were recorded using a PerkinElmer Lambda 750S instrument, equipped with an integrating sphere. Spectra were measured across the range of 300 nm to 2500 nm, and used to determined sample band gaps with the method outlined by Poeppelmeier.16Preliminary photocatalytic studies were conducted on a sample of Ba3Sc2O2Ag2Se2, using the dichloroindophenol (DCIP) dye degradation test.17 A co-catalyst of cobalt oxide was deposited on the oxyselenide powder using a method modified from the literature.18 In brief, 25 mg of Ba3Sc2O2Ag2Se2 was placed in sample vial to which was added 10 mL of a 4.24 mol dm−3 Co(NO3)2·6H2O (Sigma Aldrich, 98%) solution in acetone. The acetone was allowed to evaporate overnight, and the cobalt nitrate impregnated powder sample was transferred to an alumina boat which was placed in a tube furnace. The sample was heated to 700 °C under a flow of ammonia, and annealed for 1 hour. The sample was cooled under ammonia flow, and then heated to 200 °C for 1 hour under air exposure. This process initially reduced the cobalt nitrate to metallic cobalt, and then partially oxidised to the desired cobalt oxide co-catalyst, with an approximate loading of 2% cobalt by mass.
The CoOx@Ba3Sc2O2Ag2Se2 powder was assessed for photocatalytic activity by taking a 10 mg sample in a sample vial and adding 5 mL of a 6.44 × 10−5 M DCIP dye and 2.28 × 10−3 M glycerol solution. The test was conducted by placing a crown stirrer in the vial to disperse the powder, and exposing the sample vial to a 5 sun solar simulator (LS0104 150 W Xenon lamp) equipped with a UV filter. At 60 min intervals, for 180 min, an aliquot was taken out of the vial, and then centrifuged to separate the dye from the powder. The visible light absorption spectrum of the dye solution was collected in the range of 300 nm to 800 nm using a PerkinElmer lambda 750S spectrometer. The solution and powder were recombined, and the aliquot returned to the sample vial prior to continued light exposure. Changes in dye concentration caused by photocatalytic degradation were determined using the Beer–Lambert law relationship between absorption and concentration of the dye. Control tests were conducted using dye and glycerol solution under solar simulator illumination without the catalyst present, and the solution with and without catalyst stirred in the dark.
Room temperature conductivity measurements were carried out on a sample of Ba3Sc2O2Cu2Se2. The compound was pressed into a pellet using a 5 mm die under 2 tons of pressure. The pellet was then annealed in an alumina crucible in a vacuum sealed silica tube at 800 °C for 12 hours. Wire contacts were attached to the annealed pellet (1.5 mm thick, 5 mm diameter) of Ba3Sc2O2Cu2Se2 using silver epoxy, and four point resistance measurements collected using a Keithley DMM6500 multimeter.
Computational methodology
First-principles calculations were carried out using the Vienna Ab initio Simulation Package (VASP).19,20 Each of the modelled structures was relaxed using the PBEsol functional21 with a Hubbard-like U correction of 5.17 eV on Cu and Ag.1 The Projector-Augmented Wave (PAW) method was used to describe interactions between the core and valence electrons.22 A plane-wave cut-off of 550 eV was used to avoid Pulay stress23 and a Γ-centred mesh with a k-point density of at least 70 Å3 was used to sample the Brillouin zone. Net forces on ions were reduced to less than 0.01 eV Å−1.Competing phases for all Ba3M2O5M′2Ch2 compounds were identified using the Materials Project API,24 limiting the search to those compounds that appear in the ICSD and resulting in a total of 172 compounds as listed in Table S1 (ESI†). These competing phases were relaxed using the same calculation parameters described above, with a denser k-point mesh of at least 100 Å3 used for metallic phases. All PBEsol calculations were carried out using the Fireworks python package25 and final values for energy above the convex hull of the compositional phase diagram (Ehull) were calculated using the Pymatgen python package.26 Bond distances and angles were extracted automatically using the CrystalNN algorithm.27
The screened hybrid exchange correlation functional, HSE06,28,29 has been shown in previous work to accurately calculate the electronic structure of this family of compounds,3 and to calculate accurate band gaps across a wide range of materials,30 so was used here to calculate band structures of the stable compounds. A plane-wave cutoff of 550 eV was used and a Γ-centred mesh with a k-point density of at least 70 Å3 was used to sample the Brillouin zone.
Band structures and optical absorption plots were produced using the Sumo python package.31 The optical absorption spectra were calculated using the real and imaginary parts of the dielectric constant calculated through a Kramers–Kronig transformation and a summation over the unoccupied bands, respectively, using a method by Adolph and co-workers.32 Within this formalism, intra-band and indirect absorptions are not accounted for and only direct valence to conduction band transitions are considered.33 Charge carrier effective masses were calculated using the curvature and electronic band extrema:
 | (2) |
Results and discussion
We have investigated 32522 compositions of the type Ba3M2O5M′2Ch2 where M = Sc, In, Y or La; Ch = S or Se; and M′ = Cu or Ag, and can report the successful formation of four novel layered oxychalcogenide phases: Ba3Sc2O5Cu2Se2, Ba3Sc2O5Ag2Se2, Ba3In2O5Ag2Se2 and Ba3Y2O5Cu2Se2 with a confirmed purity for each of at least 97% as determined by Rietveld refinement of powder XRD data. These materials were annealed twice at 800 °C under vacuum but were found to be air stable once cooled to room temperature, with the exception of Ba3Y2O5Cu2Se2 which decomposed after several days exposure to air.Rietveld refinement
The powder X-ray diffraction patterns of the four novel materials were modelled using the Rietveld method with the structure of the previously reported Ba3Sc2O5Cu2S2 as a starting point,3 using appropriate ion substitutions to match the expected composition in each case. For each phase the lattice, atomic position, and isotropic displacement parameters were refined to optimise the structural model. The sample background was also refined, while the instrumental peak profile parameters were fixed with values derived from refinement of the diffraction pattern of a highly crystalline LaB6 sample collected on the same diffractometer. To account for the effect of the sample on Bragg peak broadening, the uniaxial size and strain parameters were refined. Where impurity peaks were identified, variously BaSe, BaCO3 and In2O3, these were modelled using standard crystal structures identified from the inorganic crystal structure database (ICSD),34–36 with lattice, particle size, and phase fraction parameters refined.Using this approach good fits to the powder diffraction patterns were found for the samples where the observed Bragg peaks could be assigned to the expected target phase, with wRp values of less than 4.66% for these four samples. The Rietveld refinement fits to the data are shown in Fig. 2. Ba3Sc2O5Cu2Se2 was found with 99.0% purity by mass, with the remaining Bragg peaks assigned to small amounts of BaSe and BaCO3. The sample of Ba3Sc2O5Ag2Se2 was 97.9% pure with only BaCO3 as an impunity; Ba3In2O5Ag2Se2 was found alongside 2.4% BaCO3 and 0.6% In2O3; finally, Ba3Y2O5Cu2Se2 had no identifiable crystalline impurity. A summary of the key refinement fit and lattice parameters for these four samples can be seen in Table 1.
