
Tuning the antiaromatic character and charge transport of pentalene-based antiaromatic compounds by substitution†
Jianglin
Wu
ab,
Yao
Chen
 b,
Jueshan
Liu
a,
Zhenguo
Pang
a,
Guoping
Li
b,
Zhiyun
Lu
a,
Yan
Huang
*a,
Antonio
Facchetti
*bc and
Tobin J.
Marks
*b
b,
Jueshan
Liu
a,
Zhenguo
Pang
a,
Guoping
Li
b,
Zhiyun
Lu
a,
Yan
Huang
*a,
Antonio
Facchetti
*bc and
Tobin J.
Marks
*b
aKey Laboratory of Green Chemistry and Technology (Ministry of Education), College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: huangyan@scu.edu.cn
bDepartment of Chemistry, Center for Light Energy Activated Redox Processes (LEAP), Materials Research Center (MRC), Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: t-marks@northwestern.edu; a-facchetti@northwestern.edu
cFlexterra Inc., 8025 Lamon Avenue, Skokie, IL 60077, USA
First published on 27th September 2021
Abstract
Understanding the structure–property relationships in antiaromatic molecules is crucial for controlling their electronic properties and designing new organic optoelectronic materials. Here we report the design, synthesis, and characterization of three new antiaromatic molecules (Pn, n = 1–4) based on the pentalene (P) antiaromatic core, to investigate how electron-donating and electron-accepting substituents affect P1–P4 properties. As expected, the optical, HOMO and LUMO energy levels and electronic structure are greatly modulated by core substitution. Compared to the unsubstituted compound (P1), P3 and P4 containing strong electron-withdrawing units reduced antiaromaticity as assessed by nucleus-independent chemical shift (NICS) calculations compared with P2, which is functionalized with strong electron-donating units, showing that substitution strongly tunes local antiaromaticity. Organic field-effect transistors (OFETs) fabricated using these materials indicate that P2 has an average hole mobility of ∼10−4 cm2 V−1 s−1 while P3 has an average electron mobility of up to 0.03 cm2 V−1 s−1, versus FET-inactive P1. Therefore, introduction of strong π-extended electron-withdrawing or electron-donating substituents onto an antiaromatic core is an effective strategy to switch-on charge transport capacity.
1 Introduction
Aromaticity is a fundamentally important and intriguing property of numerous organic chemical structures and has stimulated a myriad of experimental and theoretical investigations.1–3 Since Hückel first codified the (4n + 2) π-electron rule for monocyclic rings as the guideline for aromaticity,4 additional aromatic mono/oligocyclic rings have been reported5–7 and investigated in research fields as diverse as materials science,8–10 photonics,11–13 agriculture, pharmaceuticals,14 and biomedicine.15 Antiaromaticity, where (4n) π-electrons are present in a conjugated cyclic structure, as the inverse concept of aromaticity, was first proposed by Breslow in 1967.16 Unlike aromaticity, antiaromaticity leads to structural destabilization, paratropic ring currents, and π-bond localization.17–19 Importantly, it has been suggested that antiaromaticity can compress the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) energy gap and potentially promote carrier mobility in organic semiconductors, which is desirable for application in a number of organic electronic device classes,20–22 such as organic solar cells (OSCs)23,24 and organic field-effect transistors (OFETs).20,25–28 Through important design contributions both from synthetic and theoretical chemists, diverse antiaromatic molecule classes have been developed, including pentalenes,29,30 indacenes,31,32 cyclooctatetraenes,33,34 and their derivatives.35–37 However, compared to the literature of aromatic compounds, optoelectronic investigations of antiaromatic materials remain far more limited, primarily due to the modest stability of typical antiaromatic compounds38 and challenges in their chemically modification, which has typically involved fusion with aromatic rings,19,39 replacement of C–C bonds with isosteric B–N bonds,40 and aromatic ring functionalization.41,42Tuning the antiaromatic character and substitution pattern in antiaromatic molecules via structural modification is essential for achieving specific functions. For example, Xia et al. tuned the aromatic character of fused cyclobutadienoids by changing the fusion pattern43 while Yasuda et al. enhanced the antiaromatic character of pentalene by benzoannulation.44 Yamaguchi et al. reported that the antiaromaticity and electronic properties of the pentalene core could be widely tuned by the degree of aromatic character of the peripherally fused rings.29 Nevertheless, the above-mentioned molecular strategies required considerable demanding synthetic efforts.
