
Application of time-resolved electron paramagnetic resonance spectroscopy in the mechanistic study of thermally activated delayed fluorescence (TADF) materials
Xi
Chen
 ,
Xiao
Xiao
and
Jianzhang
Zhao
*
,
Xiao
Xiao
and
Jianzhang
Zhao
*
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, E-208 West Campus, 2 Ling Gong Road, Dalian 116024, P. R. China. E-mail: zhaojzh@dlut.edu.cn
First published on 14th December 2021
Abstract
Triplet exciton harvesting is crucial in organic light emitting diodes (OLEDs) because the triplet states produced by electron and hole recombination account for up to 75% of the total excitons, whereas the singlet states account for 25%. However, the triplet state of organic molecules is usually either non-emissive at room temperature or gives very long phosphorescence lifetimes, both of which are detrimental to OLEDs. Thermally activated delayed fluorescence (TADF) emitters are one answer to this challenge. Typical TADF emitters are based on an electron donor–acceptor structure motif, and the low-lying states include the charge transfer singlet (1CT) state and triplet (3CT) state, and a closely-lying localized triplet (3LE) state. Although many efficient TADF emitters have been developed for OLEDs, the underpinning photophysical processes of TADF, for instance, the charge separation, the forward intersystem crossing (ISC) and the reverse ISC (rISC), and the coupling of the excited states of these emitters, are far from clear. Herein, we introduce recent developments in the study of the photophysical processes of TADF emitters using the time-resolved electron paramagnetic resonance (TREPR) spectroscopy method. This spectral tool supplies unique information on the dynamics of the transient paramagnetic species involved in the TADF processes, for instance, the 3CT and the 3LE states, as well as the ISC mechanisms. Physical insights have been obtained with TREPR spectra on the TADF mechanism, such as simultaneous observation of the 3CT and 3LE states, vibrational and spin-vibronic coupling mediated ISC, and the electronic configuration and spatial delocalization of the 3CT and 3LE states’ wave functions. Factors beneficial to TADF obtained via theoretical computations are also briefly introduced.
1. Introduction
Organic light emitting diodes (OLEDs) have attracted much attention in recent years.1–7 One of the critical issues in the research of OLEDs is to develop efficient emitters to harvest both the singlet and the triplet excitons for efficient light emission. According to the electron spin statistics, the singlet and triplet states are formed at the rates of 25% and 75%, respectively, from the electron–hole recombination in the emitting layer of the OLED devices. For organic fluorescence emitters, the maximal internal quantum efficiency is 25% (IQE, without any other processes considered, such as triplet–triplet-annihilation, TTA). Therefore, phosphorescence emitters were developed as second generation emitters for OLEDs, for which the maximal IQE is 100%, because both the singlet and triplet excitons can be harvested for light emitting. In ordinary phosphorescent transition metal complexes, the ultrafast intersystem crossing (ISC) transforms the singlet state to the emissive (phosphorescent) triplet excited state.5,8–11 However, the phosphorescent transition metal complexes, for instance those contain Pt(II) or Ir(III) atoms,12–14 suffer from high cost, toxicity, etc. Therefore, it is desired to develop new emitting materials for OLEDs to address the above challenges, for instance, neat organic emitting materials without any precious metal atoms. This is a challenge in molecular photochemistry, because organic molecules usually do not exhibit phosphorescence at room temperature or they exhibit long phosphorescence long lifetimes.15Recently, Adachi et al. proposed to use organic compounds showing thermally activated delayed fluorescence (TADF) as light emitting materials for OLEDs.16,17 Different from the normal organic fluorescence emitters, both the singlet and the triplet excitons can be harvested in TADF emitters for light emission. In other words, for TADF molecules, both the singlet and the triplet states of the emitters are formed in the OLED devices; although the triplet excitons are usually non-emissive, it can be transformed to the emissive singlet states via reverse ISC (rISC), which is thermally activated. The small energy gap between the singlet and triplet states was believed to make the rISC efficient; thus the non-emissive triplet excited state is harvested for light emission and the luminescence lifetime is short (in the range of a few microseconds), which are beneficial for OLEDs. The maximal IQE for the TADF materials is up to 100%. Therefore, these novel materials have attracted much attention in recent years for the development of efficient OLEDs.3–7,18–25 Moreover, from the photochemistry point of view, TADF materials are also intriguing in the study of fundamental photophysics of charge transfer (CT) and ISC, as well as excited state dynamics.
Upon photoexcitation (note this is different from the scenario in operating OLED devices, in which the TADF materials are electronically excited i.e. by electron–hole recombination), charge separation, ISC, rISC, radiative and non-radiative decay of the singlet (normally a charge transfer singlet state, i.e.1CT) and triplet states are involved in the photophysical processes of TADF emitters. It is fundamentally important to unravel these entangled photophysical processes. However, till now no conclusive understanding of this kind of emissive material is achieved. For instance, there is still much room to unravel the excited states involved in the TADF process (is it two states or more), the molecular structural factors (rigid or flexible) that enhance the ISC in these heavy atom-free molecules, and to enhance the overall TADF efficiency.
The photophysical processes of the TADF emitters are complicated. In the early stage of research, only the lowest-lying S1 and T1 states were considered, and the studies focused on reducing the energy gap between the S1 and T1 states.17,26–29 However, now there is consensus in the community that an intermediate state should be involved in the TADF process, which is critical for ISC and rISC, such as the localized triplet excited state (3LE).6,30–32 Generally speaking, upon photoexcitation, either the localized singlet excited (1LE) state or the 1CT state is firstly populated, then ISC occurs, and the 3LE and the charge transfer triplet state (3CT) will be populated. rISC will populate the emissive 1CT state again.30,31,33,34 It is not a trivial task to discriminate these transient species and to determine their dynamics using time-resolved transient optical spectroscopy; for instance, the optical spectral feature of 1CT and 3CT states should be virtually the same. Note the electron exchange energy in this case is very small; considering the negligible spatial overlap of the molecular orbitals, the spin–spin interaction of the radical anion and cation is also much weaker as compared to the LE states; as a result, the energy gap between the two states is small, and the difference between the 1CT and 3CT states is mainly the electron spin angular momentum, not the electronic configuration.35–37 In other words, the UV-vis absorption of the 1CT and 3CT states will be virtually the same. Moreover, the mechanism of the formation of the 3CT state is unclear; it should either be direct 1CT → 3CT or an intermediate 3LE state should be involved. The 3LE state of the TADF molecules may be different from the triplet state of the components of the molecule (electron donor and acceptor), because the TADF molecules are usually compact electron donor–acceptor polyads, i.e. the electron donor and acceptor are connected with a short linker, and delocalization of the wave function of the triplet state may occur.31 Direct spin orbit coupling (SOC), vibrational coupling or spin-vibronic coupling may all enhance the ISC of the TADF emitters. Transient optical spectroscopy can hardly provide direct experimental evidence to discriminate these species or the photophysical processes.
In this perspective article, we summarized the recent development in the mechanism studies on the TADF materials, we focus on the study of small organic molecules based on an electron donor–acceptor motif, by using time-resolved electron spin paramagnetic resonance (TREPR) spectroscopy. TREPR spectra may provide information on the transient paramagnetic species involved in the TADF process, such as the 3CT state, or the radical pair, and the precursor of the 3LE states, the spatial localization of the triplet state wave function, and the ISC mechanisms.37–44 Some methods based on magnetic field effect have been developed for studying organic semiconductors including magneto-electroluminescence (MEL), optically detected magnetic resonance (ODMR) and magneto-conductance (MC). However, these methods are mainly based on the change in the electroluminescence intensity or electrical conductance of the device with the magnetic field changes. They are not appropriate for organic compounds showing TADF; among the other aspects, the 3CT and 3LE states of the TADF materials are non-emissive. On the other hand, TREPR spectroscopy is a technique for the characterization of the paramagnetic transient species of TADF materials upon photoexcitation. For instance, the electron spin selectivity of ISC, the zero field splitting (ZFS) D and E parameters (the electron spin distribution) of the triplet state, and the origin of the paramagnetic species can be well characterized with the TREPR spectra. Moreover, it is difficult, if not impossible, to discriminate 1CT and 3CT states by the transient optical spectroscopic methods, such as the femtosecond (fs) or nanosecond (ns) transient absorption (TA) spectroscopy, due to the weak spin–spin interaction of the radical anion and cations, as the electronic transition is literally the same for 1CT and 3CT states. Moreover, the delocalization feature of the 3CT and 3LE states in the TADF process can be characterized by TREPR spectra, but not the transient optical spectral methods.
For TREPR spectroscopy, the paramagnetic transient species are selectively detected, such as the spin polarized radicals formed in the reactions, or the spin polarized triplet excited states. The 3CT state will give a TREPR signal, whereas the 1CT is TREPR spectral silent. The magnitude of the spin–spin interaction of the radical anion and cation of the ion pairs, and the spatial confinement of the triplet state wave function, can be directly quantified with the ZFS parameters (D and E values) of the triplet states. The sign of D depends on the shape of the spin density distribution of the triplet state. D is positive (D > 0) when the spin density distribution is in the oblate shape and D is negative (D < 0) when the electron spin distribution is in the prolate shape. Hyperfine coupling parameter determined using the Electron–Nuclear DOuble Resonance (ENDOR) technique allows precise determination of the extent of delocalization of the triplet state.45,46 Moreover, the electron spin dynamics can be studied with the TREPR spectra, for instance, the precursor of the 3LE state can be identified or in other words, the ISC mechanism involved in the TADF emitters can be discriminated by the TREPR spectra.47,48 The ordinary SOC enhanced ISC, the hyperfine interaction (HFI) enhanced ISC, vibrational enhanced ISC or the ISC facilitated by the spin-vibronic coupling may be all involved in TADF. These different ISC mechanisms can be readily discriminated by the specific electron spin polarization (ESP) phase pattern of the triplet TREPR spectra of the TADF molecules upon photoexcitation (in some cases the analysis should be aided by theoretical computation).40 TREPR spectroscopy has been intensively used in study of the transient, spin-polarized free radicals and the spin correlated radical pairs (SCRP), the 3CT states and the 3LE states.36,39–41,49,50 Recently, TREPR spectroscopy has also been used in study of TADF molecules; unique insights on the photophysical processes have been obtained. Recent development in the application of TREPR spectra in study of the photophysical processes of TADF emitters is summarized in this article.
