
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dominant dimer emission provides colour stability for red thermally activated delayed fluorescence emitter†
Tom
Cardeynaels
 ab,
Marc K.
Etherington
*cd,
Simon
Paredis
a,
Andrei S.
Batsanov
e,
Jasper
Deckers
a,
Kleitos
Stavrou
d,
Dirk
Vanderzande
a,
Andrew P.
Monkman
d,
Benoît
Champagne
b and
Wouter
Maes
*a
ab,
Marc K.
Etherington
*cd,
Simon
Paredis
a,
Andrei S.
Batsanov
e,
Jasper
Deckers
a,
Kleitos
Stavrou
d,
Dirk
Vanderzande
a,
Andrew P.
Monkman
d,
Benoît
Champagne
b and
Wouter
Maes
*a
aHasselt University, Institute for Materials Research (IMO-IMOMEC), Design & Synthesis of Organic Semiconductors (DSOS), Agoralaan 1, 3590 Diepenbeek, Belgium and IMOMEC Division, IMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium. E-mail: wouter.maes@uhasselt.be
bUniversity of Namur, Laboratory of Theoretical Chemistry, Theoretical and Structural Physical Chemistry Unit, Namur Institute of Structured Matter, Rue de Bruxelles 61, 5000 Namur, Belgium
cDepartment of Mathematics, Physics & Electrical Engineering, Northumbria University, Ellison Place, Newcastle upon Tyne, NE1 8ST, UK. E-mail: marc.k.etherington@northumbria.ac.uk
dOEM Group, Department of Physics, Durham University, South Road, Durham DH1 3LE, UK
eDepartment of Chemistry, Durham University, South Road, Durham DH1 3LE, UK
First published on 25th March 2022
Abstract
Colour purity and stability in multi-donor thermally activated delayed fluorescence (TADF) emitters has significant implications for commercial organic light-emitting diode (OLED) design. The formation of emissive dimer states in the well-known 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) chromophore at elevated dopant concentrations has recently been confirmed both experimentally and via theoretical calculations, indicating that multi-donor emitters such as 4CzIPN might suffer from a lack of colour stability due to the presence of multiple emissive states. This poses a serious issue for OLED manufacturers. In this work, dithieno[3,2-b:2′,3′-d]pyrrole (DTP) is applied as an alternative donor unit in a TADF emitter for the first time. In combination with isophthalonitrile (IPN), the 4CzIPN analogue termed 4DTPIPN is obtained. The strong electron donating nature of the DTP moiety gives rise to a red shift of the emission with respect to that of 4CzIPN. We identify that 4DTPIPN has a very stable emission spectrum throughout all solid-state thin film concentrations and host materials. Rather interestingly, this colour stability is obtained via the formation of dimer/aggregate species that are present even at 0.01 wt% concentration. Unfortunately, the higher colour stability is paired with a low photoluminescence quantum yield, making 4DTPIPN unviable for device applications. Nonetheless, this work shows the importance of dimer contributions, even at dilute doping concentrations. This molecule and study provide important understanding of the aggregation behaviour of small-molecule emitters necessary for the successful application of doped and, especially, non-doped OLED architectures.
Introduction
Thermally activated delayed fluorescence (TADF) has emerged as the most successful triplet upconversion strategy because of its relatively simple mechanism and the possibility to achieve near-unity internal quantum efficiencies (IQEs) in organic light-emitting diodes (OLEDs), which is not possible for alternative strategies such as triplet–triplet annihilation (TTA) or via the use of hybridized local and charge-transfer (HLCT) states.1–4 TADF relies on the thermal upconversion of triplet excitons to the singlet state by bringing these two states close in energy. To achieve this, the overlap between the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals needs to be reduced. In practice, donor–acceptor (D–A) or donor–acceptor–donor (D–A–D) type compounds are constructed with large steric hindrance between the donor and acceptor moieties, leading to a strong twist. The large dihedral angle between the two subunits effectively localizes the HOMO and LUMO on the donor and acceptor moieties, respectively, affording spatial separation of the orbitals.The formation of charge-transfer (CT) states helps to minimise the singlet–triplet gap. However, we know from previous work that transitions between these CT states via spin–orbit coupling (SOC) become forbidden as there can be no change in orbital angular momentum.5,6 A nearby third state (often a locally excited triplet state) is necessary to obtain non-negligible rates of intersystem crossing (ISC) and reverse intersystem crossing (rISC).7 This is known as the spin-vibronic coupling mechanism of TADF, in which the CT excited triplet state (3CT) is vibrationally coupled with a locally excited triplet state (3LE) and this mixed state allows rISC to the singlet state (1CT).8–10
One of the most studied TADF emitters to date is 2,4,5,6-tetrakis(9H-carbazol-9-yl)isophthalonitrile, better known as 4CzIPN. First published in 2012 by Adachi et al.,114CzIPN was the emitter in a green OLED with an external quantum efficiency (EQE) of 19.3 ± 1.5%. This molecule has inspired many multi-carbazole and multi-donor TADF materials since. More recent investigations have considered the effect of concentration and intermolecular interactions for this model system. Kim et al. showed that a significant bathochromic shift in the CT state emission occurs for 4CzIPN with increasing concentration in a nonpolar host.12 They attributed this behaviour to the solid-state solvation effect (SSSE) in which the polarity of dopant 4CzIPN molecules influences the other surrounding 4CzIPN emitters, just as solvent molecules would do in solution. However, this was refuted by Northey et al. as the SSSE was shown to be less effective than the solvation effects in liquid solvents due to the inability of the solid-state molecules to rearrange.13 More recent follow-up modelling work by Yang et al. showed that SSSE may still play a larger role than initially predicted but it remains a weak effect compared to standard solution polarity effects.14 Etherington et al. investigated the photophysics of 4CzIPN in more detail, looking at potential dimer formation that could lead to the shift of the CT emission.15 Sublimation of pristine 4CzIPN led to the formation of several fractions, all of them crystal polymorphs with different emission properties in which the carbazole units of separate molecules were found to show partial or full face-to-face overlap to form dimer species. Photophysical characterization of 4CzIPN in different hosts and at different concentrations confirmed dimer formation in the solid state as the dominant effect resulting in the observed bathochromic shift.15 Molecular dynamics simulations by Cho et al. confirmed the presence of dimer species being responsible for the large concentration dependent red-shift of 4CzIPN emission in doped films.16
Salah et al.17 diversified this work by studying a 4CzIPN analogue with pyrazine as an acceptor unit instead of isophthalonitrile and concluded that the smaller dihedral angles between the acceptor and carbazole units resulted in a lower tendency to form dimers. This indicates that the carbazole dimerization behaviour can be controlled by altering a different part of the molecule and does not just require modification of the carbazole itself. More recently, Imbrasas et al. reported a multi-carbazole methyl 2,3,4,5,6-penta(carbazol-9-yl)benzoate (5CzCO2Me) emitter.18 In their concentration dependent study in bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO), they interpret the red-shift in emission of 5CzCO2Me within the SSSE model. This collection of work is part of a wider discussion on intermolecular and SSSE behaviour in a wide range of organic light-emitting materials, which are important for commercialisation prospects where colour stability is paramount.