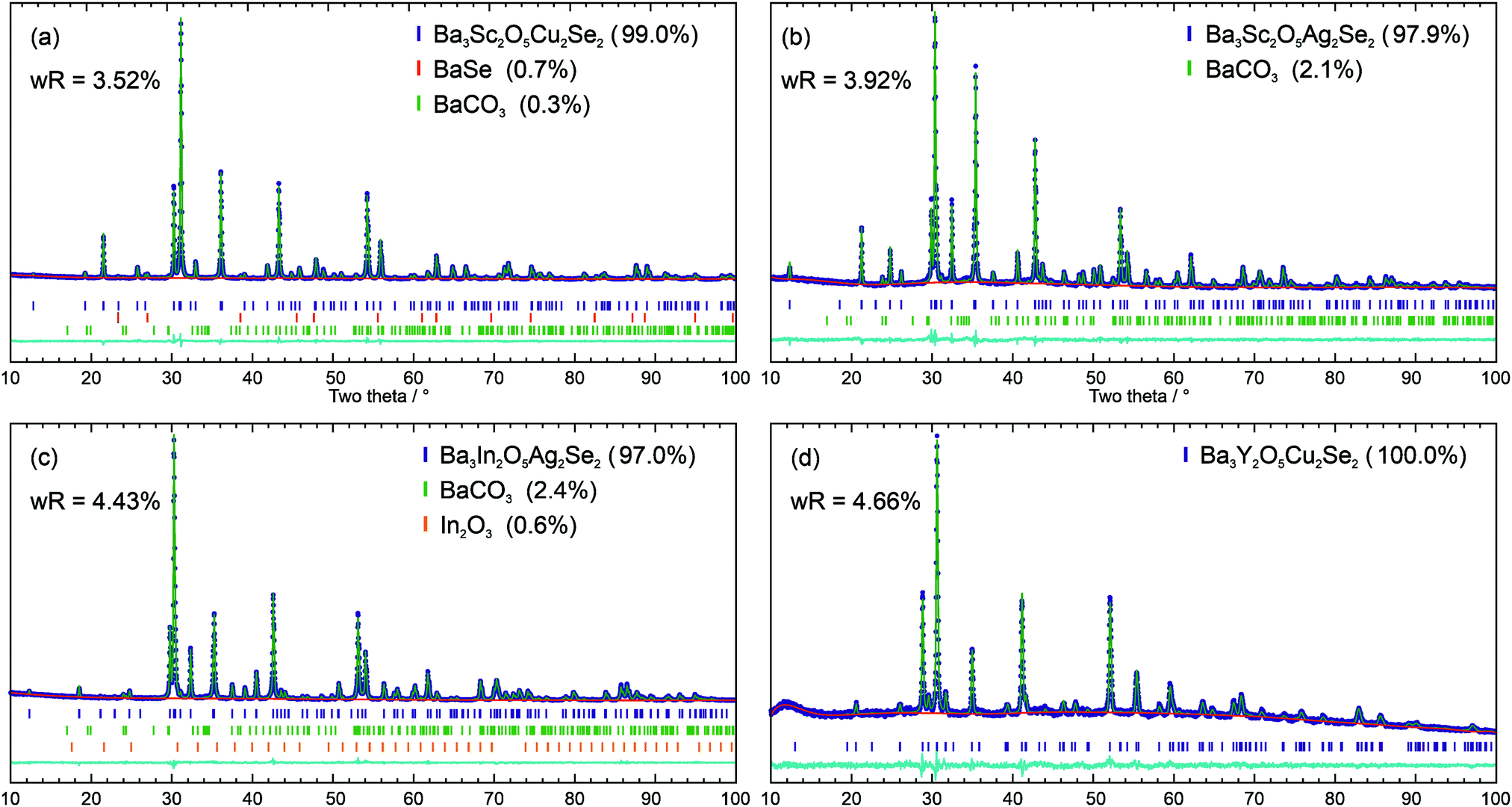 | ||
| Fig. 2 Rietveld refinement fits of the I4/mmm structure to diffraction data, with purity by mass and wRp fit values given for each data set: (a) Ba3Sc2O5Cu2Se2; (b) Ba3Sc2O5Ag2Se2; (c) Ba3In2O5Ag2Se2; (d) Ba3Y2O5Cu2Se2. In each plot the diffraction data is shown in blue, the model in in green and the background in red. Below the data are tick marks indicating the positions of the peaks, and a cyan difference curve. | ||
| Ba3Sc2O5Cu2Se2 | Ba3Sc2O5Ag2Se2 | Ba3In2O5Ag2Se2 | Ba3Y2O5Cu2Se2 | |
|---|---|---|---|---|
| Lattice parameter a/Å | 4.18127(7) | 4.21678(11) | 4.25908(8) | 4.3898(3) |
| Lattice parameter c/Å | 27.7132(6) | 28.6608(10) | 28.8780(6) | 27.474(2) |
| Volume/Å3 | 484.51(2) | 509.63(4) | 523.84(3) | 529.42(9) |
| Data points | 4443 | 4443 | 4443 | 4443 |
| Reflections (main phase) | 106 | 112 | 112 | 116 |
| Parameters | 37 | 34 | 35 | 46 |
| Purity | 99.0% | 97.9% | 97.0% | 100% |
| wRp | 3.52 | 3.92 | 4.43 | 4.66 |
| RF2 | 2.20 | 3.76 | 2.25 | 4.90 |
| χ 2 | 1.80 | 1.64 | 1.68 | 1.41 |
| Colour | Pale brown | Yellow-green | Black | Brown |
| Ba1 (0.5, 0.5, z) | 0 | 0 | 0 | 0 |
| Ba2 (0.5, 0.5, z) | 0.1424(1) | 0.1367(1) | 0.1384(1) | 0.1462(4) |
| M; (0, 0, z) | Sc; z = 0.0718(3) | Sc; z = 0.0695(3) | In; z = 0.0710(1) | Y; z = 0.0766(6) |
| O1 (0.5, 0, z) | 0.0814(5) | 0.0783(5) | 0.0804(4) | 0.089(2) |
| O2 (0, 0, z) | 0 | 0 | 0 | 0 |
| M′; (0.5, 0, z) | Cu; z = 0.25 | Ag; z = 0.25 | Ag; z = 0.25 | Cu; z = 0.25 |
| Se (0, 0, z) | 0.198(1) | 0.1885(2) | 0.1892(1) | 0.2002(7) |
In addition to the four novel Ba3M2O5M′2Ch2 compounds discussed above, there are a further four that have been previously reported: Ba3Sc2O5Cu2S2, Ba3InO5Cu2S2, Ba3In2O5Cu2Se2, and Ba3Y2O5Ag2Se2, although for the final example only the lattice parameters have been provided, with no further structural details.3,4,9 These stable layered oxychalcogenides provide a suite of homologous materials with which to compare our novel compounds, in the discussion below. We did attempt to repeat the synthesis of Ba3Y2O5Ag2Se2 for this work but found the material to be extremely air sensitive and were unable to collect diffraction data of sufficient quality to refine the atomic positions in order to provide a detailed structural analysis, however we are able to confirm the reported lattice parameters (ESI,† Fig. S1). We found that the related yttrium containing copper selenide, Ba3Y2O5Cu2Se2, is also air sensitive, but the decomposition is much slower, which allowed us to collect diffraction data of sufficient quality for Rietveld refinement, as described above.