2. TREPR spectroscopy: detection of the transient paramagnetic species and the ISC mechanisms involved in TADF
2.1 Observation of localized triplet states: different ISC mechanisms
In 2015, Ikoma et al. studied a few representative TADF emitters using TREPR spectroscopy (Fig. 1).51 The molecular structures of the compounds are with carbazole or phenoxazine as the electron donor, and cyanobenzene or triazine as the electron acceptor. Note the π-conjugation planes of the donor and acceptor units adopt orthogonal geometry as a result of the conformation restriction of the bulky moieties and the congested microenvironments of the moieties.51 Experiments show that the external quantum efficiencies (EQEs) of these TADF emitters in OLED devices are 19.3%, 12.5%, 6.0% and 5.3% for 4Cz-IPN, PXZ-TRZ, Cz-T and PIC-TRZ, respectively. | ||
| Fig. 1 Molecular structures of the typical TADF emitters. | ||
The TREPR spectra of the randomly oriented molecules of these TADF emitters in frozen solution (solvent: toluene) at 77 K were recorded (Fig. 2). For the compounds showing high EQE, i.e.4Cz-IPN and PXZ-TRZ, the ESP at the six canonical orientation are (a, e, e, a, a, e) and (a, a, e, a, e, e), respectively. For the compounds showing lower EQE, i.e.Cz-T and PIC-TRZ, the ESP of (e, e, e, a, a, a) was observed for the triplet TREPR spectra. Moreover, the TREPR spectra of Cz-T and PIC-TRZ have a larger width than the those of 4Cz-IPN and PXZ-TRZ. For Cz-T and PIC-TRZ, a single component based on the normal SOC-induced ISC is sufficient to simulate the experimental spectra. For the compounds showing a high EQE, triplet states formed by two ISC mechanisms, i.e. the SOC ISC and the HFI enhanced ISC, are required to attain a satisfactory simulation. These two different ISC mechanisms are illustrated in Fig. 3. Moreover, simulation shows that the ZFS D parameters of 4Cz-IPN and PXZ-TRZ (46 mT and 49 mT) are much smaller than the triplet state of the native cyanobenzene (|D| = 147 mT) or the triplet state of triphenyl-s-triazine (|D| = 133 mT). The decreased ZFS D parameter is an indication that the transient species is with significant delocalization or CT character, although the authors proposed that the transient species is the delocalized triplet state (the argument is the non-vanishing ZFS E parameter). The population ratios of the triplet state formed by HFI and the SOC ISC mechanisms, respectively, are 0.50 and 0.36 for 4Cz-IPN and PXZ-TRZ, respectively. Recently Brédas and Adachi et al. studied 4Cz-IPN using experimental and theoretical methods, and they proposed that an upper triplet state localized on partial molecular structure is involved in the TADF process (supported by transient absorption spectral study, and the uphill feature of ISC), and the energy of these states localized on partial molecular structure is lower than that of the carbazole triplet state (ca. 3.08 eV).34 They also found that the low-lying triplet state of 4Cz-IPN has a significant CT character, the triplet state is neither localized on the carbazole nor the cyanobenzene moieties, but delocalized on the carbazole and cyanobenzene parts.34 A TREPR spectral study on the reference compounds (partial structure of 4Cz-IPN) will be useful to fully unravel the photophysical process.
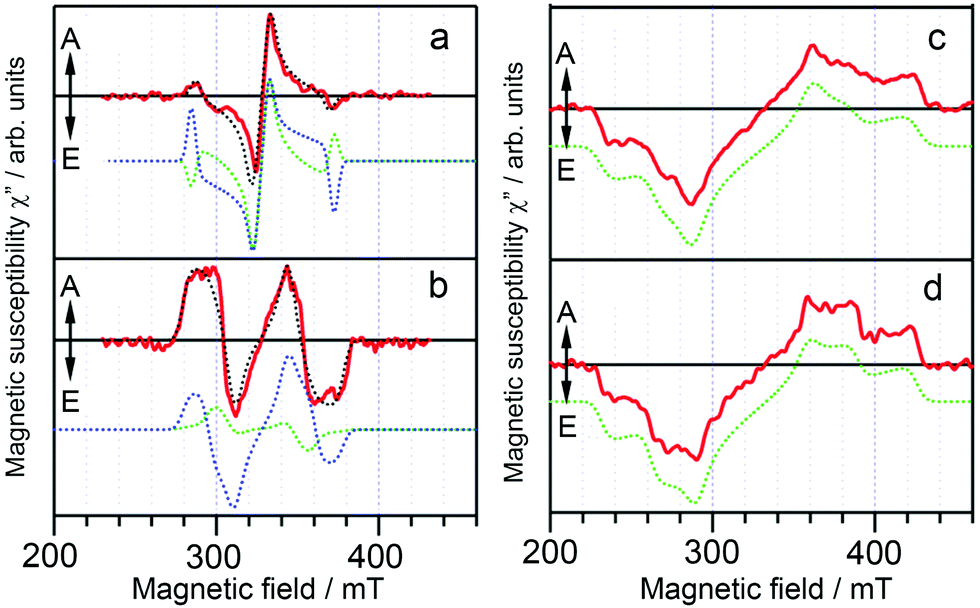 | ||
| Fig. 2 Transient EPR spectra for (a) 4Cz-IPN, (b) PXZ-TRZ, (c) Cz-T, and (d) PIC-TRZ in toluene detected at 550 ns after a laser flash at 77 K (red solid lines). The dotted lines are the simulation spectra: green, SOC-induced polarization; blue, hyperfine coupling (HFC)-induced polarization; and black, SOC plus HFC-induced polarization. Reproduced with permission.51 Copyright 2015, American Chemical Society. | ||
 | ||
| Fig. 3 Spin polarization mechanisms for the T1 state due to ISC from the S1 state under an external magnetic field along the z axis of the molecule. (a) The SOC-induced ISC. (b) HFC-induced ISC. Reproduced with permission.51 Copyright 2015, American Chemical Society. | ||
For Cz-T and PIC-TRZ, a SOC ISC induced triplet state was observed, with the TX and TY sublevels overpopulated. The ZFS D parameter of the triplet state of these compounds is 97 mT, very close to the triplet state of triphenyl-s-trazine (|D| = 133 mT). Thus it was proposed that the triplet state of Cz-T and PIC-TRZ is spatially confined. Due to the clear trend correlation between the EQE value and the triplet state TREPR features, the authors proposed that efficient TADF molecules should undergo radical pair ISC (RP-ISC).51 The difference between the SOC-induced ISC and the HFI induced ISC is illustrated in Fig. 3. For the SOC ISC mechanism, the in plane TX and TY sublevels are mainly populated, then the transfer of population rates to the T+ and T− sublevels under external magnetic fields occurs, and the (e, e, e, a, a, a) ESP phase pattern is observed for the triplet state. For the HFI induced ISC mechanism (i.e. the RP-ISC mechanism), mainly the T0 sublevel is populated, because of the smaller energy gap between this sublevel and the S1 state (in CT feature, Fig. 3). Under this circumstance, a special (a, e, e, a, a, e) or (e, a, a, e, e, a) ESP phase pattern will be observed for the triplet state. This kind of unique ESP phase pattern is inaccessible for the triplet state formed via the normal SOC ISC mechanism.
Considering the high T1 state energy of the carbazole unit (ca. 3.06 eV),52 and the CT state energy (ca. 2.69 eV, approximated by the crossing point of the normalized UV-vis absorption and the CT emission spectra),51 it is likely that the CT state, or 3LE state with significant delocalization, should be observed in the TREPR spectra. This is supported by the unusually small ZFS D parameter observed for 4Cz-IPN and PXZ-TRZ. More evidence was presented by femtosecond and nanosecond transient absorption (fs-TA and ns-TA),53 which show that a delocalized CT state and 3LE state were observed for 4Cz-IPN (the charge resonance effect), i.e. the positive charge is delocalized in the carbazole moieties, indicated by the near IR absorption band in the transient optical absorption spectra. However, it should be noted that the TREPR spectra of the triplet state is usually measured in a frozen solution at cryogenic temperature (e.g. 80 K or even lower), to suppress the tumbling of the molecules and spin lattice relaxation (SLR); the solvation effect in this case is missing, which makes the CT energy higher than that in fluid solution at room temperature.