In an attempt to overcome the dimer formation in 4CzIPN, the 9H-carbazole units were replaced here by dithieno[3,2-b:2′,3′-d]pyrrole (DTP) to create the 4DTPIPN counterpart. DTP was chosen as it is a widely used building block for the development of push–pull (small molecule and conjugated polymer) materials for organic photovoltaics19–22 and it has also been used for the design of fluorescent materials.23 While thiophene derivatives have been used for the development of fluorescent emitters, the photoluminescence quantum yield (PLQY) of such materials is generally rather low. Locking the flexible nature of 2,2′-bithiophene by ring fusion between the two thienyl units with an N-alkyl moiety leads to a significant increase in the PLQY as non-radiative decay pathways are restricted.21,23 Furthermore, the photochemical stability of the DTP unit renders DTP-based emitters one of the most successful thiophene-based fluorescent materials.23–25
In contrast to our initial goal, a more persistent form of dimer species that provided enhanced colour stability at the expense of PLQY was obtained. 4DTPIPN is put forward as a contrast to the materials studied by Salah et al.17 and shows how control and enhancement of dimer formation can be used to obtain colour stability, which may be especially relevant for non-doped OLEDs, an emerging field of investigation that will be heavily reliant on solid-state interactions.
Results and discussion
A two-step procedure was used for the preparation of 4DTPIPN, involving the synthesis of a tert-butyloxycarbonyl-protected DTP and subsequent deprotection (Scheme 1). This synthesis protocol was reported by Förtsch et al. and gives an overall yield of 62% after the ring-closure and deprotection steps.26 Despite having a lower overall yield than other procedures reported in the literature, the two-step synthesis affords better control over the reaction conditions and allows the intermediate to be isolated and purified before the final deprotection step. Another method with higher reported yield, also published by Förtsch et al.,27 was also attempted but finally abandoned as we were unable to obtain the target product via this route. The final nucleophilic substitution reaction between DTP and tetrafluoroisophthalonitrile was performed according to the synthesis procedure for 4CzIPN (see Table S2 for structure, ESI†).11 The detailed synthetic procedure and characterisation data can be found in the ESI† and Fig. S1 and S2 (ESI†). Lastly, the photo- and thermal stability of 4DTPIPN were investigated. Monitoring of the emission under constant illumination reveals only a slight decay (<5%) of the total emission intensity, whereas this is much larger for 4CzIPN (Fig. S3 and Table S1, ESI†). Thermogravimetric analysis revealed degradation with significant weight loss as a result at temperatures of 414 °C and above (Fig. S4, ESI†), which is very comparable to the values obtained for 4CzIPN in previous work.15 The 4DTPIPN structure was confirmed by single crystal X-ray crystallography (Fig. S5, ESI†). | ||
| Scheme 1 Synthesis pathway toward 4DTPIPN: (i) CuI, tert-butyl carbamate, N,N′-dimethylethylenediamine, K2CO3, toluene, 110 °C, 24 h; (ii) K2CO3, methanol, room temperature, 5 h; (iii) tetrafluoroisophthalonitrile, NaH, THF, 50 °C, 16 h. | ||
Using the Gaussian16 quantum chemistry package,28 density functional theory (DFT) calculations were performed to optimize the geometry of 4DTPIPN (M06/6-311G(d); Fig. 1).294CzIPN was included in these calculations using the same methods to allow straightforward comparison of the theoretical properties of both molecules. All frequencies were found to be real, indicating that the optimized geometries correspond to minima on the potential energy surfaces. The calculated geometry of 4DTPIPN shows dihedral angles of around 57° for the DTP units with respect to the IPN core, with only small differences between the individual DTP units. These values are in excellent correspondence with those obtained from single-crystal X-ray diffraction (Fig. S5 and S6, Tables S2 and S3, ESI†). 4CzIPN shows slightly larger dihedral angles for the carbazole units of around 62°. Nevertheless, there is a higher degree of localization of the HOMO on the donor (DTP/Cz) unit (Fig. 1) for 4DTPIPN than for 4CzIPN. This is expected to give a very small ΔEST and large CT character for the HOMO → LUMO transitions. Further calculations can be found in Fig. S7 and Table S4 (ESI†).