The realization of organic molecules having optimally placed selected donor (D)–acceptor (A) moieties/functionalities is a proven design strategy to achieve organic semiconductors for opto-electronics since the electronic structure, optical absorption, energy levels, and solid-state packing can be tuned to the greatest extent. Indeed, numerous high-performance organic semiconductors for OSCs,45–47 OFETs,48–50 organic light-emitting diodes (OLEDs),51 and organic sensors52,53 have such D–A cores and/or substituent patterns.
In marked contrast, D–A antiaromatic compounds are extremely rare.54–58 Furthermore, to the best of our knowledge, there have been no systematic investigations of semiconducting D–A antiaromatic structures. Pentalene, consisting of two fused cyclopentadiene rings with 8π electrons, is one of the most important antiaromatic rings.59 Although pentalene itself is thermally unstable and rapidly dimerizes above −196 °C,60 a growing list of aryl-fused pentalenes are known to be quite stable owing to the delocalization of the pentalene double bonds into the adjacent aromatic subunits, such as dibenzo[a,e]pentalene (DBP),61,62 dithieno[a,e]pentalene (DTP),63 naphthopentalene (NPP).64 In this study, we synthesized the new molecular structures P2–P4 using the DTP skeleton as the antiaromatic core, benzene rings as π-bridge units, and N,N-dimethylamine, dicycanovinylindan-1-one (IC), and 5,6-difluroinedicycanovinylindan-1-one (IC2F) as the terminal donor and acceptor substituents, respectively (Fig. 1a). Our results indicate that introducing electron-donating units into the DTP core enhances the antiaromaticity of the pentalene ring, while functionalization with electron-withdrawing units reduces pentalene antiaromaticity as accessed by NICS calculations. The new compounds were characterized by single-crystal X-ray diffraction analysis, ultraviolet-visible (UV-Vis) spectrophotometry, and cyclic voltammetry (CV) measurements. Furthermore, the D/A substituents significantly modulate the HOMO and LUMO energy levels from (−5.16)–(−3.39) eV to (−4.64)–(−3.73) eV, respectively, as well as tune optical absorption maxima by more than 120 nm. Finally, the charge transport properties of P3 were measured in TFT devices, demonstrating that these compounds can function as n-type semiconductors (vide infra).
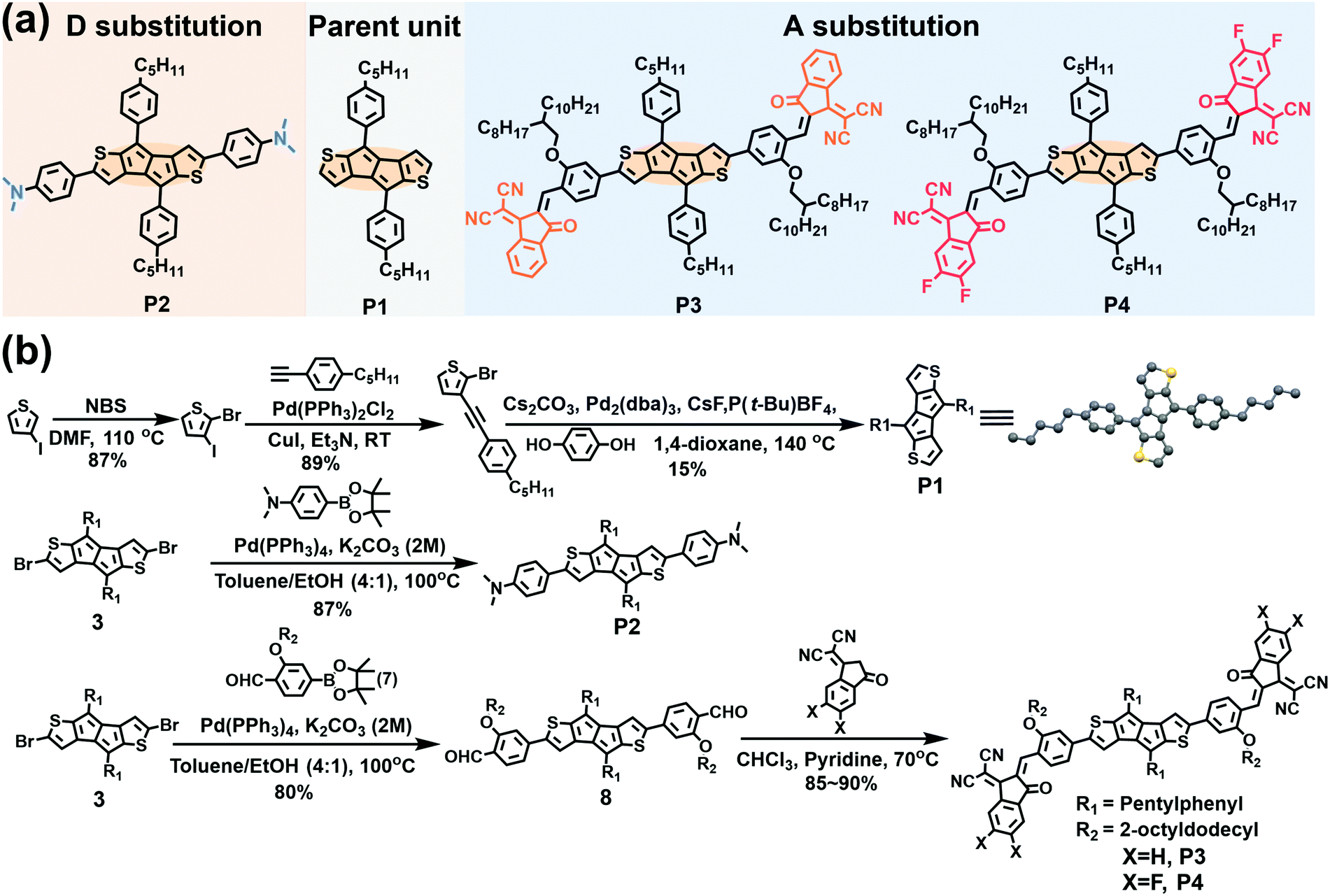 | ||
| Fig. 1 (a) The molecular structures and (b) the synthetic routes to the compounds discussed in this study. | ||