Lattice parameter trends
Plots of the cell volume and a and c lattice parameters are shown in Fig. 3, for the four novel and four previously reported layered oxychalcogenides containing the [Ba3M2O5]2+ layer, as a function of the ionic radius of the M3+ ion. For a given oxide block, increasing the size of the ions in chalcogenide layer also leads to an increase in both a and c lattice parameter, as would be expected. The magnitude of the change between chalcogenide layers is surprisingly consistent independent of the oxide block in question, an increase of 0.9% in a and 2.0% in c with substitution of copper sulfide to copper selenide and 0.9% in a and 3.3% in c with substitution of copper selenide to silver sulfide. Considering the trends for a given chalcogenide layer, we observe the expected increase in the a lattice parameter with increasing M3+ ion radius. For example, for the copper selenides, the a lattice parameters are 4.18 Å, 4.22 Å and 4.39 Å for samples containing Sc3+, In3+ and Y3+ respectively. In contrast for changes in the c lattice parameter something more unusual is observed. There is an increase with the substitution of indium for scandium, but substitution for the larger yttrium ion leads to a decrease in the c lattice parameter. Overall however, the trends in cell volume are fully consistent with increasing ion size leading to increased cell volume for substitution in either the oxide or chalcogenide layers. Therefore, the unexpected decrease in c lattice parameter in Ba3Y2O5Cu2Se2 and Ba3Y2O5Ag2Se2 is compensated for by the greater increase in the a lattice parameter in each case. To fully explain these trends we must consider the structural changes within each layer. | ||
| Fig. 3 Plot of the lattice parameters for the I4/mmm structured Ba3M2O5M′2Ch2 phase plotted against the ionic radius of the M3+ ion present in each sample. Guidelines indicate and connect the samples with the same heavy anion layer. The a parameter is shown in the lower graph with squares, and the c lattice parameter in the middle graph with triangles./the top graph depicts the cell volume versus M ionic radii. Open symbols indicate previously reported materials, from ref. 3, 4 and 9, closed symbols represent novel materials reported in this paper. | ||
Structural details
Key bond angles, lengths and structural distances are shown in Fig. 4, and tabulated in Table S2 of the ESI.† These can be used to explain the observed changes and trends in the lattice parameters. We observe that for a given oxide block there is an expansion in both the a and c lattice parameter as a function of the heavy anion layer. This is clearly driven by the increasing length of the chalcogenide bond in order from Cu–S to Cu–Se to Ag–Se, due to the increasing ion sizes. The expansion in the a lattice parameter is partially constrained by the static oxide layer, so the longer chalcogenide bond is predominantly accommodated by the flexibility of chalcogenide layer,3 with a smaller bond angle and greater chalcogenide block height. This can be observed by consideration of the three samples with the [Ba3Sc2O5]2+ layer; with copper sulfide the S–Cu–S bond angle is 114° and the chalcogenide block height is 2.71 Å. The angle tightens to 111° with a block height of 2.87 Å for the copper selenide, the sequence ending with an angle of 100° and block height of 3.51 Å, for the much longer silver selenide bond. The height of the oxide bock, as measured by the Ba–Ba distance, remains relatively constant, varying by only about 0.01 Å from the copper sulfide to the silver selenide as the apical M–O bond length which controls this distance is almost independent of the chalcogenide layer. | ||
| Fig. 4 Left schematic diagram of the unit cell of the Ba3M2O5M′2Ch2; [(M = Sc, In, Y), (M′ = Cu, Ag) and (Ch = S, Se)]. Centre and right plots of the key angles, bond lengths and distances within the structure for the oxide and chalcogenide blocks. Values plotted as a function of the M ion in the oxide block, with chalcogenide layer distinguished by black squares for [Cu2S2]2−, red circles for [Cu2Se2]2−, and blue triangles for [Ag2Se2]2−. Filled symbols are for new materials reported here, empty symbols are from prior literature, ref. 3, 4 and 9. The lines shown connect samples with the same chalcogenide layer as a guide to the eye. Where they are not shown, the error bars are smaller than the symbols used. | ||
For a given chalcogenide layer the expansion in a lattice parameter with the replacement of Sc by In is of course driven by the increased size of the M3+ ion, which can be seen in the observed increase in the M–O bond length and especially the axial bond lengths, with an increase of 0.08 Å. This leads to the expansion of the oxide block height seen in Fig. 4 in the increase in the Ba–Ba distance. The response of the chalcogenide layer is only a limited change in the chalcogenide bond length, with the flexibly of the layer leading to an increase of the cis-bond angle instead, and a correspondingly reduced chalcogenide layer block height. For example, comparing Ba3Sc2O5Ag2Se2 to Ba3In2O5Ag2Se2 the silver selenide bond angle increases by 1° and the heavy layer block height decreases by 0.015 Å. Despite this decrease in the height of the chalcogenide layer with exchange of scandium for indium in each of the chalcogenide series, the much larger increase in the height of the oxide layer predominates and accounts for the observed increase in c lattice parameter.
These trends in bond length and angle changes in the oxide and chalcogenide blocks extend to Ba3Y2O5Cu2Se2, yet despite this we observe the anomalous decrease in the c lattice parameter. This can be rationalised as the decrease in the cell height comes from a significant drop in the interlayer spacing controlled by the square cuboid environment of the interlayer barium. The significant expansion in the a lattice parameter, when comparing Ba3In2O5Cu2Se2 to Ba3Y2O5Cu2Se2, caused by the much larger yttrium ion results in a flattening of the cuboid barium environment, as seen in the sharp decrease in the O–Ba–Ch angle from 79.3° to 74.6°, and a significant decrease in the interlayer spacing, as measured by the M–Ch distance which spans this environment and accounts for a 0.49 Å decrease across the unit cell. This can be thought of as the chalcogenide and oxide layers expanding in the basal direction around the inter-layer barium and towards each other in the 001 direction, as the barium ion is no longer the ideal size for the site when compared to the pairings with smaller ions.