 | ||
| Fig. 1 Optimized geometries and HOMO and LUMO for 4DTPIPN and 4CzIPN. Isosurface values of 0.02 a.u. were used for all orbitals. | ||
To provide a benchmark to the previous studies and ascertain the behaviour of 4DTPIPN, the steady-state absorption and (time-resolved) photoluminescence spectra of the novel compound were measured in both solution—methylcyclohexane, toluene, chloroform, and 2-methyltetrahydrofuran—and in the solid state—zeonex, polystyrene, bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO), 1,3-bis(triphenylsilyl)benzene (UGH-3), and as neat film (Fig. 2, 4, 5, and Fig. S8–S20, ESI†). The solid-state experiments were performed at different concentrations for zeonex and polystyrene (0.01, 0.1, 1, 10 wt%) to analyse the aggregation behaviour (vide infra).
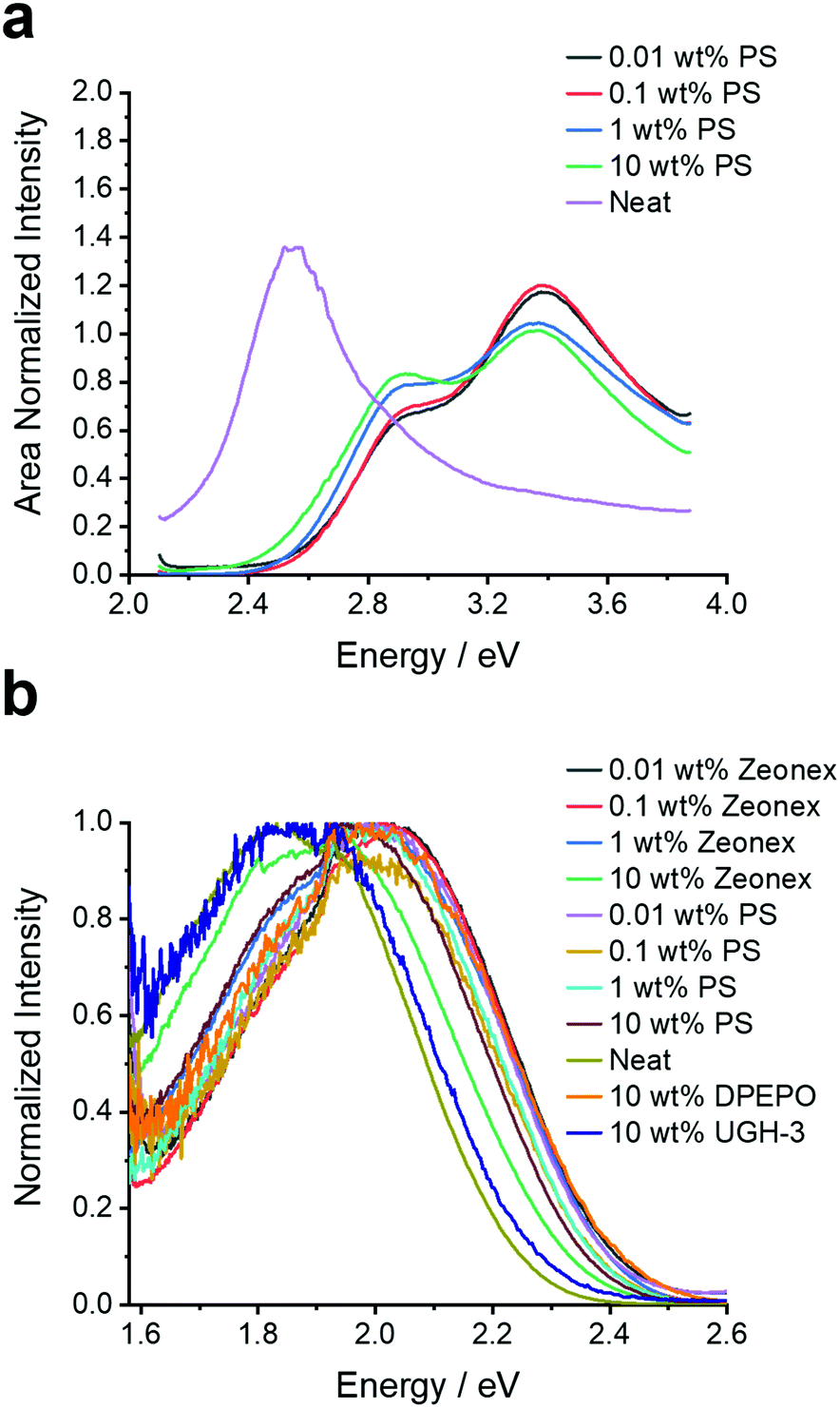 | ||
| Fig. 2 (a) Excitation spectra monitored at the emission peak of 2.05 eV and (b) fluorescence emission spectra for 4DTPIPN in various host materials (polystyrene, zeonex, UGH-3, DPEPO, and neat) and at different concentrations (for polystyrene and zeonex). N.B. For the excitation scan of the neat film the amplification of the sub bandgap absorption is a result of the measurement technique and is not a reflection of the oscillator strength of the dimer/aggregate absorption. This makes the technique effective for highlighting low absorbance bands, but the true absorption profile should always be interpreted from Fig. S8 (ESI†). | ||
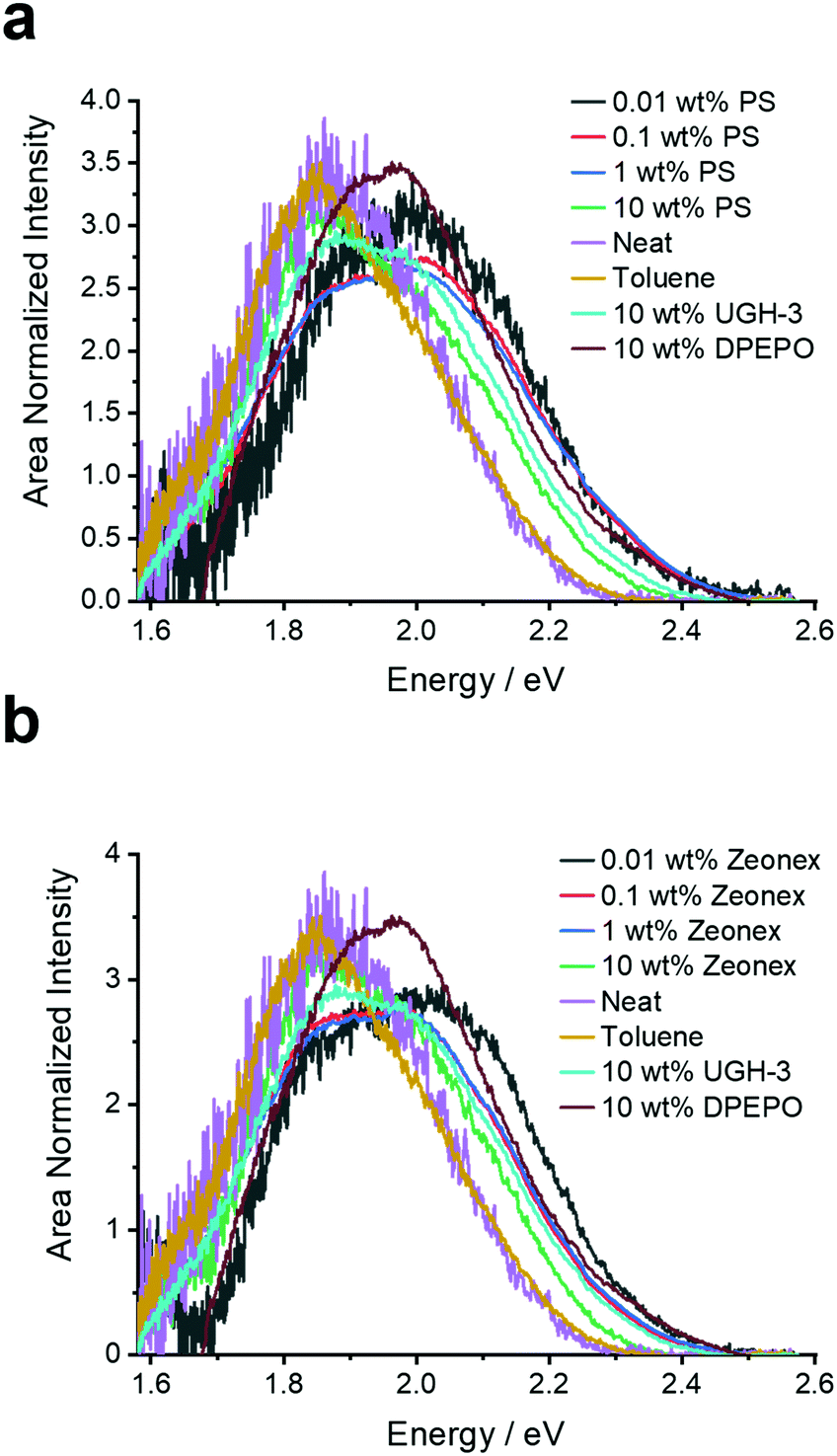 | ||
| Fig. 3 Prompt fluorescence spectra (0.8 ns after excitation) extracted from the time-resolved emission decays of 4DTPIPN in the various solid films and in toluene solution at room temperature. | ||
 | ||
| Fig. 4 Emission spectra extracted from the time-resolved emission decay of 4DTPIPN in a 1 wt% doped polystyrene film at room temperature (left) and 80 K (right). | ||
 | ||