2 Results and discussion
2.1 Molecular design and synthesis
The parent P1 unit and new antiaromatic molecules P2–P4 (Fig. 1a) were synthesized according to the routes shown in Fig. 1b using the stable and easily accessible DTP unit as the central antiaromatic core, the phenyl ring as a π-bridge, and strong electron-donating N,N-dimethylamine and strong electron-withdrawing IC and IC2F as terminal groups in the para-phenyl position. To ensure processability in common organic solvents, long alkoxy side chains (2-octyldodecyl) were introduced at the phenyl meta-position of the π-extended molecules P3 and P4. P1 was synthesized according to the literature by replacing the hexylphenyl with a pentylphenyl moiety.63 The key intermediates 3 and 7 were synthesized according to literature methodologies.63,65 Suzuki coupling reactions of compounds 7 and 4-aminophenyl boronic acid pinacol ester with compound 3 afforded compounds 8 and P2 in 85% and 80% yields, respectively. Finally, compounds P3 and P4 were obtained as black solids through Knoevenagel condensation of compound 8 with IC and IC2F (85% and 90% yields), respectively. The molecular structures of compounds P1–P4 were accessed by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (see Section S7 in the ESI†). The thermal properties of pentalene compounds were characterized by Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) (Fig. S7 and S8, Table S2, ESI†). TGA demonstrates that the thermolysis onset temperatures of P1, P2, P3 and P4 are 300, 300, 320, and 306 °C, respectively (Fig. S7 and Table S2, ESI†). In addition, DSC analysis indicates clear melting points for P1, P2, and P4 (171, 261 and 189 °C), respectively, but not for P3 (Fig. S8 and Table S2, ESI†). The TGA and DSC data indicate that P1–P4 possess good thermal stability, necessary for OFET and OSC fabrication.2.2 Single-crystal structural analysis
We attempted to grow single crystals of all compounds using various solvent systems and succeeded in obtaining single crystals suitable for X-ray diffraction analysis for P1 and P2. The crystal structures of P1 and P2, shown in Fig. 2 and Fig. S2 (ESI†), indicate that both compounds are substantially planar with the pentylphenyl group having moderate dihedral angles (∼30° for P1 and P2) with respect to the DTP core. Both P1 and P2 pentalene cores exhibit the presence of double and single-bond alternation, a common