The molecular structure features of the compounds presented in Fig. 1 deserve more analysis. For 4Cz-IPN, the carbazole moieties are in a sterically congested microenvironment; this will make the donor and the acceptor take an orthogonal (perpendicular) geometry. A study on analogue molecules shows that the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are spatially separated.17,20,54 In this case, the 1CT → 3LE may be enhanced by spin orbit charge transfer ISC (SOCT-ISC).37,55–58 With the purpose of design of heavy atom-free triplet photosensitizers based on the electron donor–acceptor molecular structure, the orthogonal geometry is beneficial for the SOCT-ISC.37,56,57,59 This is also applicable to TADF emitters.31 Moreover, the electron exchange integral for the electrons in the frontier molecular orbitals (MOs) of the CT state is small, which results in an extremely small 1CT/3CT energy gap. This small energy gap is beneficial for the HFI induced ISC (RP-ISC); however, the slow kinetics of HFI concerning ISC, and the weak coupling of the two states (1CT and 3CT) with same electronic configuration but different spin multiplicity (which is against El Sayed's rule for ISC), makes 1CT → 3CT an unlikely channel to populate the triplet state significantly,31,48i.e. the population of the 3CT state is probably mediated by the 3LE state. For PXZ-TRZ, the phenothiazine and the triazine units are separated by an intervening phenyl linker, which is also beneficial for attaining small J values. For Cz-T and PIC-TRZ, the steric hindrance is not significant; the molecules may adopt a more planar geometry, which is detrimental to the RP-ISC and the SOCT-ISC.
In the early stage of the study of TADF emitters based on the electron donor–acceptor molecular structure motif, the research was focused on reducing the energy gap between the lowest-lying S1 and T1 states, and increasing the molecular rigidity; it was believed both are beneficial to TADF. However, recent experimental and theoretical studies show that this is not true, and an intermediated dark state, i.e.3LE state, should be involved in the TADF process.30,31,33,60 For instance, it was found that electron donor–acceptor TADF emitters with similar S1/T1 state energy gaps may show significant variation in the rISC rate constants, and the rigid molecular structure (or hindered rotation) leads to suppression of rISC and thus TADF, causing the phosphorescence to turn on.61 With vibronic coupling between the 3CT and 3LE states considered, much faster ISC and population of 3LE states were observed. On the other hand, the HFI effect on the ISC rate constants is negligible (in the ns range).30 Calculations also show that the smaller energy gap between the S1 and T1 states is beneficial for ISC to produce the 3LE state. Computation on the rISC process shows that the vibronic coupling between the 3LE and 3CT states plays a significant role in the rISC kinetics. These results indicates that not only the energy gap of the S1/T1 states is important, but also the energy gap and the spin-vibronic coupling between the 3CT and 3LE states. For the latter case, restricting the molecular conformation does not necessarily enhance TADF as previously thought, it may inhibit rISC thus reducing the TADF performance.30
Monkman et al. confirmed that the energy gap between the 3LE state and the CT state of the donor–acceptor–donor triad imposes a significant effect on the rISC kinetics and thus the TADF performance, using photo-induced absorption spectra with varying host rigidities and polarities (the CT state energy level or the intramolecular motion).33 This model states that the coupling between the 1CT and 3CT states is mediated by non-adiabatic coupling (the non-Born–Oppenheimer effect; second order perturbation) between the 3LE and 3CT states, which is spin-vibronic coupling for the latter; both the 1CT → 3CT ISC and the rISC of 3CT → 1CT are greatly enhanced. However, it should be noted that these principles are probably not applicable for compounds showing TADF, but have a single chromophore molecular structure profile, i.e. not based on the electron donor–acceptor structure profile.16,62
In 2018, Greenham et al. reported experimental evidence for vibrationally assisted ISC in TADF emitters with TREPR spectra.63 They studied the TREPR spectra of two benchmark TADF emitters 4Cz-IPN and 2Cz-PN (Fig. 4), and made a different interpretation of the TREPR spectra and the photophysics of 4Cz-IPN. The key finding is that the SOC-mediated ISC is triggered by thermally populated torsion vibrational modes of the emitters, i.e. the vibronic coupling plays a significant role in TADF, and no HFI effect is involved in the ISC of the TADF emitters of 2Cz-PN and 4Cz-IPN (Fig. 5). The authors found that the SOC matrix elements’ (SOCMEs) magnitudes of the ISC from S1 to the TX, TY and TZ sublevels of the T1 state do not match with the experimentally observed population rates of the three sublevels. Thus factors other than the pure electronic model play a role in enhancing the ISC. Moreover, satisfactory simulation of the TREPR spectra of both 2Cz-PN and 4Cz-IPN can be achieved without invoking the HFI effect, while vibronic coupling plays a signal role in ISC. If only the direct SOC is considered, according to the calculated SOC matrix element magnitude, the population of T1,a and T1,b should be negligible, and the population of T1,c should be close to 1. However, this prediction does not agree with the experimental observations that show the three sublevels are almost homogenously populated. Instead, considering the geometry fluctuation, caused by the rotation freedom of the carbazole moieties driven by thermal energy at 300 K, the SOCMEs are increased, and ISC is enhanced. Computations also show that the dynamic SOCMEs of 4Cz-IPN are almost homogeneous for ISC to the three sublevels of the T1 state; experimentally the population rates of the three sublevels are Pa = 0.35, Pb = 0.18, and Pc = 0.47, respectively. Similar results were observed for 2Cz-PN.63 Thus, the authors proposed that ISC is enhanced by vibration; mainly the rotation about the linker. Thus, increasing the molecular rigidity of TADF emitters may be counterproductive to their emission properties. It should be pointed out that in this model, only two states were considered (S1 and T1).
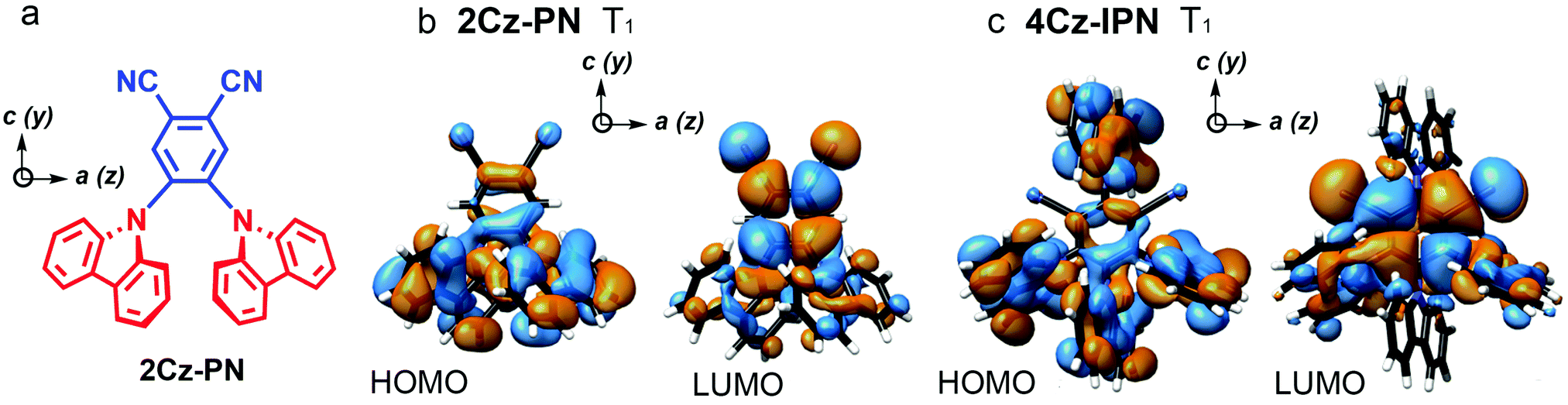 | ||
| Fig. 4 (a) Molecular structures of 2Cz-PN. UKS-DFT-calculated canonical orbitals for the T1 states of (b) 2Cz-PN and (c) 4Cz-IPN, using the PBE0 functional and EPR-II basis set. a, b, and c and x, y, and z denote the symmetry and ZFS axes, respectively. Reproduced with permission.63 Copyright 2018, American Chemical Society. | ||
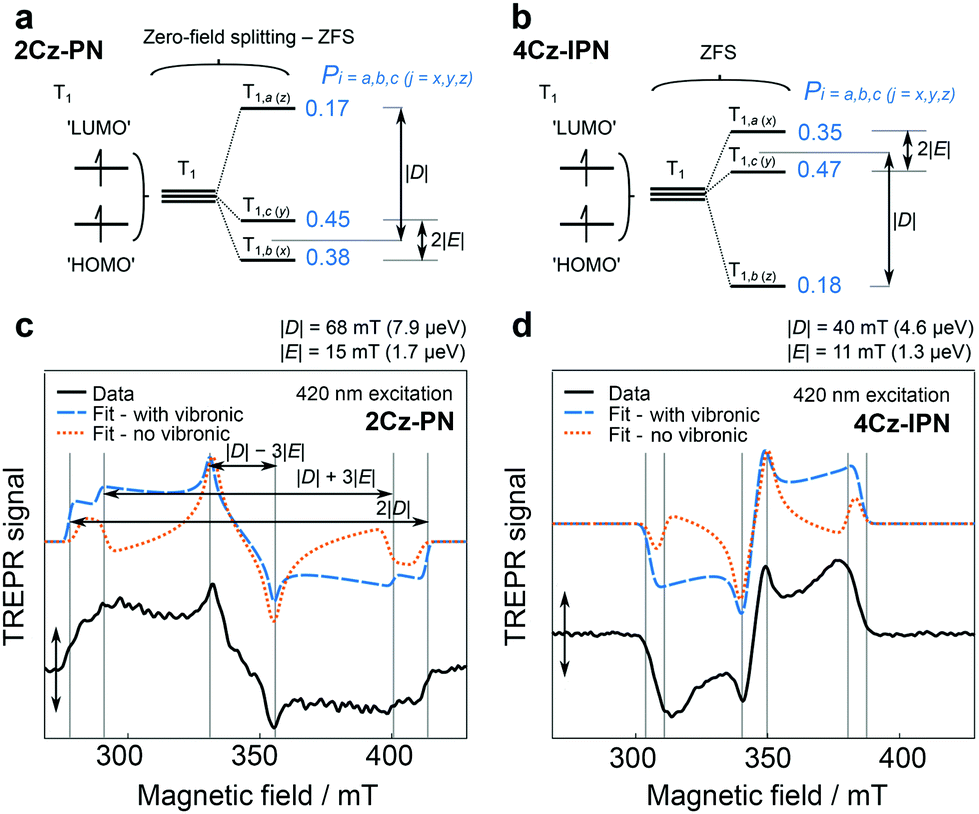 | ||