| Fig. 5 Ground-excited state electron density differences for the first and second excited singlet and triplet states. Isosurface values of 0.0004 a.u. were used for all densities. The cyan regions indicate a decrease in charge density, whereas the purple regions indicate an increase in charge density. | ||
While the UV-VIS absorption spectra are very similar both in solution and in the solid state, showing none of the significant red-shifting with concentration observed for 4CzIPN, this does not rule out dimer formation as absorption spectroscopy is less sensitive for changes in aggregation than photoluminescence spectroscopy. The single-crystal X-ray structures were also consulted and did show evidence for close intermolecular S⋯S and C–H⋯N (with the nitrogen atoms of the DTP units and cyano functional groups) interactions (Fig. S6, Table S3, ESI†), linking the molecules into a 3-dimensional network. In contrast to 4CzIPN, there are no face-to-face overlaps between the donor units of different molecules. Instead, there are edge-to-face interactions as indicated in Fig. S6 (ESI†). The absence of face-to-face stacking due to ‘wedging’ between the D2/D4 and D3 donor units (Fig. S5, ESI†) is because the dihedral angles between the DTP and IPN units are smaller than 70°. A more detailed description of the various intermolecular interactions in the single crystal lattice can be found in the ESI.†
At this stage the situation for 4DTPIPN seems to be very different to that of 4CzIPN and strong experimental evidence for dimer species using the methods utilised in the original work15 proved elusive. Therefore, the excitation scan of 4DTPIPN at its emission peak of 2.05 eV (605 nm) was recorded at different concentrations in polystyrene (Fig. 2a), in which a clear increase in the absorption shoulder at 2.8 eV is observed and ultimately transitions into a completely new absorption band for the neat film. This new absorption band is attributed to a dimer/aggregated species of 4DTPIPN and provides unequivocal evidence of a dimer/aggregated species in the system.