characteristic of antiaromatic molecules,41,62 demonstrating that the intrinsic pentalene antiaromaticity is not diminished upon introducing strong electron-donating D substituents. However, the degree of bond length alternation in the pentalene unit of P2 (1.383(3) vs. 1.479(3) Å) is slightly reduced compared to that in P1 (1.373(3) vs. 1.484(3) Å), suggesting that the π-electron delocalization in the pentalene cores of P2 is greater than that in P1, which may be due to the greater π-system extension in P2. Interestingly, the pentalene cores in P1 molecules do not stack closely and exhibit only weak secondary π-edge interactions, with the minimum core–core π–π intermolecular distance being ∼3.73 Å. In contrast, the P2 molecules stack in a brick-wall fashion (Fig. 2b, c, and Fig. S3, ESI†) with a minimum π–π core–core distance of only ∼3.37 Å, demonstrating stronger intermolecular interactions, likely due to the more expansive conjugated core.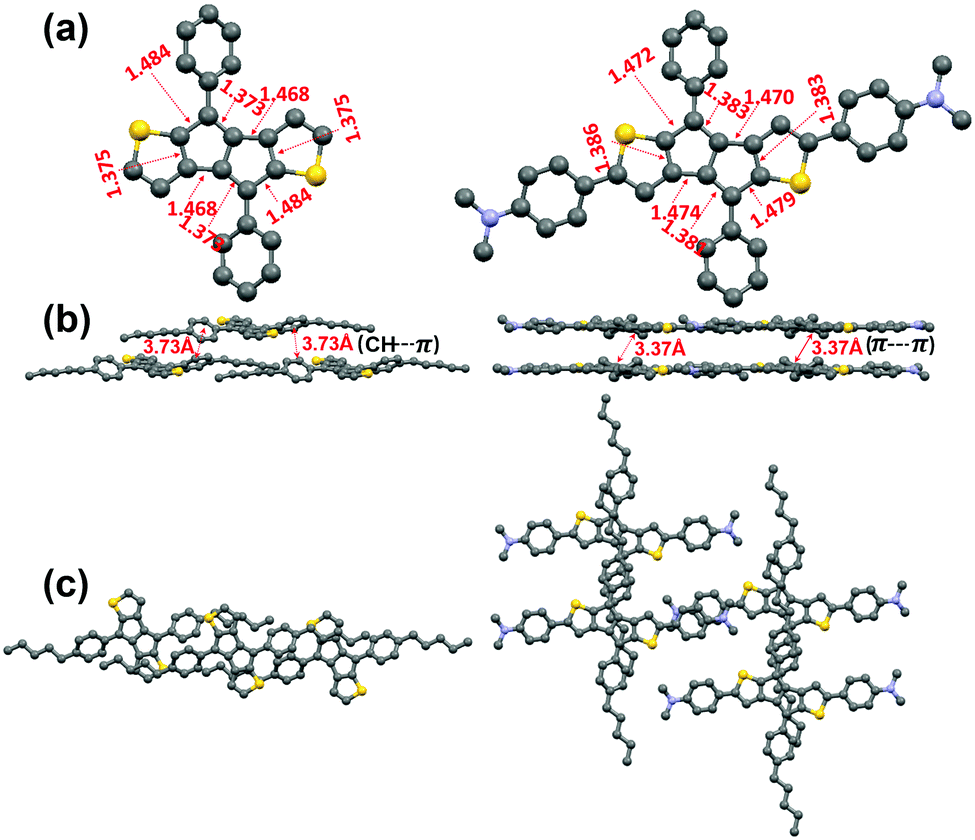 | ||
| Fig. 2 X-Ray crystal structures of P1 (left) and P2 (right). (a) Top view and (b) Packing diagram showing P1 π-edge and P2 π–π interactions (b and c). In Fig. 2a the n-pentyl chains are omitted for ease of viewing. | ||
2.3 Optoelectronic properties
The optical properties of the present compounds were investigated by ultraviolet-visible (UV-Vis) absorption spectra in both dilute CHCl3 solutions (5 × 10−6 M) and as thin-films spin-coated from chloroform solutions. The solution UV-Vis spectra in Fig. 3a indicate that all compounds have three distinct absorption bands, one at low energies, ∼500 nm for P1; 583–646 nm for P2–P4, one at intermediate energies in the 380–470 nm range, and a set at higher energies ranging from 330 to 360 nm. To assign these transitions, we carried out electronic structure computations at the B3LYP/6-311G(d) level. To reduce the computational demands, the long side chains of all molecules were replaced by methyl or methoxy groups. As shown in Fig. S9 (ESI†), the low energy absorption bands of P1–P4 are assigned to HOMO−1 → LUMO transitions. In contrast to P1 in which the low energy absorption is assigned to a locally excited (LE) transition, in P2–P4 they are assigned to charge transfer (CT) transitions (Fig. S10, ESI†). The second absorption bands from 380 to 470 nm can be assigned to the HOMO−2 → LUMO transition, while the highest energy absorption bands from 330 to 360 nm can be primarily attributed to