| Fig. 5 ZFS diagram for (a) prolate 2Cz-PN and (b) oblate 4Cz-IPN, indicating fitted |D| and |E| parameters. TREPR spectra (80 K) for toluene solutions of (c) 2Cz-PN (420 nm excitation) and (d) 4Cz-IPN (460 nm excitation), shown as black solid lines. Spectra were recorded 0.4 μs after laser excitation (0.04 μs averaged). Vertical lines overlaid on the spectra denote canonical orientations of ZFS principal axes with respect to the magnetic field, and can be used to derive |D| and |E| as shown in part c. Fits “with vibronic” and for “no vibronic” contributions to ISC are presented as blue dashed lines and yellow dotted lines, respectively. Reproduced with permission.63 Copyright 2018, American Chemical Society. | ||
2.2 Simultaneous observation of 3LE and 3CT states with TREPR spectra
TADF properties are not restricted to any specific molecular structural motif.3–6,59,64 Any compounds with closely-lying 1CT, 3CT and 3LE states may exhibit TADF properties (or more generally, close-lying S1/T1 states).65,66 Recently, we prepared a compact, orthogonal electron donor–acceptor dyad, with naphthalimide (NI) as the electron acceptor and phenothiazine (PTZ) as the electron donor (Fig. 6).67 The connection (linker or bridge) between the NI and the PTZ moieties is different in the two dyads. In NI-N-PTZ, the connection is at the N-position of the PTZ moiety, thus due to steric hindrance from the H atom at the 5-position of the NI moiety (peri-position), the π-planes of the two moieties take orthogonal geometry, and the electronic coupling of the donor and acceptor is small. For NI-C-PTZ, however, the connection is at the 3-position of the PTZ moiety; smaller steric hindrance is expected, resulting in stronger electronic coupling between the donor and the acceptor. This postulate is confirmed by the observation of a weak CT absorption band (ε = 580 M−1 cm−1 at 450 nm) for NI-N-PTZ, while a much stronger CT absorption band was observed for NI-C-PTZ (ε = 9000 M−1 cm−1 at 405 nm).67 Moreover, a larger 1CT/3CT energy gap is anticipated for NI-C-PTZ, due to the strong electronic coupling between the donor and acceptor in this dyad. CT emission bands were observed for both dyads; the 1CT state energies of the two dyads are approximated as 2.25 eV and 2.75 eV, respectively, by the on-set of the CT emission band (in hexane).67 The ISC and the rISC in NI-N-PTZ may be enhanced by the orthogonal orientation of the PTZ and the NI moieties, i.e. the SOCT-ISC mechanism.37,56–58 Since the T1 state of the native NI moiety is ca. 2.29 eV,68 we expect TADF only for NI-N-PTZ. Indeed TADF was observed only for NI-N-PTZ (in hexane at room temperature), and the luminescence decay trace shows a distinct biexponential feature, 22.6 ns (96.6%)/2.6 μs (3.4%). Normal prompt fluorescence was observed for NI-C-PTZ, with a fluorescence lifetime of 3.4 ns. This is one solid evidence that given the CT state energy is much higher than the 3LE state, no TADF will be resulted, even if 1CT and 3CT states are separated by a small energy gap. A ns-TA spectral study shows that a CT state was observed for NI-N-PTZ (Fig. 7a; in hexane).67 The strong positive absorption band in the ns-TA spectrum centered at 420 nm is assigned to the NI radical anion (NI−˙), whereas the PTZ radical cation (PTZ+˙) shows an absorption band centered at 519 nm. The lifetime of the CT state was determined as 2.6 μs, which is in good agreement with the delayed luminescence lifetimes of the dyad. It is significant to observe such a long-lived CT state in a compact electron donor–acceptor dyad. This result indicates that the CT state is lower in energy than the 3NI state, otherwise the 3NI state should be observed in the ns-TA spectra. However, the energy of the 3NI state should be very close to the CT state for the TADF emitters, and previously it was demonstrated that in some cases, only one of the two triplet states involved in triplet state equilibrium was observed with ns-TA spectra.69 For NI-C-PTZ, however, the 3NI state was observed in the ns-TA spectrum, and the triplet state lifetime was determined as 146 μs.67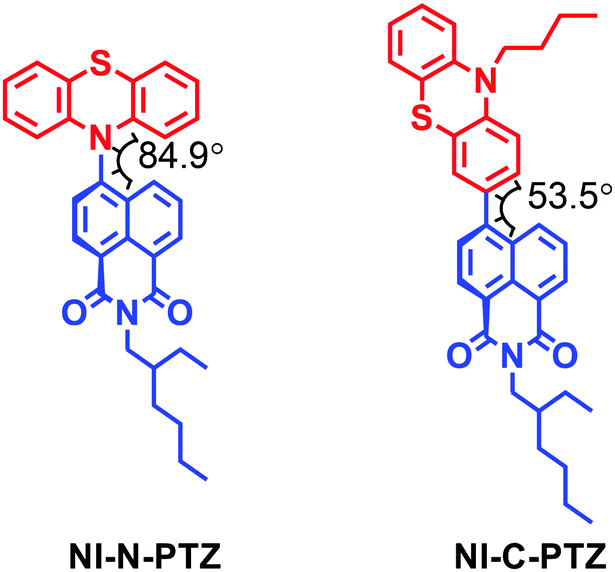 | ||
| Fig. 6 Molecular structure of NI-N-PTZ, which shows TADF properties. The molecular structure of the reference compound NI-C-PTZ is also presented. Reproduced with permission.67 Copyright 2019, American Chemical Society. | ||
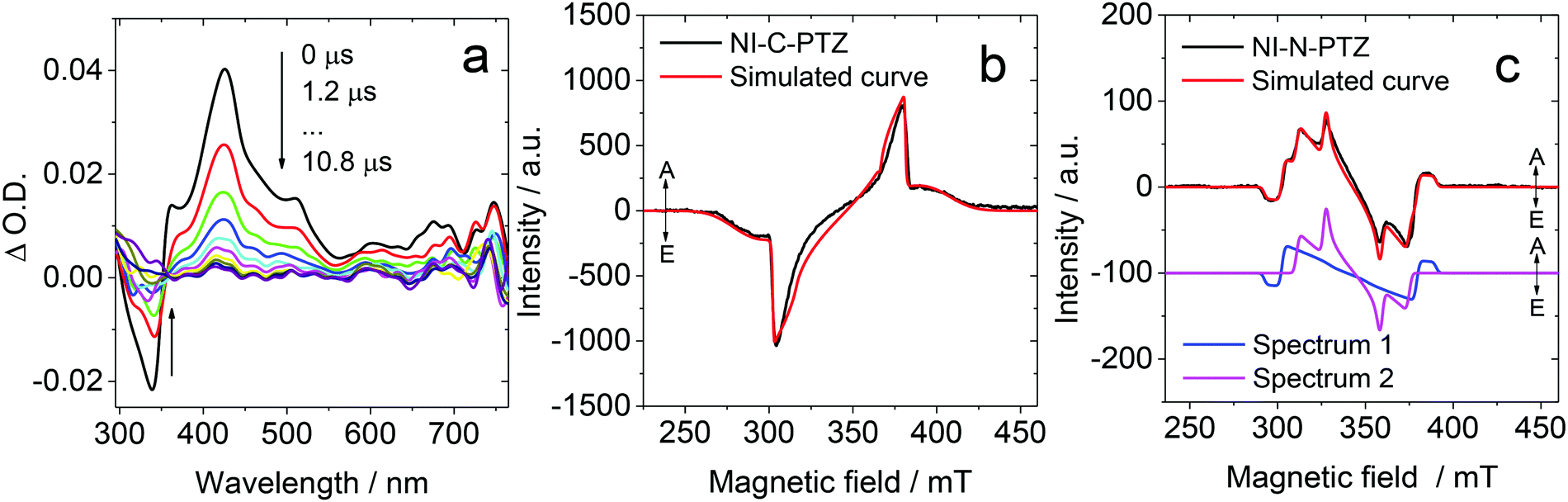 | ||
| Fig. 7 (a) Nanosecond transient absorption spectra of NI-N-PTZ excited at 330 nm. c = 1.0 × 10−5 M in deaerated hexane, 293 K. TREPR spectra of (b) NI-C-PTZ and (c) NI-N-PTZ determined using a X-band EPR spectrometer, in a frozen solution at 85 K. The delay time is 0.6–0.7 μs following a 355 nm laser pulse, c = 1.0 × 10−3 M in a mixed solvent hexane/toluene (1/2, v/v). The red lines are computer simulations of the triplet state spectra with parameters listed in Table 1 for NI-Br and NI-C-PTZ. Reproduced with permission.67 Copyright 2019, American Chemical Society. | ||
The triplet TREPR spectrum of NI-C-PTZ shows an ESP phase pattern of (e, a, e, a, e, a) (Fig. 7b), which is similar to that observed for 4-bromoNI (NI-Br, Table 1).67 Simulation of the TREPR spectrum of NI-C-PTZ gives ZFS D and E values as 64 mT and 5 mT, respectively (Table 1). Interestingly, the D value is smaller than that of the triplet state of NI-Br (92 mT), although the E parameters are similar for NI-C-PTZ (5 mT) and NI-Br (5 mT; Table 1). This result indicates that the triplet state of NI-C-PTZ is delocalized, and the spin density surface (obtained with theoretical computation) of the T1 state supports this postulate. For NI-N-PTZ, however, satisfactory simulation of the spectrum requires two different triplet states, one is with ZFS |D| and E parameters of 49 mT and 9 mT, respectively, which is assigned to the delocalized 3NI state. Another state, however, is with a much smaller |D| value of 32 mT, and the E parameter is 0 mT. This is a strong evidence that a 3CT state is observed. This is also in agreement with the ns-TA spectral observation (Fig. 7a). The compact dyad molecular structure of NI-N-PTZ, i.e. the strong spin–spin interaction between the radical anion and the cation, makes the formation of 1CT and 3CT states possible, otherwise only SCRP can be observed (in this case the electron spin–spin interaction is very weak).70–72
| Compound | |D| (mT) | E (mT) | P x | P y | P z |
|---|---|---|---|---|---|
| a Component 2 (spectrum 2 in Fig. 7c) of the TREPR spectrum of NI-N-PTZ, recorded with a delay time of 0.6–0.7 μs after laser excitation. b Component 1 (spectrum 1 in Fig. 7c) of the TREPR spectrum of NI-N-PTZ, recorded with a delay time of 0.6–0.7 μs after laser excitation. c The population rate due to the SO-ISC or SOCT-ISC mechanism contribution; main contribution from RP-ISC is not shown. | |||||