The photoluminescence of 4DTPIPN (Fig. S8b, ESI†) shows clear solvatochromism going from methylcyclohexane (MCH) to 2-methyltetrahydrofuran (2-MeTHF), with the concomitant drop in emission intensity with increasing polarity. The solvatochromic behaviour of the emission is indicative of CT character. This was confirmed via time-dependent density functional theory (TD-DFT) calculations (Fig. 3, Table 1) using a modified LC-BLYP30 range-separated exchange–correlation functional with the 6-311G(d) basis set under the Tamm–Dancoff approximation31 and applying the polarizable continuum model (cyclohexane).32 The range-separating parameter ω was set to 0.17 bohr−1, in accordance with the findings of previous works.33,34
| Compound | S1 | S2 | T1 | T2 | ||||
|---|---|---|---|---|---|---|---|---|
| Nature | q CT | Nature | q CT | Nature | q CT | Nature | q CT | |
| a This transition additionally has 20% H → L and 22% H → L + 1 character as the other dominant natures. | ||||||||
| 4DTPIPN | H → L (97%) | 1.00 | H−1 → L (95%) | 1.00 | H → L (87%) | 1.00 | H−1 → L (77%) | 0.84 |
| 4CzIPN | H → L (93%) | 0.84 | H → L + 1 (61%) | 0.97 | H → L (91%) | 0.83 | H−4 → La (39%) | 0.65 |
Apart from the spatial separation of the molecular orbitals apparent in Fig. 1, the CT character was confirmed via calculation of the ground-excited state electron density differences following the work of Le Bahers et al. (see Fig. 3 and Table 1).35 Looking at the differences between the ground and excited state electron densities (Fig. 3), both 4DTPIPN and 4CzIPN show predominant CT character, as indicated by the localized nature of the diminishing (cyan) and increasing (purple) charge density regions on the donor and acceptor units, respectively. This is quantified as the amount of charge that is transferred (qCT) during the transition in Table 1. 4DTPIPN shows full CT character (qCT = 1.0) for its first and second singlet excited states and also for the first triplet excited state, whereas the second triplet excited state shows slightly less CT character. Overall, 4CzIPN has less CT character, indicated by the reduced qCT values (Table 1). This is likely due to the better overlap between the frontier orbitals, as illustrated in Fig. 1 and Fig. S7 (ESI†).
Having confirmed the nature of the emissive states, their (time-resolved) emission behaviour and concentration dependency in the solid state was investigated. In Fig. 2b, the steady-state emission spectra in the solid state are shown. A red-shift in the emission onset is seen for 4DTPIPN (1 wt% zeonex; 2.41 eV) with respect to the reported value for 4CzIPN (1 wt% zeonex; 2.78 eV),15 confirming the stronger electron donating nature of the DTP unit. This is in good correspondence with the values obtained from the TD-DFT calculations (Table 2). Furthermore, a gradual red-shift is visible upon increasing dopant concentration, with a relatively large shift from 1 to 10 wt% zeonex and finally to the neat film, which is effectively 100 wt% 4DTPIPN. This is another indication of possible dimer/aggregate formation. The blue-shift in DPEPO with respect to UGH-3 most likely arises due to packing effects as a result of the different dipole moments and polarizability of the host materials. DPEPO has a larger dipole moment than UGH-3 and can therefore mix better with 4DTPIPN, which will lead to fewer aggregates and a blue-shifted emission.
| Compound | ΔES1 (eV) | f S1 | ΔES2 (eV) | f S2 | ΔET1 (eV) | ΔET2 (eV) | ΔET2−T1 (eV) | ΔES1−T1 (eV) |
|---|---|---|---|---|---|---|---|---|
| 4DTPIPN | 2.63 | 0.001 | 2.76 | 0.009 | 2.57 | 2.63 | 0.06 | 0.06 |
| 4CzIPN | 2.84 | 0.095 | 3.11 | 0.156 | 2.68 | 2.82 | 0.14 | 0.16 |
To further probe the aggregation effects of 4DTPIPN, time-resolved fluorescence experiments were performed on the same series of films and a dilute solution (20 μM) in toluene (Fig. 4, 5 and Fig. S10–S20, ESI†).
In all doped films, the prompt fluorescence has an onset between 2.20 and 2.45 eV (Fig. 4). Notably, for polystyrene and zeonex, there is very little change in the emission onset from 0.01 to 1 wt% doping. Only when going to 10 wt% doping, the emission slightly red-shifts, without changes in the emission peak shape. The emission in 10 wt% UGH-3 is also slightly red-shifted with respect to the 0.01, 0.1 and 1 wt% emission in polystyrene, whereas the emission in the 10 wt% DPEPO film is not. This is identical to the findings from the steady-state emission spectra. The emission in the neat film is red-shifted with respect to the emission in solid doped films. With the neat film having the highest possible concentration of 4DTPIPN, one would expect any dimer emission to be visible here. The fact that the emission across all film concentrations and hosts seems to be extremely similar, draws us to the conclusion that the aggregated/dimer species is present at all concentrations. The small shifts in onset are likely arising from a distribution of dimer conformations, a similar situation was observed in 4CzIPN when comparing the crystals to amorphous films and demonstrated during the mechanochromic studies.15 This consistency in the emission spectra across all films and the presence of an aggregated species in dilute films of 0.01 wt% show that this molecule has significant propensity to form dimers.