DFT calculations
DFT calculations were carried out for all of the 16 proposed Ba3M2O5M′2Ch2 compositions, and their stability compared to binary and ternary competing phases. Full details of the results of these calculations can be found in Table S3 (ESI†). Eight of these were found to have an energy above the convex hull (Ehull) of zero, indicating that they are stable compared to the competing phases. These eight stable compounds matched with the four previously reported samples and the four novel compounds prepared here. The eight compounds determined to be unstable with respect to the competing phases were those that also could not be experimentally prepared, which will be discussed in more detail below. Overall, it highlights for these layered oxychalcogenide systems that energy above the convex hull is an excellent predictor for experimental stability.The calculations also allowed the structures to be predicted, including the material for which we could not refine a structure, Ba3Y2O5Ag2Se2. There was a good match between the structures derived from Rietveld modelling of experimental data and the structures derived from computational methods, with the lattice parameters of the models matching experimental results within 1% for all samples. Comparing the calculated bonds and angles, the observed differences between the two models were: barium–barium distance, less than 0.4%, barium to anion bonds less than 2%, M–O bonds <1.5% and O–M–O angle <2% (M = Sc, In, Y). Previous work has shown that the PBEsol functional slightly overestimates the chalcogenide bond angle compared to experiment, and this is reflected again here with both copper and silver chalcogenide angle. The computational model over predicts the Ch–M′–Ch angle by up to 3.5%, and underpredicts the bond length by 3%.
Overall, the accurate match between the DFT calculations and the empirically derived structural models provides confidence that we can reliably interpret the model derived for Ba3Y2O5Ag2Se2 for which it was not possible to collect Rietveld quality data. For Ba3Y2O5Ag2Se2, the DFT structural model indicates a continuation of the structural trends observed in the rest of the series. The reduction in the length of c parameter compared to Ba3In2O5Ag2Se2, which we can observe experimentally, is due to a significant decrease in the inter-layer spacing, as the expansion in the a lattice parameter driven by the larger yttrium ion forces a flattening of the square cuboid environment of the inter layer barium. There is almost no expansion in the oxide block in the 001 direction despite the larger yttrium cation, and only the expected slight decrease in the chalcogenide block height.
Electronic structure and optical properties
The full electronic band structures of the eight stable compounds are shown in Fig. 5 and these are used to calculate the light hole effective masses (m*hole). Three of these band structures have been previously reported (Ba3Sc2O5Cu2S2, Ba3In2O5Cu2S2 and Ba3In2O5Cu2Se2) and are reproduced here for completeness, marked with an asterisk.3,4 The values of m*hole in the [Cu2Ch2]2−/[Ag2Se2]2− planes are found at Γ–X and Γ–N and are relatively low. The values for each sample are reported in Table 2, but across all samples the intraplane hole masses are found in the range 0.37 me to 0.65 me, with lower values for selenide containing materials and higher values for the sulfide compounds. Overall the hole masses are comparable to the values predicted for Sr3Sc2O5Cu2S2 of 0.35 me to 0.45 me,1 and in which high hole mobility has been experimentally verified. The Γ–Z reciprocal space direction corresponds to hole transport in the interplanar direction, and the modelled band structures show very flat bands, indicating much higher hole masses and limited mobility, as expected.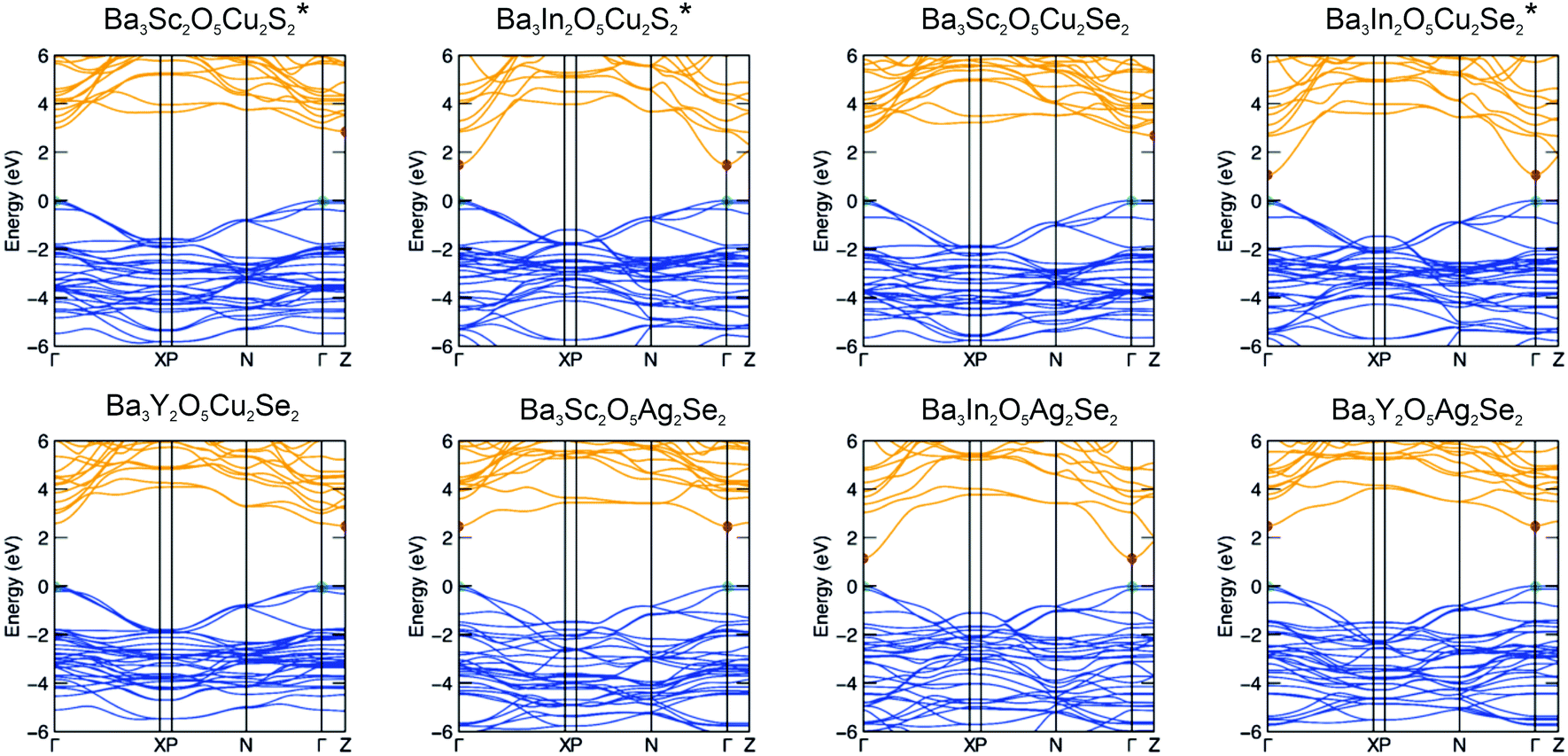 | ||