| NI-Br | 92 | 5 | 0 | 0.67 | 0.33 |
| NI-C-PTZ | 64 | 5 | 0 | 0.3 | 0.7 |
| NI-N-PTZ | 32a | 0a | 1.0 | 0 | 0 |
| 49b | 9b | 0c | 1c | 0c | |
Moreover, we observed an evolution of the ESP phase pattern for NI-N-PTZ, the ESP is switched at a longer delay time for the 3CT state. This phenomenon is probably due to the interconversion of the 3CT and 3LE states, and rotation of the ZFS principal axes of the 3CT ↔ 3LE internal conversion, and the selective depopulation of the sublevels of the 3CT via TADF emission.73 This phenomenon needs more investigation. For NI-C-PTZ, the TREPR signal decays at a longer delay time, without any ESP change. This is an interesting experimental evidence that the 3CT and 3LE states are both involved in the TADF process. Although the 1CT state is silent in TREPR spectral measurement, it is clear that 1CT state is involved in TADF for obvious reason: it is the emissive state in TADF process of the electron donor–acceptor emitters. Determination of the 1CT/3CT states energy gap precisely is not a trivial task. However, since the MOs are separated spatially for 1CT/3CT states, we assume the energy gap between the two is small.74 Observation of the 3CT and 3LE states simultaneously is a strong evidence that the TADF is not only dependent on the 1CT/3CT state gap, i.e. rISC is strongly dependent on another intermediate states.53 Given that the CT/3LE state energy is large, as for NI-C-PTZ, no TADF will be observed. Moreover, these results show that the 3LE state involved in the TADF process may have a significant CT character, or delocalization.63
Besides the TADF approach, another method is to use the TTA to break the limitation of the 25% IQE for the fluorescence emitters in OLEDs. Recently, the exploitation of hot exciton was proposed, i.e. the use of Tn → S1 ISC in OLED devices to increase the quantum efficiency.7 This mechanism usually involves the lowest singlet excited state S1 and a close-to-resonant triplet excited state (Tn), both of which display a hybrid charge transfer-locally excited (HLCT) character to ensure the best compromise between high singlet radiative decay rates, small energy separation and fast triplet-to-singlet conversion.7 However, the Tn state is difficult to be detected due to its short lifetime.
Oliver, Samuel and Zysman–Colman studied two dyads showing TADF based on the approach of exploitation of hot exciton (Fig. 8).75 Both compounds show luminescence decay traces with biexponential feature, for instance, DPA-AnCN shows a luminescence lifetime of 26.3 ns (98%)/858 ns (2%) in hexane. Since the T1 state (1.76 eV) is significantly lower than the T2 state (2.52 eV) and the emissive S1 state (2.50 eV), it is obvious that the normal TADF is not responsible for the delayed fluorescence of the compound.
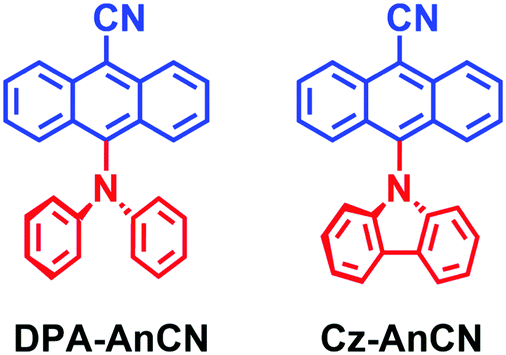 | ||
| Fig. 8 Anthracene based donor–acceptor derivatives showing delayed fluorescence based on exploitation of hot exciton. | ||
The TREPR spectra of the two compounds in a frozen solution in acetonitrile at 80 K were recorded (Fig. 9). For DPA-AnCN, two triplet states were required for satisfactory simulation of the experimental TREPR spectra, the first triplet state has a ZFS D value of 61 mT, and the second triplet state shows a narrower TREPR spectrum; a ZFS D parameter of 25 mT was obtained by simulation. The first one is attributed to the T1 state localized on the anthryl moiety, i.e. it is the 3LE state. Note the ZFD D parameter is very close to the value of the triplet state of the native anthracene (ca. 77–78 mT).48,76 The second narrower spectrum is attributed to the T2 state, which is with a significant CT character. This is supported by a much smaller D parameter (25 mT) and the vanished ZFS E parameter for the T2 state (E = 0). This postulate is also supported by the molecular orbital analysis; for T1 state, the overlap index of the HOMO and LUMO molecular orbitals is 0.8, whereas for T2, the overlap index decreases to 0.58; thus T2 state has a more significant CT character. For Cz-AnCN, a 3LE state was observed in the TREPR spectrum, which has a ZFS D value of 69 mT (Fig. 9b).
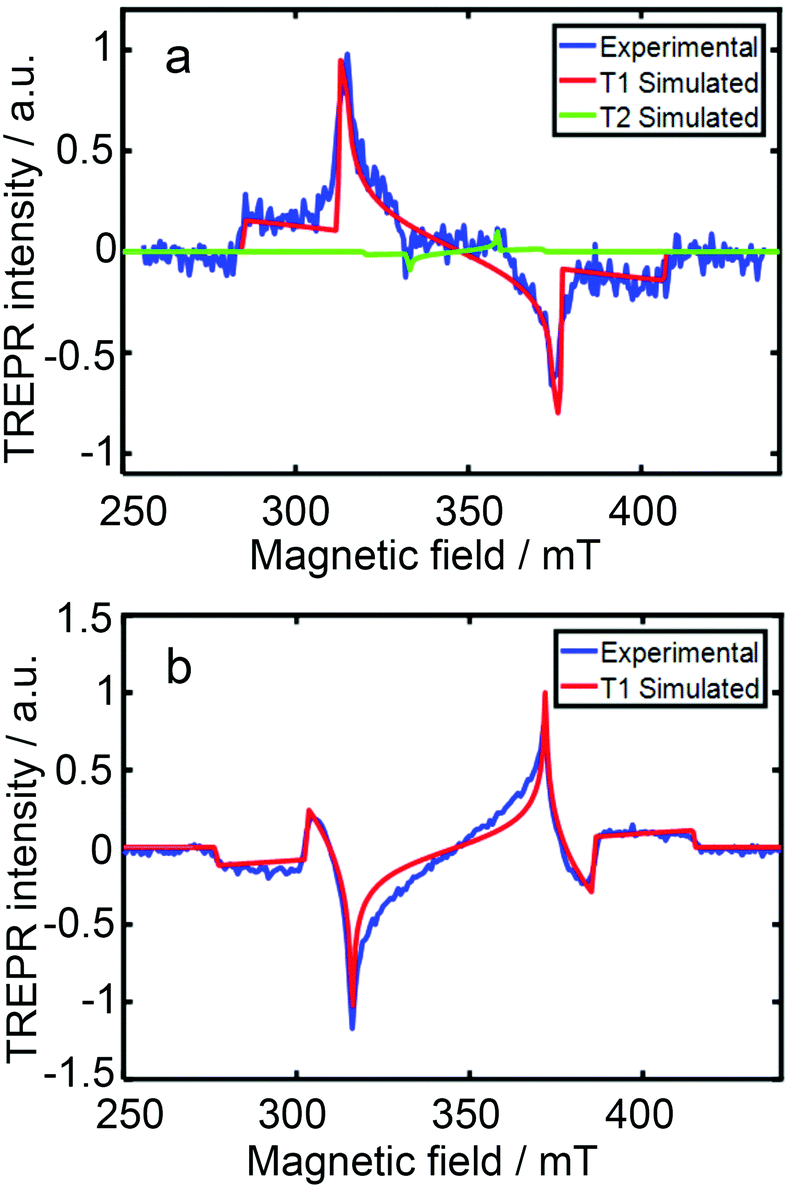 | ||
| Fig. 9 TREPR spectra at 80 K (blue line) of (a) DPA-AnCN and (b) Cz-AnCN in toluene solution (10 mg mL−1) recorded 1.5 μs after 532 nm and 410 nm laser pulses, respectively. The best-fit spectral simulations of T1 (red line) and T2 (green line) are presented. Reproduced with permission.75 Copyright 2020, Royal Society of Chemistry. | ||
The ISC of these two compounds is probably via the SOCT-ISC. The molecular geometry supports this postulate. The dihedral angle between the diphenyl moiety or carbazole and the cyanoanthryl moiety is 64°, and it is 81.1° in DPA-AnCN. The orthogonal geometry is beneficial for the conservation of the angular momentum during ISC.31 The ESP of the triplet state, which is localized on the anthryl moiety, also supports the SOCT-ISC mechanism. For DPA-AnCN, the ESP phase pattern is (a, e, a, e, a, e), for Cz-AnCN, it is (e, a, e, a, e, a). These ESP phase patterns are neither the triplet state formed by the ordinary SOC-ISC, which should show (e, e, e, a, a, a) or (a, a, a, e, e, e), nor the triplet state formed by the RP-ISC, for which the ESP phase pattern of the triplet state is (a, e, e, a, a, e) or (e, a, a, e, e, a).40 Previously with the julolidine–anthryl dyad, the 3An state formed via the SOCT-ISC mechanism shows ESP of (e, a, e, a, e, a).48 With phenothiazine–anthryl compact orthogonal dyad, we observed the 3An state with ESP of (a, e, a, e, a, e).76
Although a simplified two states model was used to rationalize the TADF,17,27 now it is a consensus that an intermediate triplet state, e.g.3LE state, should be involved in the TADF process,34,55 because the direct ISC between the states of 1CT/3CT is non-efficient, a spin-vibronic ISC mechanism should be involved in ISC between the 1CT and 3CT states.31,55,73
Recently Evans shows that indirect, not direct, coupling of the 3CT state to the singlet manifold facilitates ISC. Vibronic coupling plays a significant role in the ISC of 1CT → 3LE.73 With a few TADF emitters, Evans et al. confirmed that both the 3CT and 3LE states were observed in the TREPR spectra. This is the result of the small energy gap between the 3CT and the 3LE states. With a larger energy gap between the two, and high-lying 3CT state, only the 3LE state was detected in the TREPR spectra.73
The authors propose that TADF is facilitated by SOC, based on three possible mechanisms:
(I) Direct SOC between 1CT and 3LE states;
(II) Vibrationally enhanced SOC between 1CT and 3LE states;
(III) Spin-vibronic coupling between 1CT and 3CT via intermediate 3LE state.