The dilute toluene solution of 4DTPIPN gives a similar peak shape and position as in the neat film. However, this is coincidental and not indicative of the same physical processes but rather due to the significant difference in polarity effects of solvents versus the polarizability in the solid state. Separation of these effects allows us to assign the neat film (and other thin films) to an aggregated species and the toluene to a relaxed monomer CT state.
Further evidence of the dimer species in the thin films are the isoemissive points present in the RT spectra at certain delay times (vide infra). Isoemissive points are commensurate with the presence of two distinct species in a system.36 The higher energy emission that appears at longer delay times is assigned to monomer CT emission arising from molecules that are isolated within the polymer or small molecule matrix.
Since the compound behaves similarly in all host materials, the results for the 1 wt% polystyrene film are discussed here in detail, making reference to the isoemissive points and key onset energies that appear across all of the solid-state films (Fig. 5 and Fig. S10–S20, ESI†). Following the excitation (λexc = 355 nm), emission with an onset of around 2.38 eV (521 nm; Table 3) is observed at room temperature until several microseconds. This is assigned to the dimer/aggregate species and is a consistent presence across all films. Slight deviations in onset energy and peak emission arise from conformational effects and different proportions of monomer CT prompt emission, which may cause a small blue-shift.
| Sample | ΔESa (eV) | ΔETb (eV) | ΔESTc (eV) | PLQYaird | PLQYinertd |
|---|---|---|---|---|---|
| a Taken from the onset of the prompt fluorescence emission. b Taken from the onset of the phosphorescence emission at millisecond times at 80 K. c Calculated as ES–ET. d Photoluminescence quantum yields of the various samples measured under air and inert atmosphere. e Triplet energy and singlet–triplet energy gap could not be determined accurately due to low emission intensity at long times at 80 K. f PLQYs in UGH-3 and DPEPO films were not measured. | |||||
| 0.01 wt% PS | 2.40 | 2.34 | 0.06 | 4.9 | 2.2 |
| 0.1 wt% PS | 2.39 | 2.38 | 0.01 | 8.5 | 6.6 |
| 1 wt% PS | 2.38 | 2.38 | <0.01 | 7.6 | 9.8 |
| 10 wt% PS | 2.36 | 2.35 | 0.01 | 1.5 | 1.6 |
| 0.01 wt% zeonex | 2.40 | 2.36 | 0.04 | 5.3 | 5.9 |
| 0.1 wt% zeonex | 2.40 | 2.34 | 0.06 | 8.2 | 10.3 |
| 1 wt% zeonex | 2.41 | 2.35 | 0.06 | 3.5 | 4.5 |
| 10 wt% zeonex | 2.34 | 2.34 | <0.01 | 1.1 | 1.1 |
| 10 wt% UGH-3 | 2.28 | —e | —e | —f | —f |
| 10 wt% DPEPO | 2.42 | 2.43 | 0.01 | —f | —f |
| Neat | 2.24 | 2.13 | 0.11 | 0.6 | 0.5 |
After several microseconds, the emission blue-shifts and an isoemissive point becomes apparent (Fig. 5b). This blue-shifted delayed fluorescence is attributed to the isolated monomer CT and appears in different amounts that will be proportional to the number of isolated molecules. To uncover whether the delayed fluorescence is unimolecular or bimolecular, a laser energy measurement was conducted since TADF scales with the laser energy as a power 1, whereas TTA scales as a power 2.3 This is especially valid at very low laser powers where the triplet generation is not saturated by the laser energy. As can be seen in Fig. S21 (ESI†), the plot of the integrated emission intensity versus laser energy yields a linear correlation with a slope of 1.02 and an R2 value of 1, indicative of a unimolecular process for the delayed fluorescence of the dimer and monomer species.
At 80 K (Fig. 5d–f), the emission at 0.8 ns is very similar to that at room temperature at 0.8 ns. Most notably, the delayed fluorescence arising from the monomer CT species is no longer present as the thermally activated nature of the mechanism is switched off at low temperature. The slight blue-shift at significantly longer times is attributed to monomer phosphorescence. It is notable that the monomer species emission at RT and 80 K are no longer present in the neat film, an observation that is assigned to the increased quenching by the dimer species in the neat film (Fig. S19, ESI†).
In the previous report by Etherington et al.,154CzIPN showed a pronounced red-shift after several tens of nanoseconds and this shift is heavily influenced by the dopant concentration in zeonex. This is not the case for 4DTPIPN, as outlined above, and shows the enhanced propensity of 4DTPIPN to form dimers even at dilute concentrations of 0.01 wt%.