the HOMO → LUMO+1, HOMO−4 → LUMO+1, HOMO−14→ LUMO, and HOMO−16 → LUMO transitions for P1 to P4, respectively. Note that according to Fig. S9 (ESI†), the S0 → S1 transition bands of P1–P4, assignable to the HOMO → LUMO transition are symmetry–forbidden, which is typical of antiaromatic molecules.64,66 Compared with the P1 S0 → S2 transition, P2–P4 exhibit red-shifted low-energy absorption ranging from 113 nm to 176 nm, which is assigned to intramolecular charge transfer (ICT) between the central core and the D/A substituents, a common feature of D–A structured molecules.67 In addition, as shown in Fig. 3a, the P4 low-energy absorption is redshifted by ∼20 nm vs. the P3 in S0 → S2 transition, which is attributed to the stronger ICT effect caused by the fluoro-substitution.68 Interestingly, compared to the P1 S0 → S2 transition, the introduction of electron-deficient units into the DTP core in P3 and P4 enhances molar absorption coefficient, consistent with the DFT results (Fig. S9, ESI†). These data demonstrate that introducing D–A motifs into an antiaromatic molecular unit is effective in overcoming the low absorptivity of antiaromatic molecules.29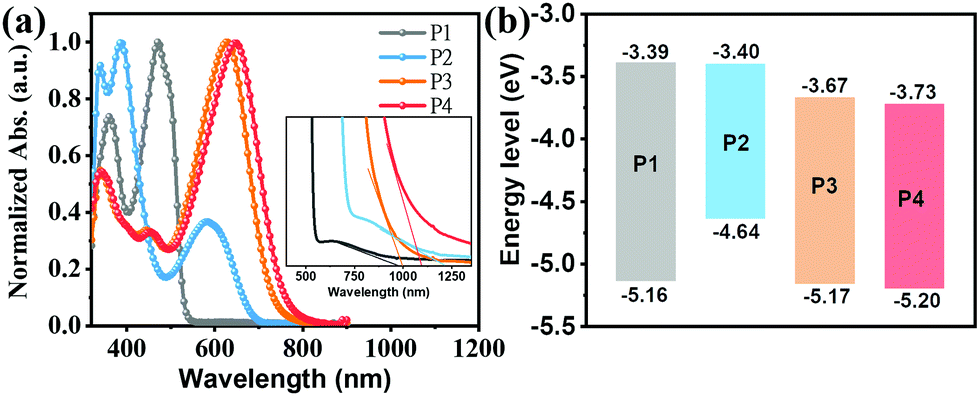 | ||
| Fig. 3 (a) Solution optical absorption spectra (inset shows a magnified view at long wavelengths) and (b) energy level diagram of the indicated compounds. | ||
The redox properties of P1–P4 were investigated by cyclic voltammetry (CV) measurements in CH2Cl2 (Fig. 3b and Fig. S6 (ESI†); Table 1). From the CV data, the frontier molecular orbital energy levels were estimated from the redox potentials. Thus, the oxidation and reduction potentials of P1–P4 were found to be +0.36, −0.16, +0.37, and +0.40 eV, and −1.41, −1.40, −1.13, −1.03 eV, respectively. Thus, the HOMO energy levels of P1, P3, and P4 remain in a narrow range (∼−5.16 eV) while that of P2 is substantially higher (−4.64 eV). In contrast, the LUMO energy levels of P1 and P2 (∼−3.40 eV) and those of P3 and P4 (∼−3.70 eV) are similar, but far deeper. These results are in agreement with the presence of the electron-donating substituents (N(CH3)2) in P2 and the strong electron-withdrawing substituents (IC and IC2F) in P3 and P4, respectively. The optical bandgap substantiates this picture as well (Table 1). These data indicate that P2 could be a hole transporting/donor semiconductor while P3 and P4 could be electron transporting/acceptor semiconductors in OFET/OSC devices.