These three ISC processes can be described by the following equation of the SOC Hamiltonian (include three non-zero terms).
| ĤSO = 〈ψ1CT|ĤSO|ψ3LE〉 | (1) |
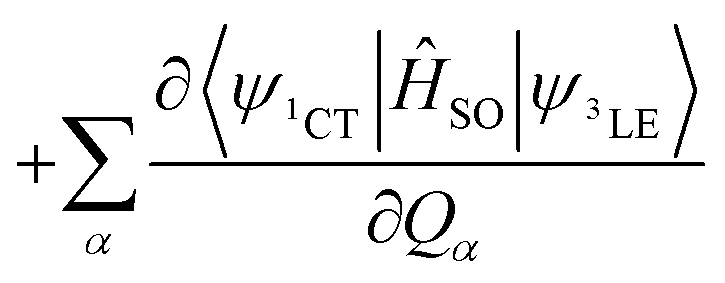 | (2) |
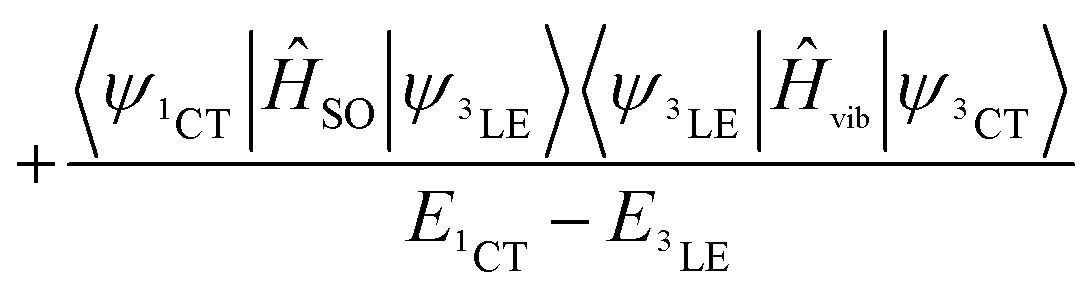 | (3) |
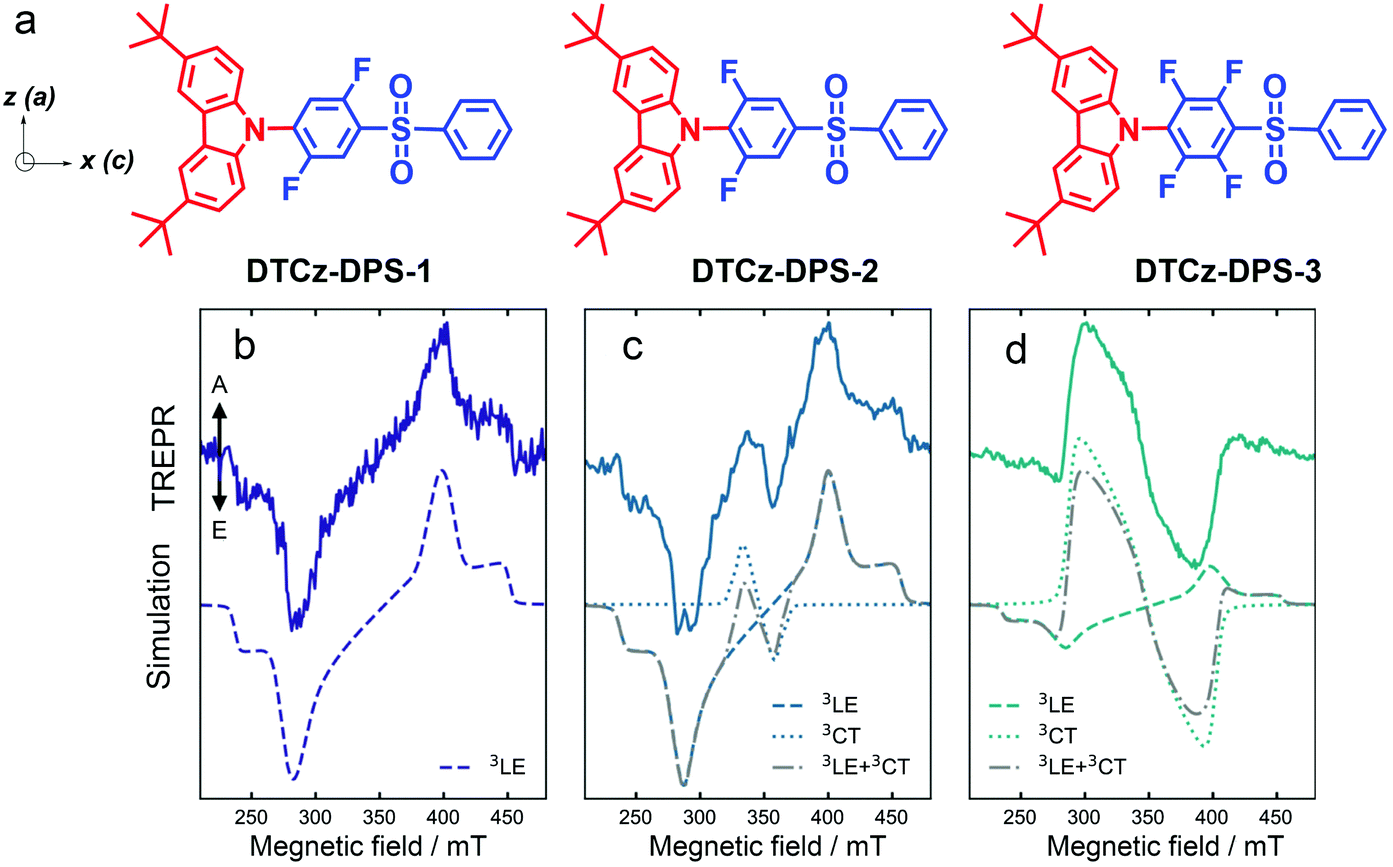 | ||
| Fig. 10 (a) Molecular structures of DTCz-DPS-1, DTCz-DPS-2 and DTCz-DPS-3. The in-plane molecular axes a and c and the 3LE ZFS axes z and x are shown. Spin-polarized TREPR signals collected at 30 K in toluene for (b) DTCz-DPS-1, (c) DTCz-DPS-2 and (d) DTCz-DPS-3. Solid lines show the TREPR signal recorded 2 μs after 355 nm laser excitation and integrated over 1 μs. Dashed and dotted lines are the simulated local excitation triplet state (3LE) and charge transfer triplet state (3CT) polarization patterns, respectively. Dash-dot grey lines are the weighted sums of the 3LE and 3CT simulations. The spin-polarized patterns are characterized by absorptive (A) and emissive (E) features. Reproduced with permission.73 Copyright 2021, Springer Nature. | ||
The compounds in Fig. 10 are with variable CT state energy, because of the difference in the electron withdrawal ability of the fluorinated phenyl linker, but the 3LE state energy (localized on the carbazole units) is kept consistent. From DTCz-DPS-1 to DTCz-DPS-3, the electron acceptor becomes stronger, thus the CT state energy decreases in order of 3.42 eV, 3.40 eV and 3.32 eV, respectively, whereas the 3LE state energy of the three compounds is kept as 3.05 eV (Table 2). Biexponential decay was observed for the luminescence of all the compounds (Table 2), but the contribution of the delayed fluorescence to the total luminescence is different for the three compounds. The energy gaps between the low-lying 3LE state and the 3CT state are 104 meV, 81 meV and 33 meV, respectively.
