DOI:
10.1039/D1TC03421A
(Paper)
J. Mater. Chem. C, 2022,
10, 2718-2723
An n-type narrow-bandgap organoboron polymer with quinoidal character synthesized by direct arylation polymerization†
Received
22nd July 2021
, Accepted 24th September 2021
First published on 6th October 2021
Abstract
Narrow bandgap conjugated polymers have received great attention due to their wide applications in optoelectronic devices. Because of the synthetic difficulty, it is greatly challenging to develop n-type conjugated polymers with a narrow bandgap with quinoidal character. Herein, we report an n-type narrow-bandgap organoboron polymer with quinoidal character, PBN-TP. PBN-TP is designed with alternating units of BNBP (double B ← N bridged bipyridine) and quinoidal TP (thieno[3,4-b]pyrazine). Direct arylation polymerization of the di-bromo BNBP unit and the di-hydro TP unit affords the organoboron polymer with a high number-average molecular weight of 28.3 kDa. Compared with the control polymer with alternating units of BNBP and thiophene, PBN-T, the replacement of the thiophene unit with a quinoidal TP unit reduces the bond length alternation (BLA) of carbon–carbon bonds and planarized the polymer backbone of PBN-TP, leading to an enhanced contribution of the polymer quinoidal resonance structure. As a result, compared with PBN-T, PBN-TP shows a much reduced bandgap of 1.50 eV, which is among the lowest reported for n-type organoboron polymers based on BNBP. Moreover, due to the low LUMO energy level, PBN-TP could be readily n-doped and used as an n-type polymer thermoelectric material. This work provides a new n-type narrow bandgap conjugated polymer with quinoidal character.
Introduction
Narrow bandgap conjugated polymers have received great attention because of their wide applications in organic photovoltaics (OPV),1–4 organic photodetectors (PDs)5,6 and organic field-effect transistors (OFETs).7,8 Generally, there are two strategies to design conjugated polymers with a low bandgap, i.e. the electron donor–acceptor (D–A) structure and quinoidal structure.9,10 D–A type conjugated polymers have been intensively studied and have exhibited excellent organic optoelectronic device performance.11–14 In comparison, there are probably fewer conjugated polymers with quinoidal character known than polymers with a D–A structure.15,16 There are fewer n-type conjugated polymers than p-type conjugated polymers.17,18 It is interesting to design n-type narrow-bandgap conjugated polymers with quinoidal character. However, the synthesis of this kind of conjugated polymers is challenging. Di-stannyl monomer or di-boronic ester monomers of quinoid units are rarely reported because of their electronic structure and reactivity. Therefore, material chemists cannot perform Stille polymerization or Suzuki polymerization to synthesize n-type narrow-bandgap conjugated polymers with quinoidal character.19–21
Organoboron chemistry offers an important toolbox to design conjugated polymers with tunable opto-electronic properties.22,23 For example, Helten et al. have reported a boron–nitrogen analogue, poly(p-phenylene vinylene) (PPV), and provided clear evidence for π-conjugation across the B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N units along the polymer backbone.24 Jäkle and co-workers have confirmed that B–N functionalized ladder-type dipyridylfluorene monomers could be used as building blocks for the preparation of luminescent conjugated polymers with tunable emission characteristics.25 Our group has previously developed an organoboron electron-accepting building block, double B ← N bridged bipyridine (BNBP), to design n-type conjugated polymers.26 A family of D–A type conjugated polymers based on BNBP unit has been synthesized and has been successfully used in high-performance OSCs,27–29 organic indoor photovoltaics,30 OFETs,31 and organic thermoelectrics.32 However, these polymers always exhibit a medium bandgap larger than 1.8 eV and show absorption in the visible range. In this work, we aim to develop narrow bandgap n-type conjugated polymers based on BNBP by copolymerizing with quinoidal structures.
N units along the polymer backbone.24 Jäkle and co-workers have confirmed that B–N functionalized ladder-type dipyridylfluorene monomers could be used as building blocks for the preparation of luminescent conjugated polymers with tunable emission characteristics.25 Our group has previously developed an organoboron electron-accepting building block, double B ← N bridged bipyridine (BNBP), to design n-type conjugated polymers.26 A family of D–A type conjugated polymers based on BNBP unit has been synthesized and has been successfully used in high-performance OSCs,27–29 organic indoor photovoltaics,30 OFETs,31 and organic thermoelectrics.32 However, these polymers always exhibit a medium bandgap larger than 1.8 eV and show absorption in the visible range. In this work, we aim to develop narrow bandgap n-type conjugated polymers based on BNBP by copolymerizing with quinoidal structures.
Recently, direct arylation polymerization (DArP) has emerged as a new method to synthesize conjugated polymers.33–36 This method exploits the palladium-catalyzed activation of aromatic C–H bonds for the synthesis of conjugated polymers. Compared with Stille polymerization or Suzuki polymerization, DArP has the great advantage of easy and facile monomer synthesis. Thieno[3,4-b]pyrazine (TP) is a well-known quinoid-type building block with quinoidal character to construct conjugated polymers with a narrow bandgap.37–39 In this work, we report an n-type conjugated polymer (PBN-TP) based on alternating units of BNBP and TP, which is synthesized by DArP with a number-average molecular weight (Mn) of 28.3 kDa. Owing to the electron-accepting properties of the BNBP unit and the quinoidal structure of the TP unit, PBN-TP exhibits a deep-lying LUMO energy level of −3.64 eV and a narrow optical bandgap (Eoptg) of 1.50 eV. Moreover, PBN-TP can be readily n-doped and can be used as an n-type thermoelectric material. This work provides a new n-type narrow bandgap conjugated polymer with quinoidal character.