| Compd |
λ
abs
(nm) (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
λ abs (nm) | E ox (eV) | E red (eV) | HOMOc (eV) | LUMOd (eV) |
E
cvge (eV) |
HOMOf (eV) | LUMOg (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in 5.0 × 10−6 mol L−1 CHCl3 solution. b Thin films cast from CHCl3 solution onto quartz substrates. c HOMO energy levels were estimated from the equation HOMO = −(Eox + 4.80) eV. d LUMO energy levels were estimated from the equation LUMO = −(Ered + 4.80) eV. e E cvg = ELUMO − EHOMO. f HOMO energy levels computed by DFT. g LUMO energy levels computed by DFT. | |||||||||
| P1 | 470 (4.26) | 513 | 0.36 | −1.41 | −5.16 | −3.39 | 1.77 | −5.13 | −2.91 |
| P2 | 583 (4.09) | 622 | −0.16 | −1.40 | −4.64 | −3.40 | 1.24 | −4.60 | −2.86 |
| P3 | 626 (4.89) | 656 | 0.37 | −1.13 | −5.17 | −3.67 | 1.50 | −5.26 | −3.37 |
| P4 | 646 (4.98) | 683 | 0.40 | −1.03 | −5.20 | −3.73 | 1.47 | −5.30 | −3.45 |
2.4 Aromaticity studies
To investigate the effect of the D–A structure on the aromatic character of the DTP central unit, we carried out nucleus-independent chemical shift (NICS-XY) surveys and computed the induced ground state (S0) current density (ACID). Further details are shown in Section S3.1 of the ESI.† To summarize the literature, antiaromatic rings are characterized by (1) paratropic ring currents as determined by positive NICS values, and (2) anticlockwise ring currents revealed via ACID plots.19 As shown in Fig. 4 and Table S4 (ESI†), the NICS value of the pentalene core (a and a′ rings) of P1 (δ18.2 ppm) is larger than that of P3 (δ15.3 ppm). Furthermore, P4 with a more electron-deficient unit, exhibits a lower NICS value than P3 (δ14.8 vs. δ15.3 ppm). These results suggest that the introduction of an electron-deficient group on the DTP core can reduce the pentalene unit antiaromatic character, with the antiaromatic character showing a negative relationship with the degree of A unit electron-deficiency. In contrast, P2 containing a strong electron-donating group exhibits an enhanced antiaromatic character with a higher NICS value of δ20.4 ppm. However, compared to P1, the effective conjugation of both D–A P3 and P4 compounds is extended. Thus, to understand the effects of extended conjugation on the antiaromaticity of the pentalene core, we carried out additional computations for other pentalene compounds with a P1 core and having –OCH3 (P1–OCH3, a weak electron-donating group) and –NH(CH3)2+ (P1–NH(CH3)2+, a strong electron-withdrawing group) substituents. The results, shown in Fig. S11 (ESI†), indicate that P1–OCH3 exhibits similar NICS values to that of P1 despite the former having a more extended effective conjugation than P1, indicating that extended effective conjugation minimally affects antiaromaticity in these systems. However, computations on electron-deficient