
A perylene diimide dimer-based electron transporting material with an A–D–A structure for efficient inverted perovskite solar cells†

Abstract
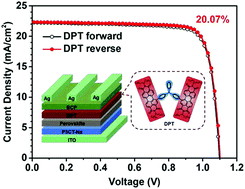
Through the introduction of two perylene diimide (PDI) moieties to the triphenylamine (TPA) core, a PDI dimer-based electron transport material has been synthesized with an A (electron acceptor)–D (electron donor)–A structure. It demonstrated a high electron mobility of 8.23 × 10−4 cm2 V−1 s−1 as a thin film and achieved a PCE of 20.07% in perovskite solar cells, higher than that of PC61BM (18.38%) as the standard reference under the same conditions. This is the highest conversion efficiency among various PDI derivatives as ETMs, providing the optimized molecular configuration by combination of twisted core (TPA) and planar peripheral moieties (PDI), as well as matched electronic properties with multiple charge transfer processes.
- This article is part of the themed collection: Special issue in honour of Daoben Zhu
 Please wait while we load your content...
Please wait while we load your content...