The photoluminescence quantum yields (PLQYs) of the various films were measured under air and inert atmosphere (Table 3) and are rather low. PLQY measurements often have significant error bars due to the process of the measurement and while there is variation with concentration across the samples this data merely confirms that across all films the PLQY is low and in the range of 1–10%. This is consistent with the emission being dominated by the dimer emission in the solid state and generally high non-radiative recombination across both solid state and solution. While the single-crystal structure showed the presence of edge-to-face interactions – as opposed to face-to-face interactions, which are generally considered to lead to larger non-radiative losses – there is photophysical evidence that face-to-face interactions must be present in this system in the host environments that we have considered. The presence of red-shifted emission and the emergence of a red-shifted absorption band in the excitation spectra with increased concentration cannot be attributed to edge-to-face interactions and must be due to π–π interactions. Furthermore, sulphur atoms are often correlated to having a higher SOC than second row elements such as carbon, nitrogen and oxygen. The presence of 8 sulphur atoms per molecule is expected to drastically increase the SOC and the non-radiative relaxation and therefore contributes, perhaps even to a larger extent, to the low PLQYs. Next to the increase in non-radiative losses, transitions such as ISC will also be enhanced by the induced SOC and this is expected to cause a high proportion of triplets. An estimate of the triplet state population was ascertained by measuring the singlet oxygen quantum yield (ΦΔ) in solution for 4DTPIPN and 4CzIPN (Fig. S22, ESI†). The singlet oxygen quantum yield of 4DTPIPN is 19%, confirming the tendency to form triplet excited states. However, the singlet oxygen quantum yield of 4CzIPN is 43%, surpassing that of 4DTPIPN. Important to note, however, are the PLQYs for 4CzIPN (94% in toluene under inert atmosphere)11 and 4DTPIPN (10% in 0.1 wt% zeonex). Since 4CzIPN is known as a good TADF emitter with a high PLQY, it is no surprise that the ISC is efficient and non-radiative losses are diminished with a high triplet formation as a result. 4DTPIPN has a much lower PLQY under inert atmosphere but manages to form a significant amount of triplet excited states. From these results we can conclude that 4DTPIPN very efficiently forms excited triplet states and is able to convert them back to the singlet state via rISC but suffers from larger non-radiative losses with respect to 4CzIPN. To verify this hypothesis, kinetic fitting of the time-resolved emission decays was performed for 4DTPIPN (and 4CzIPN for comparison) according to the procedure introduced by N. Haase et al. (Fig. S23 and S24 and Table S5, ESI†).37 It is apparent that 4DTPIPN has a slightly shorter lifetime for the prompt fluorescence (τPF), which is in line with having a slightly greater kF, and a significantly shorter delayed fluorescence lifetime (τDF), which is in line with having a much greater krISC (4×). The higher rISC can be attributed to a smaller ΔEST. Despite having overall higher rates for the fluorescence, ISC and rISC, 4DTPIPN still shows lower PLQYs and a kSnr that is an order of magnitude higher than 4CzIPN.
Unfortunately, the low PLQY of 4DTPIPN in doped films limits its application in OLEDs and no devices were constructed using this material. The dependence of the PLQY on the presence of oxygen and the inherent temperature dependence of the rISC process could be used to construct TADF-based oxygen and temperature sensors, as has been described in recent literature.38–41 The relatively high singlet oxygen quantum yield, red-shifted emission and material stability make 4DTPIPN an interesting material for (image-guided) photodynamic therapy applications.42 While many TADF emitters have relatively high triplet formation yields, they are seldomly applied as photosensitizers.42 Furthermore, the red-shifted and delayed emission can be used for time-resolved bio-imaging applications in which the delayed emission remains present after the cell autofluorescence has died out.43–45
Conclusions
We have shown the synthesis and photophysical characterisation of 4DTPIPN, using the 4H-dithieno[3,2-b:2′,3′-d]pyrrole (DTP) moiety as a novel donor unit to construct an emissive donor–acceptor type compound. 4DTPIPN afforded a red-shifted emission in comparison to the 9H-carbazole analogue 4CzIPN, which has been investigated extensively in literature prior to this study. Time-resolved emission spectroscopy showed the presence of a persistent aggregate/dimer species that undergoes delayed fluorescence. This aggregate was found to be more significant than that which was found for 4CzIPN and is present even in dilute film.15 Rather contrastingly, it has been more difficult to obtain evidence of the dimer/aggregate species through steady-state absorption measurements and analysis of the crystal structure and packing. However, clear evidence of the dimer/aggregate species has been obtained from excitation scans of the doped films showing significant red-shifts and the appearance of a low energy band in the neat film. Combined with the observation of an isoemissive point in the time-resolved fluorescence spectroscopy across almost all conditions, we conclude that the dimer/aggregate species in this compound is dominant and pervasive, so much so that the emission profiles of the compound are very similar across all film conditions.These results show that 4DTPIPN has the potential of achieving higher colour stability as a result of dimer/aggregate states. However, this colour stability comes at the cost of PLQY. Regardless, this investigation highlights the effects of dimer/aggregate contributions that may be present even at dilute doping concentrations and could play an important role in the discussion of solid-state solvation effects. Awareness of this aggregation will be important in the understanding of molecular interactions of small-molecule emitters that will be utilised in both doped and non-doped OLEDs.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the University of Namur and Hasselt University [PhD BILA scholarship T. Cardeynaels]. The authors also thank the Research Foundation–Flanders (FWO Vlaanderen) for financial support [project G087718N, G0D1521N, I006320N, GOH3816NAUHL and PhD scholarship S. Paredis]. The calculations were performed on the computers of the ‘Consortium des équipements de Calcul Intensif (CÉCI)’ (http://www.ceci-hpc.be), including those of the ‘UNamur Technological Platform of High-Performance Computing (PTCI)’ (http://www.ptci.unamur.be), for which we gratefully acknowledge the financial support from the FNRS-FRFC, the Walloon Region and the University of Namur [Conventions No. 2.5020.11, GEQ U.G006.15, U.G018.19, 1610468 and RW/GEQ2016]. M. K. Etherington and A. P. Monkman are supported by EU Horizon 2020 Grant Agreement No. 732013 (HyperOLED). K. Stavrou and A. P. Monkman acknowledge the TADFlife project funded by the European Union's Horizon 2020-MCSA-ITN Research and Innovation Programme under grant agreement no 812872.References
- C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- L. Bergmann, D. M. Zink, S. Bräse, T. Baumann and D. Volz, Top. Curr. Chem., 2016, 374, 22 CrossRef PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- B. T. Lim, S. Okajima, A. K. Chandra and E. C. Lim, Chem. Phys. Lett., 1981, 79, 22–27 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- H. S. Kim, S.-R. Park and M. C. Suh, J. Phys. Chem. C, 2017, 121, 13986–13997 CrossRef CAS.