Results and discussion
Molecular design
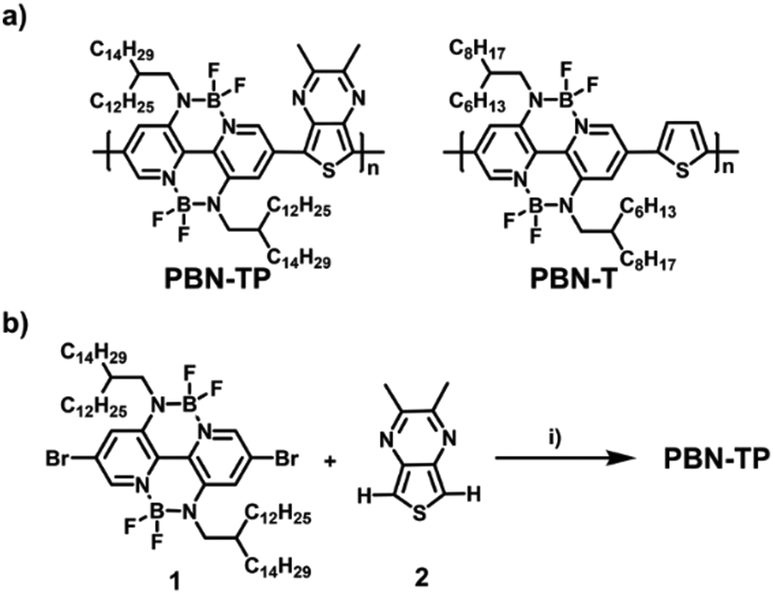
Scheme 1a shows the chemical structure of PBN-TP, which consists of BNBP units and TP units. TP is a widely used unit with quinoidal character, in which the quinoidal character of the thiophene moiety can be stabilized by the formation of an aromatic pyrazine ring in the resonance structure. In addition, the sp2 hybrid nitrogen atoms in TP bring no additional steric hindrance when copolymerizing with the BNBP unit, which would be beneficial in facilitating the coplanarity of the polymer backbone of PBN-TP. Two methyl groups are introduced onto the TP unit to protect the α-H of the pyrazine ring when performing the DArP reaction.40 Long and branched alkyl side chains of 2-dodecylhexadecyl are introduced onto the BNBP unit to ensure the solubility of PBN-TP. To illustrate the effect of quinoidal TP unit on the optoelectronic properties of PBN-TP, we select PBN-T as the control polymer, which consists of alternating units of BNBP and thiophene (Scheme 1b).26
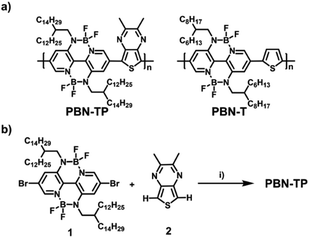 |
| | Scheme 1 (a) Chemical structure of the designed polymer PBN-TP and the control polymer PBN-T. (b) The synthetic route of PBN-TP. Reagents and conditions: (i) Pd(OAc)2 (0.05 equiv.), KOAc (6.0 equiv.), Bu4NBr (2.0 equiv.), toluene, 115 °C. | |
Synthesis and characterization
Scheme 1b shows the synthetic route of PBN-TP. The di-bromo BNBP monomer (1) and the di-hydro TP monomer (2) were synthesized following previously reported methods. Direct arylation polymerization of 1 and 2 was conducted in toluene with palladium(II) acetate/potassium acetate/tetrabutylammonium bromide as the catalytic system and afforded PBN-TP with a high molecular weight. The synthetic procedures are provided in the ESI.† The chemical structure of the polymer was confirmed by 1H NMR, 19F NMR and elemental analysis. Gel permeation chromatography (GPC) with 1,2,4-trichlorobenzene as the eluent at 150 °C indicates that the Mn and polydispersity indices (PDI) of PBN-TP are 28.3 kDa and 2.36, respectively (Table 1 and Fig. S1, ESI†). The high Mn of PBN-TP suggests high reactivity of the di-bromo BNBP monomer and the di-hydro TP monomer in direct arylation polymerization. PBN-TP is readily soluble in organic solvents, such as chloroform and chlorobenzene. PBN-TP exhibits excellent thermal stability with thermal decomposition temperature (Td) at 5% weight loss over 370 °C (Table 1 and Fig. S2, ESI†).
Table 1 Molecular weights (Mn), polydispersity indices (PDI), thermal decomposition temperatures at 5% weight loss (Td), energy levels, maximum absorption wavelength (λmax), and optical bandgap (Eoptg) of PBN-TP and PBN-T
| Polymer |
M
n [kDa] |
PDI |
T
d [°C] |
E
LUMO
[eV] |
E
HOMO
[eV] |
λ
max
[nm] |
E
optg [eV] |
|
The data are cited from ref. 26.
Calculated by cyclic voltammetry method using the equation ELUMO/EHOMO = −(4.80 + Eredonset/Eoxonset) eV, where Eredonset/Eoxonset is the reduction/oxidation onset potential vs. Fc/Fc+.