| Fig. 5 Electronic band structures of the eight thermodynamically stable Ba3M2O5M′2Ch2 compounds calculated using the HSE06 DFT functional. Red and green dots mark the conduction band minimum (CBM) and valence band maximum (VBM), respectively. Compositions marked with an asterisk have been published previously.3,4 | ||
| Band gap/eV | HSE06 optical band gap/eV | Light hole mass/me | VBM composition | CBM composition | |
|---|---|---|---|---|---|
| a Previously reported. b Uncertainty due to sample air sensitivity. Light hole effective masses for the newly reported compounds are broken down further by reciprocal space direction in Table S5 (ESI). | |||||
| Ba3Sc2O5Cu2S2a | 3.27a | 3.24 | 0.6–0.64 | S 2p | Ba 5d, Cu 4s |
| Ba3Y2O5Cu2Se2 | 3.04 | 0.46–0.5 | Se 3p | Ba 5d, Cu 4s | |
| Ba3Sc2O5Cu2Se2 | 3.09 | 2.95 | 0.45 | Se 3p | Ba 5d, Cu 4s |
| Ba3Y2O5Ag2Se2a | 2.63 | 0.46–0.48 | Se 3p | Ag 5s | |
| Ba3Sc2O5Ag2Se2 | 2.58 | 2.60 | 0.44–0.56 | Se 3p | Ag 5s |
| Ba3In2O5Cu2S2a | 1.77a | 1.84 | 0.54–0.56 | S 2p | In 5s |
| Ba3In2O5Ag2Se2 | 1.44 | 1.58 | 0.37–0.56 | Se 3p | In 5s |
| Ba3In2O5Cu2Se2a | 1.32a | 1.48 | 0.40–0.49 | Se 3p | In 5s |
The band gaps of the novel materials were experimentally determined from analysis of the diffuse reflection spectra, using the method recently outlined by Poeppelmeier, as suitable for crystalline, non-degenerate semiconductors.16 In brief, the Kubelka–Munk function was used to estimate the sample absorption from the reflectivity,37 and then a plot of the square of the absorption against the photon energy was used to extrapolate a tangent from the point of inflection to determine the band gap of the sample (Fig. 6). This was carried out for three of the novel materials, Ba3Sc2O5Cu2Se2 (band gap of 3.1 eV), Ba3Sc2O5Ag2Se2 (2.6 eV), Ba3In2O5Ag2Se2 (1.4 eV). These band gaps values are given in Table 2, alongside those previously reported for Ba3Sc2O5Cu2S2, Ba3In2O5Cu2S2 and Ba3In2O5Cu2Se2 and the values determined computationally from calculated optical absorption plots (Fig. S2, ESI†). For the yttrium containing samples experimental measurements could not be made due to their air sensitivity, so only the modelled band gaps are reported. Overall, there is good agreement between the experimental and calculated band gaps, as would be expected based on what we have shown previously for this family of materials.3
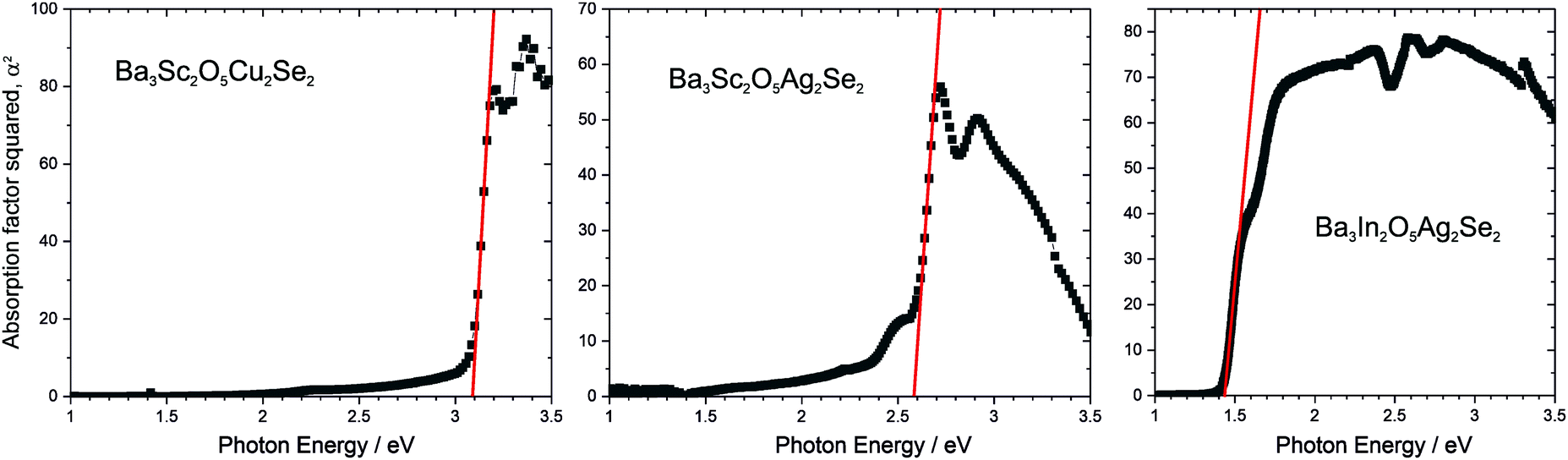 | ||
| Fig. 6 Plots of α2 against photon energy for novel compositions, Ba3Sc2O5Cu2Se2, Ba3Sc2O5Ag2Se2, and Ba3Sc2O5Ag2Se2, to allow band gaps to be determined from tangents shown in red. Absorption co-efficient determined using the Kubelka–Munk transformation. Spectra for yttrium containing samples could not be obtained due to air sensitivity. | ||
The trends in the band gaps observed for this series of compounds can be explained by considering the composition of the band edges derived from the electronic structure modelling. For all of the compounds, the valence band maximum (VBM) is comprised predominantly of the filled chalcogenide p orbitals, while the conduction band minimum (CBM) is predominantly composed of the empty coinage metal s states, except for the indium containing samples with the [Ba3In2O5]2+ block, where the In 5s states dominate then CBM. Ignoring the for the moment indium containing samples, this means that in the majority of the samples the size of the band gap is dependant only on the composition of the chalcogenide layer. We would expect sulfides to have larger band gaps than selenides, as the more electronegative sulfur lowers the VBM, while compounds with copper in the heavy layer would have a larger band gap than silver containing samples, as the more electronegative silver lowers the position of the CBM.