| Compound | λ PL (nm) | S1b (eV) | T1b (eV) | τ PF (ns) | τ DF (μs) | % delayedd | H SO (cm−1) | ΔGf (meV) |
|---|---|---|---|---|---|---|---|---|
| a Photoluminescence charge-transfer transition peak maxima in deaerated toluene at 295 K. b The energetic onset of photoluminescence (S1) and phosphorescence (T1) in deaerated toluene at 77 K. c Prompt fluorescence lifetime (±5 ns) and delayed fluorescence lifetime (±100 ns) from integrated photoluminescence kinetics, recorded in deaerated toluene at 295 K. d Contribution of delayed fluorescence component to total photoluminescence from integrated time-resolved photoluminescence measured in deaerated toluene at 295 K. e Root sum square of the x, y, z components of the calculated 〈ψ1CT|ĤSO|ψ3LE〉 at optimized 3LE geometry. f Difference in Gibbs free energy associated with 3LE and 3CT minima at their optimized geometries. | ||||||||
| DTCz-DPS-1 | 438 | 3.42 | 3.05 | 15 | 11 | 3 | 0.31 | 104 |
| DTCz-DPS-2 | 449 | 3.40 | 3.05 | 17 | 31 | 28 | 0.27 | 81 |
| DTCz-DPS-3 | 527 | 3.32 | 3.05 | 5 | 15 | 37 | 0.32 | 33 |
For DTCz-DPS-1, a triplet TREPR spectrum with the (e, e, e, a, a, a) phase pattern was observed. Simulation of the spectrum gives the ZFS D and E parameters of −108 mT and −9 mT, respectively. Thus this state can be assigned to a 3LE state confined on the carbazole moiety; this is supported by the ZFS D of the triplet state of native carbazole 107 mT.78 For DTCz-DPS-2 and DTCz-DPS-3, however, two triplet states are required for satisfactory simulation of the experimental TREPR spectra. For DTCz-DPS-2, one triplet is with ZFS D and E parameters of −108 mT and −9 mT, respectively (Table 3). This state is a carbazole-localized 3LE state, similar to that of DTCz-DPS-1. The second triplet state is with a much smaller ZFS D parameter of −23 mT, indicating it is a 3CT state. Note the ZFS E parameter is not vanishing, which is determined as −3 mT. The weight of the 3CT state in the total TREPR spectra is 10%. Similar TREPR results were observed for DTCz-DPS-3. Both 3LE and 3CT states were observed in the TREPR spectra, the weight of the 3CT state increases to 80% in the TREPR spectra.
| Compounds | 3LE parameters | 3CT parameters | ||||||
|---|---|---|---|---|---|---|---|---|
| Experimental D, E (mT) | Calculated D, E (mT) | P x,y,z | Weight | Experimental D, E (mT) | Calculated D, E (mT) | P x,y,z | Weight | |
| a The Px,y,z uncertainty is ±0.1 in all cases except for DTCz-DPS-13LE, which has an uncertainty of ±0.01. The weighing factor corresponds to the contribution of each 3LE or 3CT simulation to its total simulated spectrum. | ||||||||
| DTCz-DPS-1 | −108, −9 | −115, −10 | 0.38, 0.00,0.62 | 1.0 | — | — | — | — |
| DTCz-DPS-2 | −110, −10 | −110, −8 | 0.4, 0.0, 0.6 | 0.9 | −23, −3 | −76, −16 | 0.4, 0.6, 0.0 | 0.1 |
| DTCz-DPS-3 | −110, −8 | −112, −9 | 0.4, 0.0, 0.6 | 0.2 | −56, −19 | −94, −17 | 0.6, 0.4, 0.0 | 0.8 |
The authors propose that the ESP phase pattern of the 3LE state indicates the ISC mechanism is not via HFI, i.e. the RP-ISC mechanism does not contribute to the formation of the 3LE state. The authors stated that the 3LE state is populated via the vibrational SOC, because the in-planes sublevels are over populated. Direct SOC ISC will give different population rates of the sublevels of the 3LE state, which was not observed in the experimental TREPR spectra. Based on the ESP of the 3LE and the 3CT states, and the mutual orientation of the donor and acceptor (there should be ZFS axes rotation for the internal conversion from 3LE to 3CT), the authors propose that the 3CT state is populated by internal conversion from the 3LE state, aided by the spin-vibronic coupling effect. Spin-vibronic induced ISC in TADF emitters with a smaller 3CT/3LE state energy gap is supported by the more significant 3CT state weight in the TREPR spectra (Fig. 10c).
The insights into TADF photophysics attained with the TREPR spectra show that the 3LE state must be close to the CT state in energy. Moreover, the 1CT → 3LE must be efficient. As shown by the compact electron donor–acceptor dyads, one factor enhancing the 1CT → 3LE is the orthogonal geometry of the π-conjugation planes of the electron donor and acceptor units in the emitter molecule. The observed ESP of the 3LE state TREPR spectrum excludes the HFI induced ISC (i.e. RP-ISC mechanism). Moreover, vibration mode concerning the rotation of the linker between the electron donor and acceptor may increase the ISC of the TADF emitters (according to term (2)). Thus, previously proposed methods of increasing the molecular rigidity, to increase the TADF luminescence quantum yield and to maintain a narrow emission band, actually may reduce the TADF performance of the emitter.
3. Summary and prospect
In summary, the recent developments in the study of TADF emitters for application in OLEDs using time-resolved electron paramagnetic resonance (TREPR) spectroscopy were summarized, the transient species involved in the process of TADF molecular photophysics was discussed, and the influence of energy gap, vibrational and spin-vibronic coupling between the triplet charge transfer state (3CT) and the localized triplet excited state (3LE) on the TADF process was also analyzed. This spectroscopic method is unique in that it selectively detects the transient paramagnetic species involved in the TADF process, for instance, 3CT and 3LE states. Note in most cases these states are mixed with different extent CT/LE features; this character can be well probed by the TREPR spectra. Moreover, based on the electron spin polarization (ESP) phase patterns of the triplet state TREPR spectra, the orientation of the zero field splitting (ZFS) principal axes, different ISC mechanisms involved in the TADF process can be discriminated, i.e. the direct SOC, the vibrationally enhanced ISC, and the spin-vibronic ISC (non-Born–Oppenheimer effect). These different ISC mechanisms are difficult to be discriminated by the transient optical spectroscopic methods. Moreover, it should be pointed out that the real TADF process in the OLED device, in which the excitons are formed electronically, i.e. by electron–hole recombination (charge recombination), may be different from that under photoexcitation conditions.79 The process involving different intermediate excited states was observed under different excitation conditions, i.e. localized triplet states are only detected upon photoexcitation, but not upon electronic excitation.79 For the TADF molecules, photoexcitation will firstly produce the 1CT state (either by direct S0 → 1CT excitation, or by ultrafast charge separation, i.e.1LE → 1CT transition), then the triplet state 3LE will be populated, subsequently the 3CT state will be formed, mostly via3LE → 3CT (note these are simplified situations). In an operating OLED device, however, the 1CT and 3CT (and 3LE state) states will be produced directly from the excitons (i.e. by charge recombination), or by energy transfer from the host molecules, in which case, no ISC is required for the formation of the 3CT state, although the ISC and reverse ISC (rISC) will take place after the initial states are formed. Therefore, the photophysics of the TADF molecules may not be exactly the same for photoexcitation and electronic excitation.Moreover, one should keep in mind that TREPR spectroscopy is not a non-invasive method to probe the TADF photophysical processes, because when an external magnetic field is applied, the photophysical processes may be altered by the magnetic field, especially when the energy gap between different states is comparable to the Zeeman splitting magnitude. For the X-band EPR with an energy magnitude of ca. 10 μeV, normally the effect on the photophysical processes is negligible, although the RP-ISC efficiency can be affected by external magnetic fields. For the W-band or Q-band spectrometer (with the magnitude of the hundreds of μeV and meV, respectively), the energy of the magnetic field is large enough to influence the TADF photophysical processes significantly. These factors need to be considered in the future study of TADF materials using the TREPR spectra. Moreover, normally the study of the triplet states with TREPR spectra are performed in a frozen solution at cryogenic temperature, to suppress molecular tumbling and spin lattice relaxation, but these experimental conditions can also affect the photophysical processes, for instance the CT state energy and the kinetics of the charge transfer. Concerning these aspects, TREPR spectral study of the TADF molecules in film or liquid crystal is desired, but the related reports are rare. There is still more room to unravel the general principles governing the efficient TADF process in the electron donor–acceptor based emitters for application in OLEDs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Zhao thanks the NSFC (U2001222, 21673031 and 21761142005), the Fundamental Research Funds for the Central Universities (DUT19TD28) and the State Key Laboratory of Fine Chemicals of Dalian University for financial support.Notes and references
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH Verlag GmbH, 2008 Search PubMed.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2016, 4, 11355–11381 RSC.
- Y. Im, S. Y. Byun, J. H. Kim, D. R. Lee, C. S. Oh, K. S. Yook and J. Y. Lee, Adv. Funct. Mater., 2017, 27, 1603007 CrossRef.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- J. Zhang, F. Zhao, X. Zhu, W.-K. Wong, D. Ma and W.-Y. Wong, J. Mater. Chem., 2012, 22, 16448–16457 RSC.
- S. Wang, Z. Cheng, X. Song, X. Yan, K. Ye, Y. Liu, G. Yang and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 9892–9901 CrossRef CAS PubMed.
- Z.-Y. Wang, L.-Y. Zhang, L.-J. Xu, L.-X. Shi, J.-Y. Wang and Z.-N. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 14433–14439 CrossRef CAS PubMed.