Measured in thin films.
|
| PBN-TP |
28.3 |
2.36 |
371 |
−3.64 |
−5.44 |
714 |
1.50 |
| PBN-T |
24.6a |
2.01a |
357a |
−3.50 |
−5.77 |
619 |
1.92 |
DFT calculations
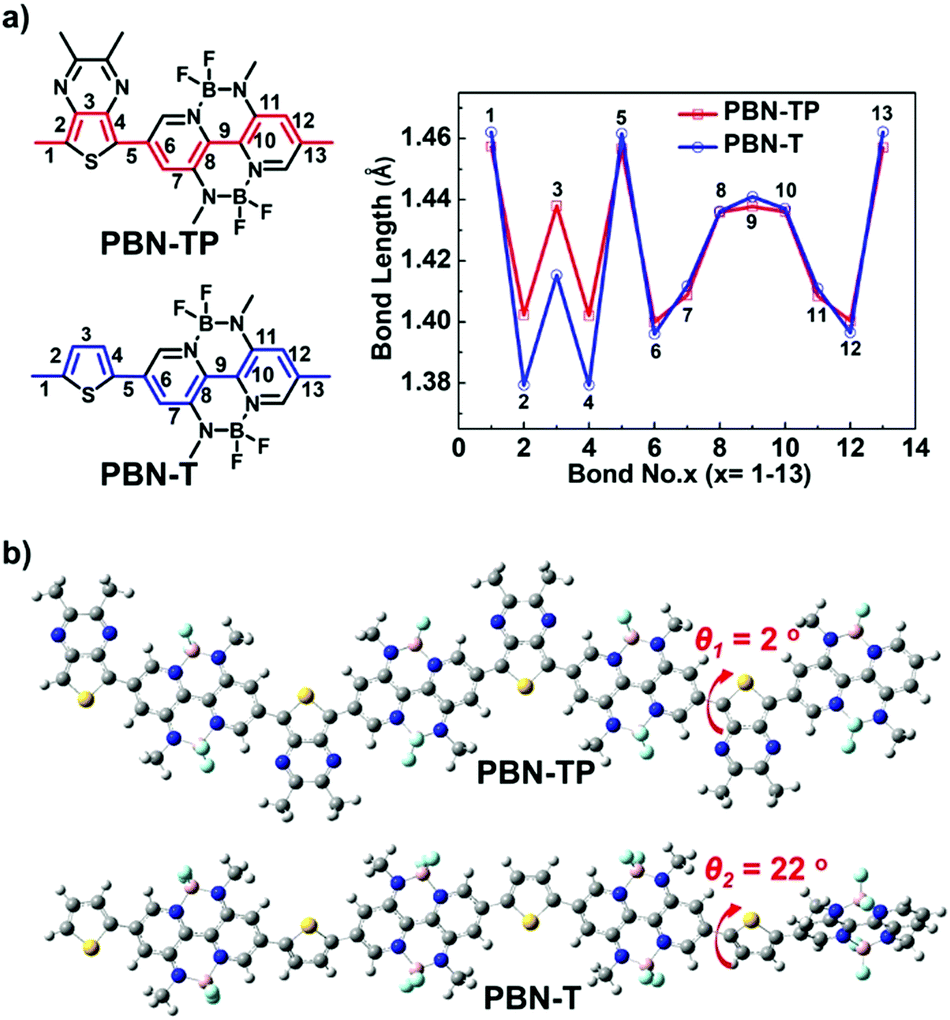
To illustrate the electronic structure and configuration of PBN-TP and PBN-T, we carried out density functional theory (DFT) calculations at the B3LYP/6-31G** level with their four repeating units. All the long alkyl chains have been replaced by methyl groups for simplification. Fig. 1a shows the chemical structures of repeat units of PBN-TP and PBN-T with carbon–carbon bond numbers and the corresponding plots of carbon–carbon bond length obtained by DFT calculations. The length of C–C bonds and CC bonds in the BNBP moiety of PBN-TP is obviously shorter and longer than those in PBN-T, respectively, indicating a smaller bond length alternation (BLA) of PBN-TP.41 This is ascribed to the introduction of a TP unit into the polymer backbone, which could enhance the contribution of the polymer quinoidal resonance structure. Fig. 1b shows the optimized geometries of polymer backbones of PBN-TP and PBN-T, respectively. The dihedral angle between the BNBP unit and adjacent TP unit (θ1) in PBN-TP is calculated to be 2°, indicating a nearly coplanar polymer backbone. In comparison, the dihedral angle between the BNBP unit and adjacent thiophene unit (θ2) in PBN-T is calculated to be 22°, indicating a twisted polymer backbone. The introduction of TP units greatly facilitates the planarity of polymer backbones, which would also be beneficial for improving the quinoidal character of PBN-TP.
