
Highly efficient blue electroluminescence based on TADF emitters with spiroacridine donors: methyl group effect on photophysical properties†

Abstract
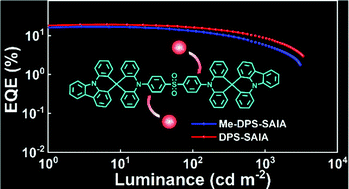
The methyl group plays an important role in the regulation of the photoluminescence and electroluminescence properties of thermally activated delayed fluorescence (TADF) emitters. In this work, a new type of methyl effect was revealed by designing donor–accepter–donor (D–A–D) type TADF systems with decorating methyl groups at the ortho-position of the donor units. Different from previous work in which the methyl groups at the ortho-position resulted in improved TADF properties, the insertion of methyl groups in this work led to the reduction of the molecular rigidity, the photoluminescence quantum yield (PLQY) and the ratio of delayed fluorescence, as well as the extension of the lifetime of delayed fluorescence. The unusual changes of the photoluminescence behavior were well explained by theoretical simulations and further verified by the performance in electroluminescence devices. The blue organic light-emitting diodes (OLEDs) based on the new emitters achieved a maximum external quantum efficiency (EQE) of 19.3%.
- This article is part of the themed collection: Materials for thermally activated delayed fluorescence and/or triplet fusion upconversion
 Please wait while we load your content...
Please wait while we load your content...