compound P1–NH(CH3)2+ indicate that it exhibits considerably reduced NICS values vs.P1, further confirming the present conclusions that the introduction of electron-deficient substitutes will decrease antiaromaticity. These results argue that the pentalene core antiaromaticity can be tuned via the electron-donation ability or degree of electron-deficiency of the D or A units in the D–A structure. To understand the ring currents in the present compounds, ACID calculations were also carried out to visualize the local paratropic and diatropic ring currents induced by local antiaromaticity and aromaticity. As shown in Fig. 5, a counterclockwise ring current is observed in both pentalene five-membered rings for all compounds, indicative of the strong paratropicity, and thus the antiaromaticity of this unit, which is consistent with the structural results of the single crystal diffraction analysis. In contrast, a clockwise diatropic ring current was visualized in the fused thiophene, in agreement with the NICS calculations. | ||
| Fig. 4 The NICS-XY scans (a) and pentalene core NICS values (b) of the indicated compounds involved in this study. | ||
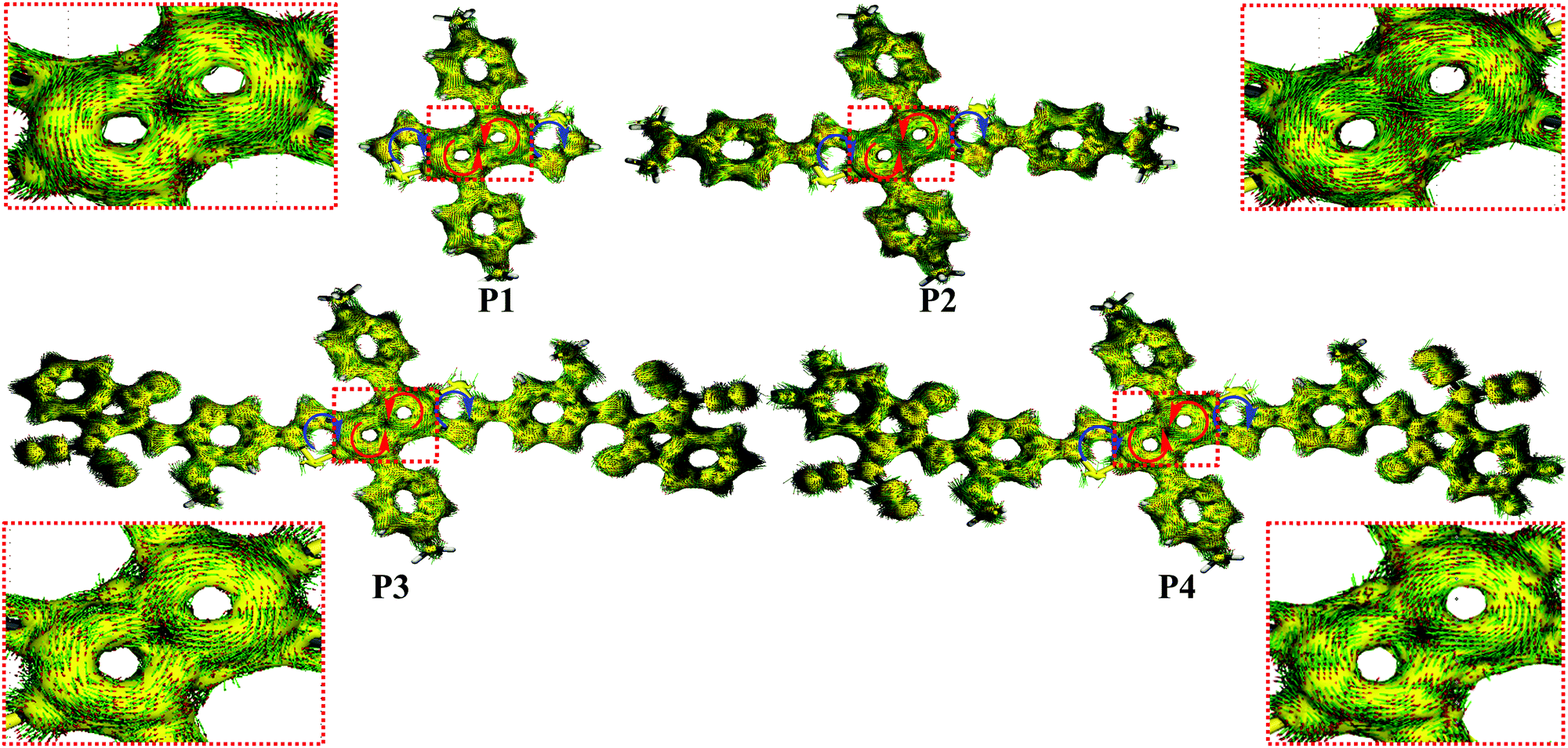 | ||
| Fig. 5 Computed ring currents for the indicated molecules. | ||
2.5 OFET transport characterization
The charge transport properties of P1–P4 were assessed by fabricating thin-film FET devices. Unfortunately, P4 is too insoluble to afford continuous films suitable for FET evaluation.69,70 For P1 and P3 top-gate/bottom-contact (TGBC) FETs were characterized in a device configuration of, substrate (glass)/source–drain (Cr/Au/thiol)/semiconductor/dielectric (PMMA)/gate (Al). Here, 4-methoxy thiophenol was used as a self-assembled monolayer (SAM) to modify the source and drain electrode surfaces to increase charge injection.71 However, since P2 has good solubility in common solvents used for depositing the PMMA dielectric, such as 2-butanone and ethyl acetate, Bottom-Gate Top-Contact (BGTC) FET structures [(Si)/dielectric (SiO2)/semiconductor/source–drain (Au)] were used for this study.Based on survey experiments, P1 was found to be FET-inactive while P2 is a p-type semiconductor with an average hole mobility of ∼10−4 cm2 V−1 s−1 (Fig. S12 and S13, ESI†). Compound P3 could be effectively processed by spin-coating from CHCl3 solutions (4 mg mL−1) and the as-cast films exhibit n-type transport with an electron mobility (μe) of 0.014 cm2 V−1 s−1. The OFET performance of P3 was further optimized by thermally annealing the semiconductor films at various temperatures; see details in Fig. 6a and S14 and Table S5 (ESI†). Thus, it was found that the μe of P3 devices annealed over the 130–200 °C range maximizes to 0.03 cm2 V−1 s−1 after thermal annealing at 180 °C for then falls to 0.025 cm2 V−1 s−1 at 200 °C.