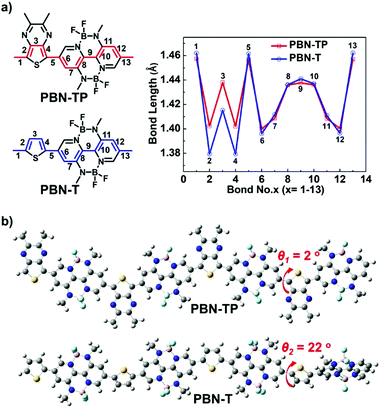 |
| | Fig. 1 (a) The chemical structures of PBN-TP and PBN-T repeat units with carbon–carbon bond numbers and corresponding plots of carbon–carbon bond length obtained by DFT calculations (B3LYP/6-31G**). (b) DFT-optimized geometry of four repeating units of PBN-TP and PBN-T, dihedral angles between the BNBP unit and TP unit (θ1), and between the BNBP unit and thiophene unit (θ2) are shown. All of the long alkyl chains have been replaced by methyl groups for simplification. | |
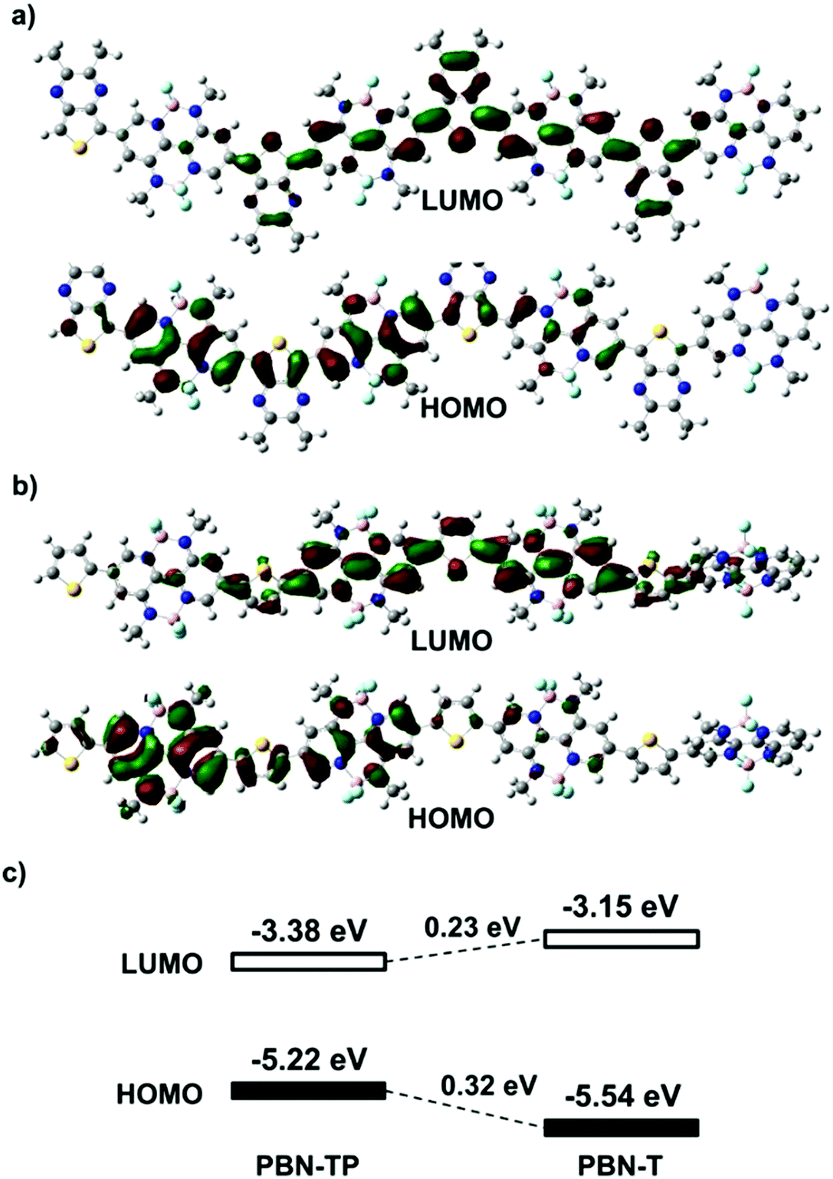
Fig. 2a and b show the calculated LUMOs/HOMOs based on the four repeating units of PBN-TP and PBN-T, respectively. Both PBN-TP and PBN-T show delocalized LUMOs/HOMOs along the polymer backbone. Fig. 2c shows the alignment of LUMO/HOMO energy levels (ELUMO/EHOMO) of PBN-TP and PBN-T based on DFT calculations. Compared with PBN-T, PBN-TP shows an obviously downshifted ELUMO and an upshifted EHOMO by 0.23 eV and 0.32 eV, respectively, thus leading to a much reduced bandgap. This can be ascribed to the quinoidal structure of PBN-TP.
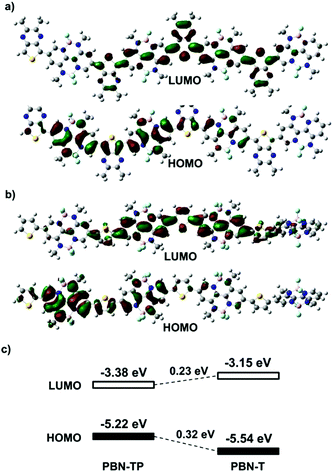 |
| | Fig. 2 Calculated Kohn–Sham LUMOs/HOMOs based on four repeating units of (a) PBN-TP and (b) PBN-T. (c) The alignment of the LUMO/HOMO energy levels of PBN-T and PBN-TP based on DFT calculations. All the long alkyl chains have been replaced by methyl groups for simplification. | |
We further studied the electronic structure at the negatively charged state of the two polymers based on DFT calculations at the UωB97XD/6-31G* level with their four repeating units.42 As shown in Fig. S3 (ESI†), the model compound of PBN-T exhibits spin density distribution on two repeating units. In comparison, the spin density distribution of PBN-TP covers nearly three repeating units, indicating a well-delocalized anion polarons. This delocalized electronic structure of PBN-TP might be ascribed to its quinoidal character and planar polymer backbones.