- H. Tian, D. Liu, J. Li, M. Ma, Y. Lan, W. Wei, R. Niu and K. Song, New J. Chem., 2021, 45, 11253–11260 RSC.
- D. Jacquemin and D. Escudero, Chem. Sci., 2017, 8, 7844–7850 RSC.
- W. C. Chen, C. Sukpattanacharoen, W. H. Chan, C. C. Huang, H. F. Hsu, D. Shen, W. Y. Hung, N. Kungwan, D. Escudero, C. S. Lee and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002494 CrossRef CAS.
- W. Liu, L. Zhou, L. Y. Jin, W. Xie, C.-M. Che and G. Cheng, J. Mater. Chem. C, 2021, 9, 3384–3390 RSC.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, CA, 2009 Search PubMed.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- Q. Peng, D. Fan, R. Duan, Y. Yi, Y. Niu, D. Wang and Z. Shuai, J. Phys. Chem. C, 2017, 121, 13448–13456 CrossRef CAS.
- M. Li, Y. Liu, R. Duan, X. Wei, Y. Yi, Y. Wang and C.-F. Chen, Angew. Chem., Int. Ed., 2017, 56, 8818–8822 CrossRef CAS PubMed.
- T. T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Beilstein J. Org. Chem., 2018, 14, 282–308 CrossRef CAS PubMed.
- X. Zhang, D. Jacquemin, Q. Peng, Z. Shuai and D. Escudero, J. Phys. Chem. C, 2018, 122, 6340–6347 CrossRef CAS.
- Z. Cheng, Z. Li, Y. Xu, J. Liang, C. Lin, J. Wei and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 28096–28105 CrossRef CAS PubMed.
- L. Wang, Q. Ou, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2021, 125, 1468–1475 CrossRef CAS PubMed.
- Y. Mei, D. Liu, J. Li, H. Li and W. Wei, J. Mater. Chem. C, 2021, 9, 5885–5892 RSC.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CrossRef CAS.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- Z.-W. Li, L.-Y. Peng, X.-F. Song, W.-K. Chen, Y.-J. Gao, W.-H. Fang and G. Cui, J. Phys. Chem. Lett., 2021, 12, 5944–5950 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- J. W. Verhoeven, H. J. van Ramesdonk, M. M. Groeneveld, A. C. Benniston and A. Harriman, ChemPhysChem, 2005, 6, 2251–2260 CrossRef CAS PubMed.
- J. W. Verhoeven, J. Photochem. Photobiol. C, 2006, 7, 40–60 CrossRef CAS.
- Y. Hou, X. Zhang, K. Chen, D. Liu, Z. Wang, Q. Liu, J. Zhao and A. Barbon, J. Mater. Chem. C, 2019, 7, 12048–12074 RSC.
- H. Levanon and J. R. Norris, Chem. Rev., 1978, 78, 185–198 CrossRef CAS.
- C. Hintze, U. E. Steiner and M. Drescher, ChemPhysChem, 2017, 18, 6–16 CrossRef CAS PubMed.
- S. Richert, C. E. Tait and C. R. Timmel, J. Magn. Reson., 2017, 280, 103–116 CrossRef CAS PubMed.
- S. Weber, eMagRes, 2017, 6, 255–270 CAS.
- T. Biskup, Front. Chem., 2019, 7, 10 CrossRef CAS PubMed.
- M. Imran, X. Zhang, Z. Wang, X. Chen, J. Zhao, A. Barbon and V. K. Voronkova, Phys. Chem. Chem. Phys., 2021, 23, 15835–15868 RSC.
- Z. Wang, X. Zhang and J. Zhao, J. Phys. Chem. C, 2021, 125, 19097–19109 CrossRef.
- S. Richert, G. Bullard, J. Rawson, P. J. Angiolillo, M. J. Therien and C. R. Timmel, J. Am. Chem. Soc., 2017, 139, 5301–5304 CrossRef CAS PubMed.
- C. Hintze, P. Korf, F. Degen, F. Schütze, S. Mecking, U. E. Steiner and M. Drescher, J. Phys. Chem. Lett., 2017, 8, 690–695 CrossRef CAS PubMed.
- Z. E. X. Dance, Q. Mi, D. W. McCamant, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. B, 2006, 110, 25163–25173 CrossRef CAS PubMed.
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed.
- N. J. Turro, M. H. Kleinman and E. Karatekin, Angew. Chem., Int. Ed., 2000, 39, 4436–4461 CrossRef PubMed.
- A. Kawai and K. Shibuya, J. Photochem. Photobiol. C, 2006, 7, 89–103 CrossRef CAS.
- T. Ogiwara, Y. Wakikawa and T. Ikoma, J. Phys. Chem. A, 2015, 119, 3415–3418 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 2006 Search PubMed.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, 1603282 CrossRef PubMed.
- R. Ishimatsu, T. Edura, C. Adachi, K. Nakano and T. Imato, Chem. – Eur. J., 2016, 22, 4889–4898 CrossRef CAS PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- D. J. Gibbons, A. Farawar, P. Mazzella, S. Leroy-Lhez and R. M. Williams, Photochem. Photobiol. Sci., 2020, 19, 136–158 CrossRef CAS PubMed.
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, J. Mater. Chem. C, 2021, 9, 11944–11973 RSC.
- I. Bhattacharjee, N. Acharya, H. Bhatia and D. Ray, J. Phys. Chem. Lett., 2018, 9, 2733–2738 CrossRef CAS PubMed.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- E. W. Evans, Y. Olivier, Y. Puttisong, W. K. Myers, T. J. H. Hele, S. M. Menke, T. H. Thomas, D. Credgington, D. Beljonne, R. H. Friend and N. C. Greenham, J. Phys. Chem. Lett., 2018, 9, 4053–4058 CrossRef CAS PubMed.
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- S. Kuila, A. Ghorai, P. K. Samanta, R. B. K. Siram, S. K. Pati, K. S. Narayan and S. J. George, Chem. – Eur. J., 2019, 25, 16007–16011 CrossRef CAS PubMed.
- M. Hussain, A. M. El-Zohry, Y. Hou, A. Toffoletti, J. Zhao, A. Barbon and O. F. Mohammed, J. Phys. Chem. B, 2021, 125, 10813–10831 CrossRef CAS PubMed.
- G. Tang, A. A. Sukhanov, J. Zhao, W. Yang, Z. Wang, Q. Liu, V. K. Voronkova, M. Di Donato, D. Escudero and D. Jacquemin, J. Phys. Chem. C, 2019, 123, 30171–30186 CrossRef CAS.
- A. Demeter, L. Biczok, T. Berces, V. Wintgens, P. Valat and J. Kossanyi, J. Phys. Chem., 1993, 97, 3217–3224 CrossRef CAS.
- X. Zhang, Y. Hou, X. Xiao, X. Chen, M. Hu, X. Geng, Z. Wang and J. Zhao, Coordin. Chem. Rev., 2020, 417, 213371 CrossRef CAS.
- S. Suzuki, R. Sugimura, M. Kozaki, K. Keyaki, K. Nozaki, N. Ikeda, K. Akiyama and K. Okada, J. Am. Chem. Soc., 2009, 131, 10374–10375 CrossRef CAS PubMed.
- Y. Kobori, M. Fuki and H. Murai, J. Phys. Chem. B, 2010, 114, 14621–14630 CrossRef CAS PubMed.
- A. Karimata, H. Kawauchi, S. Suzuki, M. Kozaki, N. Ikeda, K. Keyaki, K. Nozaki, K. Akiyama and K. Okada, Chem. Lett., 2013, 794–796 CrossRef CAS.
- B. H. Drummond, N. Aizawa, Y. Zhang, W. K. Myers, Y. Xiong, M. W. Cooper, S. Barlow, Q. Gu, L. R. Weiss, A. J. Gillett, D. Credgington, Y.-J. Pu, S. R. Marder and E. W. Evans, Nat. Commun., 2021, 12, 4532 CrossRef CAS PubMed.
- J. Karpiuk, A. Majka, E. Karolak and J. Nowacki, J. Phys. Chem. Lett., 2017, 8, 4659–4667 CrossRef CAS PubMed.
- N. Sharma, M. Y. Wong, D. Hall, E. Spuling, F. Tenopala-Carmona, A. Privitera, G. Copley, D. B. Cordes, A. M. Z. Slawin, C. Murawski, M. C. Gather, D. Beljonne, Y. Olivier, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2020, 8, 3773–3783 RSC.
- Y. Hou, T. Biskup, S. Rein, Z. Wang, L. Bussotti, N. Russo, P. Foggi, J. Zhao, M. Di Donato, G. Mazzone and S. Weber, J. Phys. Chem. C, 2018, 122, 27850–27865 CrossRef CAS.
- D. Liu, A. M. El-Zohry, M. Taddei, C. Matt, L. Bussotti, Z. Wang, J. Zhao, O. F. Mohammed, M. Di Donato and S. Weber, Angew. Chem., Int. Ed., 2020, 59, 11591–11599 CrossRef CAS PubMed.
- I. S. M. Saiful, P. Heinze, Y. Ohba, S. Yamauchi, M. Yamamoto, Y. Tohda and K. Tani, Mol. Phys., 2006, 104, 1535–1542 CrossRef CAS.
- N. Bunzmann, S. Weissenseel, L. Kudriashova, J. Gruene, B. Krugmann, J. V. Grazulevicius, A. Sperlich and V. Dyakonov, Mater. Horiz., 2020, 7, 1126–1137 RSC.
| This journal is © The Royal Society of Chemistry 2022 |