This explains why Ba3Sc2O5Cu2S2 has the largest band gap (3.24 eV, modelled value), due to the electronegative sulphur lowering the VBM, and the relatively electropositive copper raising the CBM. As both Ba3Y2O5Cu2Se2 and Ba3S2O5Cu2Se2 share the copper selenide heavy layer they have similar modelled band gaps (3.04 eV and 2.95 eV), due the VBM and CBM being composed of the same orbitals, but smaller than Ba3Sc2O5Cu2S2 due to the contribution of the more electropositive selenium to the VBM. The silver selenides; Ba3Y2O5Ag2Se2 and Ba3S2O5Ag2Se2 both have smaller band gaps of 2.63 eV and 2.60 eV, due to presence of the lower lying silver 5s states. The indium containing compounds have much smaller band gaps, as indium 5s states are much lower in energy than either copper or silver ns states giving band gap energies of 1.84 eV, 1.58 eV and 1.48 eV for the copper sulfide, silver selenide and copper selenide, respectively. The small differences observed in the modelled band gaps for the pairs Ba3Sc2O5Cu2Se2 and Ba3Y2O5Cu2Se2 (2.95 eV to 3.04 eV); Ba3Sc2O5Cu2Se2 and Ba3Y2O5Cu2Se2 (2.60 eV to 2.63 eV); and Ba3In2O5Cu2Se2 and Ba3In2O5Ag2Se2 (1.48 eV to 1.58 eV), despite each pair having identical band edge compositions, is due to a secondary effect where an increase in the basal lattice parameter reduces overlap and decreases the dispersion in the bands leading to the slight increase in band gap.8
From the novel compounds reported in this work we can conclude from the electronic structure data that Ba3Sc2O5Cu2Se2 is of most interest as a possible p-type transparent conductor. It has a calculated hole mobility equivalent to Sr3Sc2O5Cu2S2, and a sufficient large band gap to prevent visible light absorption and is air stable. We can also conclude that Ba3Sc2O5Ag2Se2 might be a functional visible light photocatalyst, based on its reasonable transport properties and 2.6 eV band gap.
The visible light photocatalytic potential of Ba3Sc2O5Ag2Se2 was assessed using a dye degradation test, where a solution of dichloroindophenol (DCIP) dye undergoes reductive decolouration in the presence of glycerol as a sacrificial oxidant, enhanced by the photocatalyst. This is an effective and simple test to assess visible light photocatalysts.17 The sample of Ba3Sc2O5Ag2Se2 was loaded with a cobalt oxide co-catalyst to enhance its activity,18 and allow for measurements to be conducted in a 3 hour time window. A 5 sun solar simulator with a UV light filter was used as the light source. This found that 10 mg of CoOx@Ba3Sc2O5Ag2Se2 in 5 mL of 6.44 × 10−5 M dye solution was able to degrade 15.1% of the dye during the three hour experiment. No degradation of the dye was observed without the catalyst present, or in the presence of the catalyst without the light source. This confirms that Ba3Sc2O5Ag2Se2 is a visible light photocatalyst, comparable to other layered oxychalcogenides tested using the same procedure, with a similar rate to Ba3In2O5Cu2S2 (14.5% dye degradation in three hours), but lower than Ba3Sc2O5Cu2S2 (27.0% degradation).4
Additionally, room temperature conductivity measurements were conducted on a pellet of Ba3Sc2O5Cu2Se2, which was found to have a density of 73% relative to the theoretical density derived from diffraction data. Four point probe measurements found that the pellet had a conductivity of 5.0(1) × 10−5 S cm−1, which is comparable to previous reports on other promising for pristine undoped layered oxychalcogenides such as LaOCuS (1 × 10−4 S cm−1),10 and Sr2GaO3CuS (2.2 × 10−4 S cm−1),38 although not as high as values reported for LaOCuSe (24 S cm−1) or Sr3Sc2O5Cu2S2 (2.8 S cm−1).2,39 However, it should also be considered that the value reported here for Ba3Sc2O5Cu2Se2 are for an annealed pellet, rather than a plasma sintered one.
Discussion of the unstable compositions
Above we discussed in detail the structural and electronic properties and relationships of the eight compounds that were found to be stable and could be synthesised from the 16 possible Ba3M2O5M′2Ch2 compounds, where M = In, Sc, Y, Ga and M′ = Cu, Ag. It is valuable to consider, however, the factors which underlie why the remaining eight materials are not stable.We have not been able to form any compound containing lanthanum, i.e. those with a [Ba3La2O5]2+ layer. All Bragg peaks observed in the diffraction patterns of our attempts at these samples could be indexed to elemental or binary by-products, as summarised in Table S4 (ESI†), with no indication of the expected peaks of the target phases. These syntheses were speculative, as there are no prior examples of lanthanum in the 5 coordinate M3+ ion environment in the 32522-structure type. The lanthanum ion, with an ionic radius of 1.03 Å, is considerably larger than the yttrium ion (0.90 Å) which is the largest ion found on the M site in the known examples of the structure type. We hypothesise that the limiting size for ions in the oxide block can be predicted based on the Goldschmidt tolerance factor, t, originally derived for predicting the formability of the perovskite structure, using the relationship of the relative ion sizes rA, rB and rO for the 12 coordinate cation site, the 6 coordinate cation site and the anion site, as shown in eqn (3). Although not always a guarantee of stability, it is successful as a filter for unstable compositions in the perovskites.40 A survey of all of the known 32522 structure types (and related 42622 structure) finds that these all have oxide layers with tolerance factors in the range of 0.9 to 1.0, which matches that observed for undistorted, cubic perovskites, implying that these oxychalcogenide structure types have a relative tight tolerance for the size of the ions. Calculating the tolerance factor for [Ba3La2O5]2+ give a value of 0.88, which falls outside of this stable range, and indicates that the barium ion is too small for the lanthanum ion. The tolerance factors for the oxide layers can be found in Table 3. Perovskite type compounds can be made with tolerance factors less than 0.9, but these undergo distortion through polyhedral tilting, to reduce the size of the 12-coordinate A site. It is likely that such distortions are not possible in the 32522 structure due to the need for commensurate bonding to the chalcogenide layer.