Opto-electronic properties
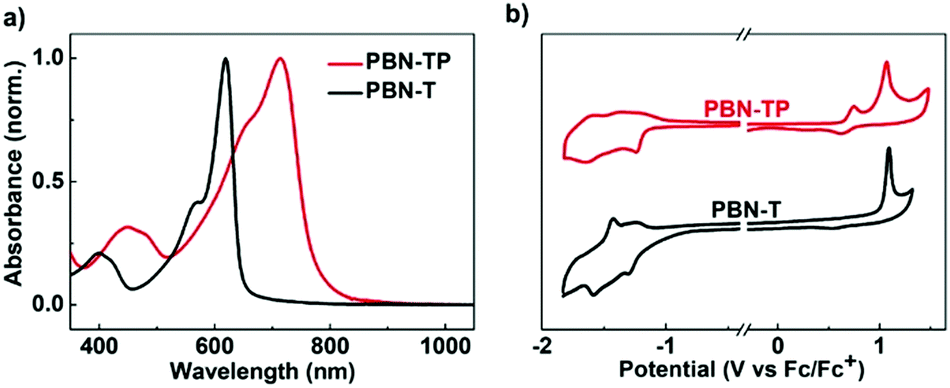
The UV-vis-NIR absorption spectra of PBN-TP and PBN-T in thin films are shown in Fig. 3a, and the data are summarized in Table 1. PBN-T shows one main absorption band in the visible region with a maximum absorption (λmax) of 619 nm. According to the onset absorption wavelength, the optical bandgap (Eoptg) of PBN-T is estimated to be 1.92 eV. In comparison, PBN-TP shows a great redshift of the absorption band with a λmax of 714 nm. The Eoptg of PBN-TP is estimated to be 1.50 eV, which is much smaller than that of PBN-T. This is ascribed to the quinoidal character of PBN-TP. The Eoptg of PBN-TP is among the lowest reported for organoboron polymers based on BNBP. To estimate the ELUMO/EHOMO of PBN-TP and PBN-T, we performed cyclic voltammetry (CV) measurements with the two polymer thin films. The cyclic voltammograms are shown in Fig. 3b. Both the two polymers exhibit obvious reduction and oxidation waves. According to the onset potential of the reduction/oxidation waves, the ELUMO/EHOMO values of PBN-TP and PBN-T are estimated to be −3.64/−5.44 eV and −3.50/−5.77 eV (Table 1), respectively. Compared with PBN-T, PBN-TP shows an obviously reduced ELUMO and an increased EHOMO, corresponding to a narrower bandgap. The CV results are consistent with the DFT calculations.
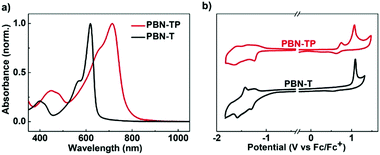 |
| | Fig. 3 (a) UV-vis-NIR absorption spectra and (b) cyclic voltammograms of PBN-TP and PBN-T in thin films. | |
n-Doping and electrical conductivity
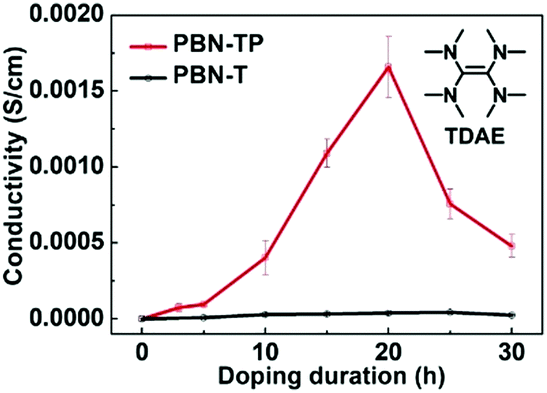
The low ELUMO of PBN-TP motivates us to investigate its electrical conductivity after n-doping. We used tetrakis(dimethylamino)ethylene (TDAE) (see the inset of Fig. 4; commercially available) as the n-dopant and exposed PBN-TP thin films to the TDAE vapor for different time periods to perform n-doping.43,44 We also doped PBN-T films for comparison. The electrical conductivity (σ) of these doped films was measured with the two-probe or four-probe method (ESI,† Fig. S5). Fig. 4 shows the electrical conductivities of PBN-TP and PBN-T thin films doped at different doping durations. Both the pristine PBN-TP and PBN-T films show a very low electrical conductivity of about 1.0 × 10−8 S cm−1. This is attributed to the low carrier concentration in the polymer films before n-doping. After exposure to TDAE vapor, PBN-TP films show a dramatically increased conductivity and gives a maximum of 1.9 × 10−3 S cm−1 at an exposure time of 20 h. Then, the conductivity of PBN-TP begins to drop after 20 h, which is probably ascribed to the disruption of polymer films by excess TDAE molecules. The maximum electrical conductivity of doped PBN-TP films is greatly improved by more than 5 orders of magnitude compared with their pristine film, implying an effective n-doping of PBN-TP. However, the electrical conductivity of PBN-T shows no obvious improvement after n-doping, indicating the less efficient n-doping process of PBN-T films upon TDAE vapor exposure. The effective n-doping and relatively high electrical conductivity of PBN-TP may be attributed to its deep-lying ELUMO, which can thermodynamically facilitate electron transfer from n-dopants to the polymer, and thus greatly increase the carrier concentration. Another possible reason is that the quinoidal character of PBN-TP backbone is beneficial for the delocalized distribution of anion polarons, which would facilitate free charge carrier generation and transportation.