- T. Northey, J. Stacey and T. J. Penfold, J. Mater. Chem. C, 2017, 5, 11001–11009 RSC.
- L. Yang, J. T. Horton, M. C. Payne, T. J. Penfold and D. J. Cole, J. Chem. Theory Comput., 2021, 17, 5021–5033 CrossRef CAS PubMed.
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 11109–11117 CrossRef CAS PubMed.
- E. Cho, M. Hong, V. Coropceanu and J.-L. Brédas, Adv. Opt. Mater., 2021, 2002135 CrossRef CAS.
- L. Salah, M. K. Etherington, A. Shuaib, A. Danos, A. A. Nazeer, B. Ghazal, A. Prlj, A. T. Turley, A. Mallick, P. R. McGonigal, B. F. E. Curchod, A. P. Monkman and S. Makhseed, J. Mater. Chem. C, 2021, 9, 189–198 RSC.
- P. Imbrasas, R. Lygaitis, P. Kleine, R. Scholz, C. Hänisch, S. Buchholtz, K. Ortstein, F. Talnack, S. C. B. Mannsfeld, S. Lenk and S. Reineke, Adv. Opt. Mater., 2021, 2002153 CrossRef CAS.
- Y. Geng, A. Tang, K. Tajima, Q. Zeng and E. Zhou, J. Mater. Chem. A, 2019, 7, 64–96 RSC.
- S. C. Rasmussen and S. J. Evenson, Prog. Polym. Sci., 2013, 38, 1773–1804 CrossRef CAS.
- G. Turkoglu, M. E. Cinar and T. Ozturk, Top. Curr. Chem., 2017, 375, 84 CrossRef PubMed.
- W. Vanormelingen, J. Kesters, P. Verstappen, J. Drijkoningen, J. Kudrjasova, S. Koudjina, V. Liégeois, B. Champagne, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, J. Mater. Chem. A, 2014, 2, 7535–7545 RSC.
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528–4543 RSC.
- H. Mo, K. R. Radke, K. Ogawa, C. L. Heth, B. T. Erpelding and S. C. Rasmussen, Phys. Chem. Chem. Phys., 2010, 12, 14585–14595 RSC.
- K. R. Radke, K. Ogawa and S. C. Rasmussen, Org. Lett., 2005, 7, 5253–5256 CrossRef CAS PubMed.
- S. Förtsch and P. Bäuerle, Polym. Chem., 2017, 8, 3586–3595 RSC.
- S. Förtsch, A. Vogt and P. Bäuerle, J. Phys. Org. Chem., 2017, 30, e3743 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Rev. A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- T. Cardeynaels, S. Paredis, J. Deckers, S. Brebels, D. Vanderzande, W. Maes and B. Champagne, Phys. Chem. Chem. Phys., 2020, 22, 16387–16399 RSC.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CrossRef CAS.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- A. S. R. Koti, M. M. G. Krishna and N. Periasamy, J. Phys. Chem. A, 2001, 105, 1767–1771 CrossRef CAS.
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, A. Mekic, W. Brütting and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 29173–29179 CrossRef CAS.
- S. Kochmann, C. Baleizão, M. N. Berberan-Santos and O. S. Wolfbeis, Anal. Chem., 2013, 85, 1300–1304 CrossRef CAS PubMed.
- N. R. Paisley, C. M. Tonge and Z. M. Hudson, Front. Chem., 2020, 8, 229 CrossRef CAS PubMed.
- D. Gudeika, O. Bezvikonnyi, D. Volyniuk, E. Skuodis, P.-H. Lee, C.-H. Chen, W.-C. Ding, J.-H. Lee, T.-L. Chiu and J. V. Grazulevicius, J. Mater. Chem. C, 2020, 8, 9632–9638 RSC.
- U. Tsiko, O. Bezvikonnyi, D. Volyniuk, B. F. Minaev, J. Keruckas, M. Cekaviciute, E. Jatautiene, V. Andruleviciene, A. Dabuliene and J. V. Grazulevicius, Dyes Pigm., 2022, 197, 109952 CrossRef CAS.
- A. M. Polgar and Z. M. Hudson, Chem. Commun., 2021, 57, 10675–10688 RSC.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- N. R. Paisley, S. V. Halldorson, M. V. Tran, R. Gupta, S. Kamal, W. R. Algar and Z. M. Hudson, Angew. Chem., Int. Ed., 2021, 60, 18630–18638 CrossRef CAS PubMed.
- C. I. C. Crucho, J. Avó, A. M. Diniz, S. N. Pinto, J. Barbosa, P. O. Smith, M. N. Berberan-Santos, L.-O. Pålsson and F. B. Dias, Front. Chem., 2020, 8, 404 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information on the synthesis procedures, 1H and 13C NMR spectra, thermo and photostability, time-resolved photoluminescence spectra, laser energy measurements, HOMO and LUMO topologies, single crystal X-ray analysis, singlet oxygen measurements and coordinates of the optimized geometries can be found in the electronic supplementary information. CCDC 2095357–2095358. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc04913e |
| This journal is © The Royal Society of Chemistry 2022 |