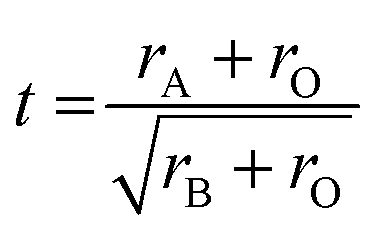 | (3) |
| Oxide layer | Tolerance factor, t |
|---|---|
| [Ba3Sc2O5]2+ | 0.99 |
| [Ba3In2O5]2+ | 0.97 |
| [Ba3Y2O5]2+ | 0.93 |
| [Ba3La2O5]2+ | 0.88 |
We were also unable to experimentally synthesise any compound containing a silver sulfide layer, nor were any predicted to be stable from computational methods. At first this is surprising, given the ability to synthesis examples of all the other copper and silver chalcogenide layers. However, in each of the attempts at forming a silver selenide, the same by-products were observed, elemental silver and barium sulphate. Our hypothesis for this instability is that silver is too readily reduced by sulfide, with BaSO4 acting as a thermodynamic sink. In contrast the silver selenides are stable because of the lower reduction potential of Se2− compared to S2−, and the consequent lower enthalpy of formation of BaSeO4. This also helps to explain the lack of prior examples of layered silver sulfides in the literature, where to our knowledge there are only two examples, these are LaOAgS,41 and Sr3Fe2.5O5Ag1.5S2; in the former there is no alkaline earth to form a sulphate and in the latter the composition is stabilised by partial substitution of silver for iron in the sulfide layer.42
The final compound that could not be synthesized was Ba3Y2O5Cu2S2. In our attempt at the synthesis of this we could not identify any of the expected peaks of the target phase in the diffraction pattern collected on the sample, which instead was found to contain Bragg peaks which could indexed to a mixture of Y2O3, BaS, Cu, BaSO4 and BaYO4. This inability to combine the [Ba3Y2O5]2+ layer with the [Cu2S2]2− layer can be rationalised based on the expected size. Yttrium is the largest of the three ions considered in the oxide block, while the [Cu2S2]2− layer contains the smallest paring of ions for the chalcogenide layer, so it is likely that the [Ba3Y2O5]2+ is simply too large to be accommodated in the structure with the copper sulfide layers. Although predicted to be unstable, it was still possible to computationally model and produce a relaxed structure for Ba3Y2O5Cu2S2, which had predicted lattice parameters of 4.34 Å and 26.48 Å. The largest previously reported a lattice parameter for a layered oxychalcogenide containing copper sulfide layers is the value of 4.18 Å in Ba3In2O5Cu2S2.4 In order to achieve this theoretical basal expansion to 4.34 Å the model requires an extreme stretching of the copper sulfide geometry, such that the S–Cu–S angle becomes 125°, even accounting for the tendency to over-predict the angle value, this is an extreme distortion from the ideal tetrahedral geometry. This would support the argument of size incompatibility as the reason for the inability to synthesis Ba3Y2O5Cu2S2, and also suggest the possible upper size limit for inclusion of the copper sulfide layer is approximately 4.20 Å and certainly no larger than 4.35 Å.
Conclusions
Our investigation of the Ba3M2O5M′2Ch2 compositions with coinage metal chalcogenide layers, and scandium, indium, yttrium and lanthanum oxide layers through both modelling and synthesis, has allowed us to identify four new compositions, Ba3Sc2O5Cu2Se2, Ba3Sc2O5Ag2Se2, Ba3In2O5Ag2Se2, and Ba3Y2O5Cu2Se2, and confirm that by targeting larger ions in the oxide layer we can promote the formation of this 32522 structure. We have identified that the Goldschmidt tolerance factor can be used as a simple guide for predicting formability, and that this must lie within a range of 0.9 to 1.0 for ions in the oxide layer in order to produce a stable 32522 structure. Additionally, computational modelling is highly effective at predicting stability for this series of compounds. We have also provided an explanation for the instability of layered oxychalcogenides with silver sulfide layers, which is due to the favourable redox reaction between sulfide and silver ions. Finally, we have identified that Ba3Sc2O5Ag2Se2 has potential as visible light photocatalyst, based on its transport properties and band gap, while Ba3Sc2O5Cu2Se2 is of interest as a possible p-type transparent conductor, and confirmed these with preliminary experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GH would like to acknowledge the financial support of the EPSRC through the grant EP/T011793/1. Via membership of the UK's HEC Materials Chemistry Consortium, which is funded by the EPSRC (EP/L000202, EP/R029431, EP/T022213), this work used the ARCHER2 UK National Supercomputing Service (www.archer2.ac.uk) and the UK Materials and Molecular Modelling (MMM) Hub (Thomas – EP/P020194 & Young – EP/T022213 supercomputers). DWD, BADW, and DOS would like to acknowledge support from the European Research Council, ERC, (Grant 758345). Finally BADW would like to acknowledge support from the Research Council of Norway (Project no. 275810).References
- D. O. Scanlon and G. W. Watson, (Cu2S2)(Sr3Sc2O5)−A Layered, Direct Band Gap, p-Type Transparent Conducting Oxychalcogenide: a Theoretical Analysis, Chem. Mater., 2009, 21(22), 5435–5442 CrossRef CAS.
- M.-L. Liu, L.-B. Wu, F.-Q. Huang, L.-D. Chen and I.-W. Chen, A promising p-type transparent conducting material: layered oxysulfide [Cu2S2][Sr3Sc2O5], J. Appl. Phys., 2007, 102(11), 116108 CrossRef.
- B. A. D. Williamson, G. J. Limburn, G. W. Watson, G. Hyett and D. O. Scanlon, Computationally Driven Discovery of Layered Quinary Oxychalcogenides: potential p-Type Transparent Conductors?, Matter, 2020, 3(3), 759–781 CrossRef PubMed.
- G. J. Limburn, M. J. P. Stephens, B. A. D. Williamson, A. Iborra-Torres, D. O. Scanlon and G. Hyett, Photocatalytic, structural and optical properties of mixed anion solid solutions Ba3Sc2−xInxO5Cu2S2 and Ba3In2O5Cu2S2−ySey, J. Mater. Chem. A, 2020, 8(38), 19887–19897 RSC.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Oxysulfide photocatalyst for visible-light-driven overall water splitting, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- M. Yashima, K. Ogisu and K. Domen, Structure and electron density of oxysulfide Sm2Ti2S2O4.9, a visible-light-responsive photocatalyst, Acta Crystallogr., Sect. B: Struct. Sci., 2008, B64(3), 291–298 CrossRef PubMed.
- S. D. N. Luu and P. Vaqueiro, Layered oxychalcogenides: structural chemistry and thermoelectric properties, J. Materiomics, 2016, 2(2), 131–140 CrossRef.
- S. J. Clarke, P. Adamson, S. J. C. Herkelrath, O. J. Rutt, D. R. Parker, M. J. Pitcher and C. F. Smura, Structures, physical properties, and chemistry of layered oxychalcogenides and oxypnictides, Inorg. Chem., 2008, 47(19), 8473–8486 CrossRef CAS PubMed.
- H. Ogino, Y. Katagi, J.-I. Shimoyama, K. Yamanoi, M. Tsuboi, T. Shimizu, N. Sarukura and K. Kishio, Luminescence properties of layered chalcogenide oxides Ba3RE2Ag2Se2O5, Opt. Mater., 2014, 36(12), 1978–1981 CrossRef CAS.
- K. Ueda, H. Hiramatsu, M. Hirano, T. Kamiya and H. Hosono, Wide-gap layered oxychalcogenide semiconductors: materials, electronic structures and optoelectronic properties, Thin Solid Films, 2006, 496(1), 8–15 CrossRef CAS.
- H. Hosono, Recent progress in transparent oxide semiconductors: materials and device application, Thin Solid Films, 2007, 515(15), 6000–6014 CrossRef CAS.