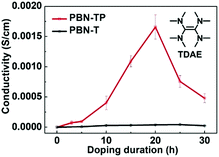 |
| | Fig. 4 Electrical conductivities of PBN-TP and PBN-T films at different doping durations. The inset shows the chemical structure of TDAE. | |
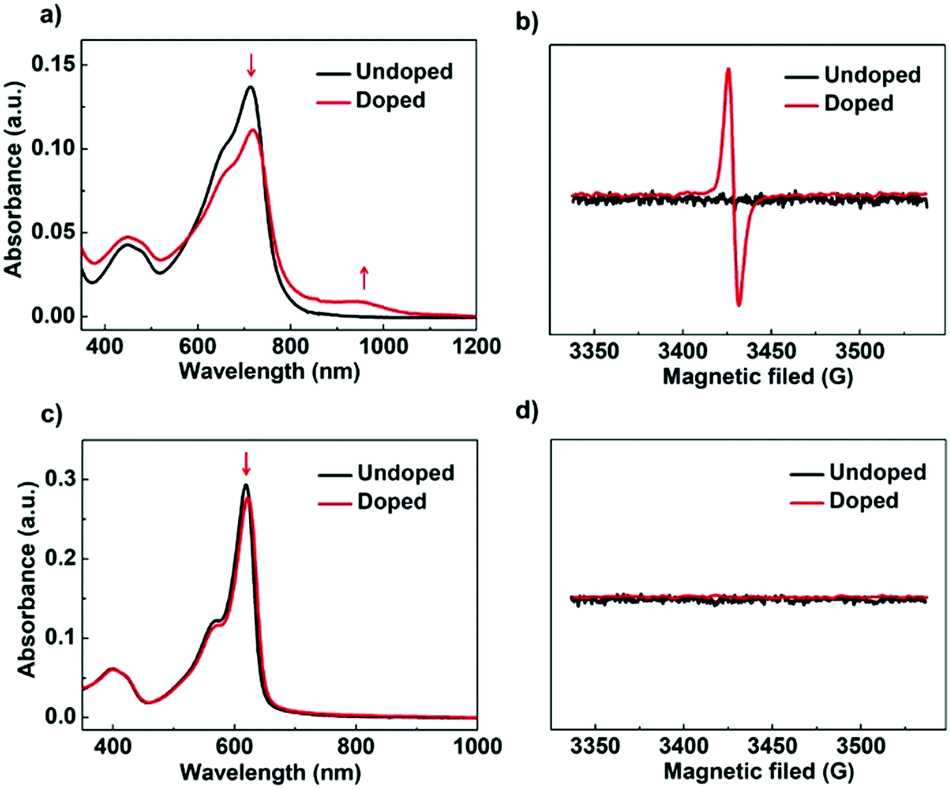
To gain further insights into the doping process of PBN-TP and PBN-T films, we performed UV-vis-NIR absorption and electron paramagnetic resonance (EPR) measurements in their pristine and doped films. As shown in Fig. 5a, the pristine PBN-TP film shows one main absorption band peak at 714 nm. After exposure to TDAE vapor for 20 hours, the film shows decreased absorbance at 714 nm accompanied by the appearance of a new absorption band covering from 800 nm to 1050 nm. The new absorption band is attributed to the polaron-induced electronic transitions after n-doping.32,45 The n-doping behavior of PBN-TP is further confirmed by the EPR spectra (Fig. 5b). There is no EPR signals in the pristine film; however, strong signals appeared in the doped film, suggesting that unpaired electrons are present in the doped film.32,46 The UV-vis-NIR absorption and EPR results confirm the effective n-doping behavior of PBN-TP. On the other hand, no obvious change was observed for PBN-T films in both UV-vis-NIR absorption and EPR spectra before and after n-doping (Fig. 5c and d), indicating the less effective n-doping process of PBN-T.
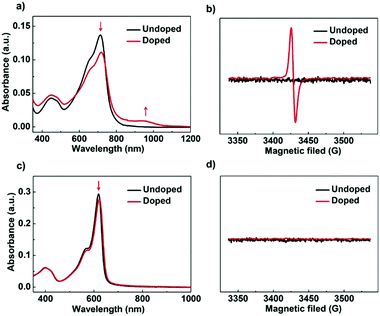 |
| | Fig. 5 UV-vis-NIR absorption spectra of (a) PBN-TP and (c) PBN-T in thin film before and after TDAE doping (tvapor = 20 h). Electron paramagnetic resonance (EPR) spectra of (b) PBN-TP and (d) PBN-T before and after TDAE doping (tvapor = 20 h). | |
Thermoelectric performance
Conjugated polymers are an important class of thermoelectric materials due to their advantages of solution processibility at low cost, light weight and flexibility. They are promising for applications in wearable electronics and sensor networks. Thermoelectric performance of conjugated polymers can be evaluated by power factor (PF), which is determined as PF = S2σ, where S is the Seebeck coefficient and σ is electrical conductivity. Thermoelectric modules require both p-type and n-type polymer thermoelectric materials. However, as the number of n-type conjugated polymers that can be readily n-doped is lower, the development of n-type polymer thermoelectric materials lags far behind that of p-type ones.47–51
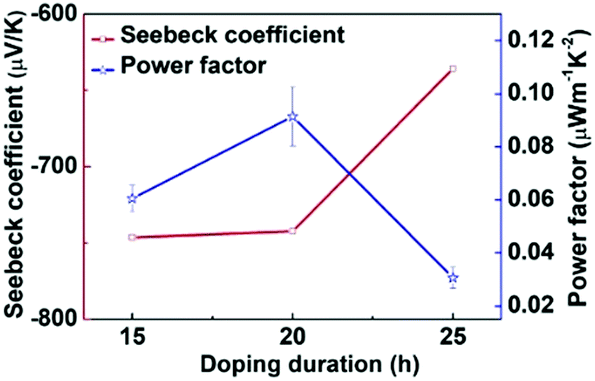
Motivated by the effective n-doping of PBN-TP, we further investigated its thermoelectric performance after n-doping. The S of the doped PBN-TP films was tested by recording the thermovoltage (Vtherm) at a certain temperature difference (ΔT) (ESI,† Fig. S6). As shown in Fig. 6, all the doped PBN-TP films show negative S values, indicating an electron-dominant charge transport. With extending the doping time, the absolute S values gradually decrease. According to these S and σ values at a certain doping time, we estimate the PF values of PBN-TP. The results are shown in Fig. 6. The maximum PF is calculated to be 0.11 μW m−1 K−2, with a Seebeck coefficient of −742.4 μV K−1 and an electrical conductivity of 1.9 × 10−3 S cm−1 at the doping time of 20 h. This result indicates that PBN-TP can be used as an n-type polymer thermoelectric material.
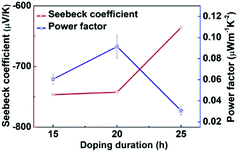 |
| | Fig. 6 Seebeck coefficients and power factors of PBN-TP films exposed to TDAE vapor for different time periods. | |
Conclusions
In summary, we reported an n-type organoboron polymer with quinoidal character, PBN-TP. DArP of the di-bromo BNBP unit and di-hydro TP unit afforded PBN-TP with a high Mn of 28.3 kDa. Compared with the control polymer with alternating units of BNBP and thiophene, the introduction of a TP unit reduced the BLA of the carbon–carbon bonds and planarized the polymer backbone of PBN-TP, leading to an enhanced contribution of the polymer quinoidal resonance structure. Compared with the control polymer, PBN-TP shows a downshifted ELUMO and an upshifted EHOMO, and thus a much reduced bandgap of 1.50 eV. The bandgap of PBN-TP is among the lowest reported for n-type organoboron polymers based on BNBP. Moreover, due to the relatively low ELUMO, PBN-TP could be readily n-doped and used as an n-type polymer thermoelectric material. This work provides a new n-type narrow bandgap conjugated polymer with quinoidal character.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support by the National Natural Science Foundation of China (No. 22075271, 21625403, 21875244, 21875241).
Notes and references
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef PubMed.
- K. Gao, Y. Kan, X. Chen, F. Liu, B. Kan, L. Nian, X. Wan, Y. Chen, X. Peng, T. P. Russell, Y. Cao and A. K. Jen, Adv. Mater., 2020, 32, e1906129 CrossRef.
- G. Li, W.-H. Chang and Y. Yang, Nat. Rev. Mater., 2017, 2, 17043 CrossRef.
- Z. Wu, Y. Zhai, H. Kim, J. D. Azoulay and T. N. Ng, Acc. Chem. Res., 2018, 51, 3144–3153 CrossRef PubMed.
- Q. Li, Y. Guo and Y. Liu, Chem. Mater., 2019, 31, 6359–6379 CrossRef.
- Y. Ren, X. Yang, L. Zhou, J. Y. Mao, S. T. Han and Y. Zhou, Adv. Funct. Mater., 2019, 29, 1902105 CrossRef.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef.
- X. Huang, N. Lan, W. Chen, Y. Yan, W. Zeng and S. Liu, J. Mater. Sci., 2021, 56, 8334–8357 CrossRef.
- M. C. Scharber and N. S. Sariciftci, Adv. Mater. Technol., 2021, 6, 2000857 CrossRef.
- E. E. Havinga, W. Ten Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119–126 CrossRef.
- M. Gon, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2018, 57, 6546–6551 CrossRef.
- J. Wakabayashi, M. Gon, K. Tanaka and Y. Chujo, Macromolecules, 2020, 53, 4524–4532 CrossRef.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- S. Tanaka, C. Kitamura and Y. Yamashita, Chem. Mater., 1996, 8, 570–578 CrossRef CAS.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC.
- H. Sun, X. Guo and A. Facchetti, Chemistry, 2020, 6, 1310–1326 CrossRef CAS.
- M. Liao, J. Duan, P. A. Peng, J. Zhang and M. Zhou, RSC Adv., 2020, 10, 41764–41779 RSC.
- C. K. Michinori Karikomi, Shoji Tanaka and Y. Yamashita, J. Am. Chem. Soc., 1995, 117, 6791–6792 CrossRef.
- Z. Zhu, D. Waller, R. Gaudiana, M. Morana, M. Scharber, D. Muhlbacher and C. Brabec, Macromolecules, 2007, 40, 1981–1986 CrossRef CAS.