- D. O. Charkin, A. V. Sadakov, O. E. Omel’yanovskii and S. M. Kazakov, Synthesis, crystal structure, and properties of novel perovskite oxychalcogenides, Ca2CuFeO3Ch (Ch = S, Se), Mater. Res. Bull., 2010, 45(12), 2012–2016 CrossRef CAS.
- Y. Inoue, Photocatalytic water splitting by RuO2-loaded metal oxides and nitrides with d0- and d10-related electronic configurations, Energy Environ. Sci., 2009, 2(4), 364–386 RSC.
- G. Hyett, Z. A. Gal, C. F. Smura and S. J. Clarke, Ba2Mn2O4Cu0.9S: a layered oxysulfide with a new perovskite-related manganese oxide fragment, Chem. Mater., 2008, 20(2), 559–566 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, GSAS-II: the genesis of a modern open-source all purpose crystallography software package, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- A. Dolgonos, T. O. Mason and K. R. Poeppelmeier, Direct optical band gap measurement in polycrystalline semiconductors: a critical look at the Tauc method, J. Solid State Chem., 2016, 240, 43–48 CrossRef CAS.
- A. Mills and M. McGrady, A study of new photocatalyst indicator inks, J. Photochem. Photobiol., A, 2008, 193(2-3), 228–236 CrossRef CAS.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, Cobalt-Modified Porous Single-Crystalline LaTiO2N for Highly Efficient Water Oxidation under Visible Light, J. Am. Chem. Soc., 2012, 134(20), 8348–8351 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. L. Zhou and K. Burke, Restoring the density-gradient expansion for exchange in solids and surfaces, Phys. Rev. Lett., 2008, 100(13), 136406 CrossRef PubMed.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- P. Pulay, Ab initio calculation of force constants and equilibrium geometries in polyatomic molecules, Mol. Phys., 1969, 17(2), 197–204 CrossRef CAS.
- S. P. Ong, S. Cholia, A. Jain, M. Brafman, D. Gunter, G. Ceder and K. A. Persson, The Materials Application Programming Interface (API): a simple, flexible and efficient API for materials data based on REpresentational State Transfer (REST) principles, Comput. Mater. Sci., 2015, 97, 209–215 CrossRef.
- A. Jain, S. P. Ong, W. Chen, B. Medasani, X. Qu, M. Kocher, M. Brafman, G. Petretto, G.-M. Rignanese, G. Hautier, D. Gunter and K. A. Persson, FireWorks: a dynamic workflow system designed for high-throughput applications, Concurr. Comput. Pract. Exp., 2015, 27(17), 5037–5059 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (pymatgen): a robust, open-source python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- H. Pan, A. M. Ganose, M. Horton, M. Aykol, K. A. Persson, N. E. R. Zimmermann and A. Jain, Benchmarking Coordination Number Prediction Algorithms on Inorganic Crystal Structures, Inorg. Chem., 2021, 60(3), 1590–1603 CrossRef CAS PubMed.
- J. Heyd and G. E. Scuseria, Efficient hybrid density functional calculations in solids: assessment of the Heyd–Scuseria–Ernzerhof screened Coulomb hybrid functional, J. Chem. Phys., 2004, 121(3), 1187–1192 CrossRef CAS PubMed.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, Energy band gaps and lattice parameters evaluated with the Heyd–Scuseria–Ernzerhof screened hybrid functional, J. Chem. Phys., 2005, 123(17), 174101 CrossRef PubMed.
- P. Borlido, J. Schmidt, A. W. Huran, F. Tran, M. A. L. Marques and S. Botti, Exchange-correlation functionals for band gaps of solids: benchmark, reparametrization and machine learning, npj Comput. Mater., 2020, 6(1), 96 CrossRef CAS.
- A. M. Ganose, A. J. Jackson and D. O. Scanlon, sumo: command-line tools for plotting and analysis of periodic *ab initio* calculations, J. Open Source Software, 2018, 3(28), 717–719 CrossRef.
- B. Adolph, J. Furthmuller and F. Bechstedt, Optical properties of semiconductors using projector-augmented waves, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63(12), 125108 CrossRef.
- M. Gajdos, K. Hummer, G. Kresse, J. Furthmuller and F. Bechstedt, Linear optical properties in the projector-augmented wave methodology, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(4), 045112 CrossRef.
- J. P. Devilliers, Crystal structures of aragonite, strontianite, and witherite, Am. Mineral., 1971, 56(5-6), 758–767 CAS.
- M. Marezio, Refinement of the crystal structure of In2O3 at two wavelengths, Acta Crystallogr., 1966, 20(6), 723–728 CrossRef CAS.
- M. K. Slattery, The Crystal Structure of Metallic Tellurium and Selenium and of Strontium and Barium Selenide, Phys. Rev., 1925, 25(3), 333–337 CrossRef CAS.
- P. Kubelka, New Contributions to the Optics of Intensely Light-Scattering Materials. Part I, J. Opt. Soc. Am., 1948, 38(5), 448–457 CrossRef CAS PubMed.
- K. Ueda, S. Hirose, H. Kawazoe and H. Hosono, Electrical and Optical Properties of Layered Oxysulfides with CuS Layers: Sr−Cu−M−O−S System (M = Zn, Ga, In), Chem. Mater., 2001, 13(5), 1880–1883 CrossRef CAS.
- M. L. Liu, L. B. Wu, F. Q. Huang, L. D. Chen and J. A. Ibers, Syntheses, crystal and electronic structure, and some optical and transport properties of LnCuOTe (Ln = La, Ce, Nd), J. Solid State Chem., 2007, 180(1), 62–69 CrossRef CAS.
- W. Li, E. Ionescu, R. Riedel and A. Gurlo, Can we predict the formability of perovskite oxynitrides from tolerance and octahedral factors?, J. Mater. Chem. A, 2013, 1(39), 12239–12245 RSC.
- M. Palazzi and S. Jaulmes, Structure of the ionic conductor (lao)ags, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1337–1339 CrossRef.
- L. Cario, A. Lafond, T. Morvan, H. Kabbour, G. Andre and P. Palvadeau, Design and magnetic properties of new compounds containing iron 2D building blocks of the perovskite type, Solid State Sci., 2005, 7(8), 936–944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: Rietveld refinement of attempt at forming Ba3Y2O5Ag2Se2. Fig. S2: Calculated optical absorption data. Table S1: List of competing phases used in calculating energy above hull. Table S2: Selected band distances and angles from refined structural models. Table S3: Structural details from computational models. Table S4: Details of refinement of compounds which could not be synthesized. Table S5: Light hole masses for newly reported compounds. All data supporting this study are openly available from the University of Southampton repository at DOI: http://10.5258/SOTON/D2122. See DOI: 10.1039/d1tc05051f |
| This journal is © The Royal Society of Chemistry 2022 |