- G. Qian, B. Dai, M. Luo, D. Yu, J. Zhan, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2008, 20, 6208–6216 CrossRef CAS.
- F. Jakle, Chem. Rev., 2010, 110, 3985–4022 CrossRef CAS.
- X. Shao, J. Wang, T. B. Marder, Z. Xie, J. Liu and L. Wang, Macromolecules, 2021, 54, 6718–6725 CrossRef CAS.
- T. Lorenz, M. Crumbach, T. Eckert, A. Lik and H. Helten, Angew. Chem., Int. Ed., 2017, 56, 2780–2784 CrossRef CAS PubMed.
- A. F. Alahmadi, R. A. Lalancette and F. Jakle, Macromol. Rapid Commun., 2018, 39, e1800456 CrossRef.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS PubMed.
- R. Zhao, J. Liu and L. Wang, Acc. Chem. Res., 2020, 53, 1557–1567 CrossRef CAS PubMed.
- X. Long, C. Dou, J. Liu and L. Wang, Chin. Chem. Lett., 2018, 29, 1343–1346 CrossRef CAS.
- Z. Zhang, Z. Ding, D. J. Jones, W. W. H. Wong, B. Kan, Z. Bi, X. Wan, W. Ma, Y. Chen, X. Long, C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2018, 61, 1025–1033 CrossRef CAS.
- Z. Ding, R. Zhao, Y. Yu and J. Liu, J. Mater. Chem. A, 2019, 7, 26533–26539 RSC.
- R. Zhao, Y. Min, C. Dou, B. Lin, W. Ma, J. Liu and L. Wang, ACS Appl. Polym. Mater., 2019, 2, 19–25 CrossRef.
- C. Dong, S. Deng, B. Meng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 16184–16190 CrossRef CAS PubMed.
- J. T. Blaskovits and M. Leclerc, Macromol. Rapid Commun., 2019, 40, e1800512 CrossRef.
- N. S. Gobalasingham and B. C. Thompson, Prog. Polym. Sci., 2018, 83, 135–201 CrossRef CAS.
- K. Guo, Y. Jiang, Y. Sui, Y.-F. Deng and Y.-H. Geng, Chin. J. Polym. Sci., 2019, 37, 1099–1104 CrossRef CAS.
- H. Xie, T. Fan, Q. Lei and W. Fang, Sci. China: Chem., 2016, 59, 1432–1447 CrossRef CAS.
- K. A. Mitchell, D. D. Kenning, T. R. Calhoun, D. J. Sattler, M. R. Funfar and S. C. Rasmussen, J. Org. Chem., 2002, 67, 9073–9076 CrossRef.
- C.-H. Chen, C.-H. Hsieh, M. Dubosc, Y.-J. Cheng and C.-S. Hsu, Macromolecules, 2009, 43, 697–708 CrossRef.
- J. P. Nietfeld, R. L. Schwiderski, T. P. Gonnella and S. C. Rasmussen, J. Org. Chem., 2011, 76, 6383–6388 CrossRef CAS.
- N. I. Abdo, A. A. El-Shehawy, A. A. El-Barbary and J.-S. Lee, Eur. J. Org. Chem., 2012, 5540–5551 CrossRef CAS.
- Y. Cai, X. Xue, G. Han, Z. Bi, B. Fan, T. Liu, D. Xie, L. Huo, W. Ma, Y. Yi, C. Yang and Y. Sun, Chem. Mater., 2018, 30, 319–323 CrossRef CAS.
- Y. Lu, Z. D. Yu, H. I. Un, Z. F. Yao, H. Y. You, W. Jin, L. Li, Z. Y. Wang, B. W. Dong, S. Barlow, E. Longhi, C. A. Di, D. Zhu, J. Y. Wang, C. Silva, S. R. Marder and J. Pei, Adv. Mater., 2021, 33, e2005946 CrossRef.
- S. Wang, H. Sun, U. Ail, M. Vagin, P. O. Persson, J. W. Andreasen, W. Thiel, M. Berggren, X. Crispin, D. Fazzi and S. Fabiano, Adv. Mater., 2016, 28, 10764–10771 CrossRef CAS.
- J. Conrad Burkholder and W. R. Dolbier, J. Org. Chem., 1998, 63, 5385–5394 CrossRef.
- Y. Lu, Z. D. Yu, Y. Liu, Y. F. Ding, C. Y. Yang, Z. F. Yao, Z. Y. Wang, H. Y. You, X. F. Cheng, B. Tang, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2020, 142, 15340–15348 CrossRef CAS.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Muller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed.
- C. Dong, B. Meng, J. Liu and L. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 10428–10433 CrossRef CAS.
- Y. Min, C. Dong, H. Tian, J. Liu and L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 33321–33327 CrossRef CAS PubMed.
- X. Yan, M. Xiong, J. T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z. Y. Wang, Z. F. Yao, J. Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS.
- Y. Lu, J.-Y. Wang and J. Pei, Chem. Mater., 2019, 31, 6412–6423 CrossRef CAS.
- Y. Sun, C. A. Di, W. Xu and D. Zhu, Adv. Electron. Mater., 2019, 5, 1800825 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, molecular weight, thermal properties, theoretical calculations, absorption spectra and thermoelectric properties. See DOI: 10.1039/d1tc03421a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab and
Lixiang
Wang
*ab and
Lixiang
Wang