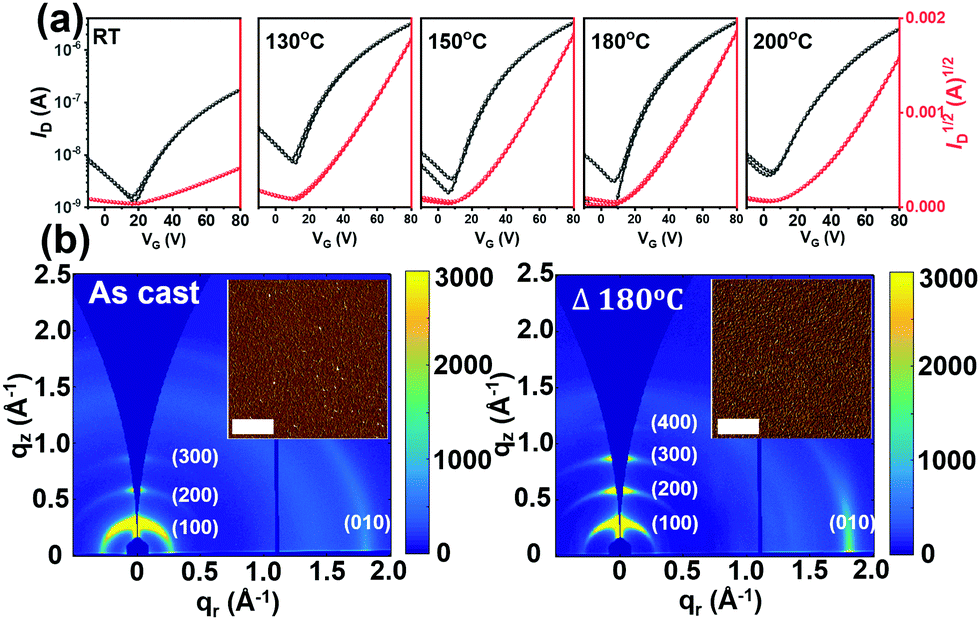 | ||
| Fig. 6 (a) Transfer plots of P3-based OFET devices of P3 upon thermal annealing at the indicated temperatures, RT, 130, 150, 180, and 200 °C. (b) 2D-GIWAXS and AFM (insert) images of P3 films as casted and upon annealing at 180 °C. The scale bar of the AFM images is 1 μm. | ||
To understand the effect of thermal annealing on the P3 semiconductor film structure and charge transport properties, AFM and two-dimensional grazing incidence wide-angle X-ray scattering (2D-GIWAXS) measurements were carried out. The AFM height images (Fig. S16, ESI†) indicate that the pristine and all thermally annealed P3 films exhibit smooth surfaces with small roughness values (σRMS) of 0.55–1.08 nm. Compared with the pristine P3 films, which exhibit small aggregates, an obvious fibrillar nanostructure is observed for all the annealed films, suggesting greater crystallinity, which was further confirmed by the 2D-GIWAXS results. For better comparison, the 2D-GIWAXS images of the pristine film and the optimized film annealed at 180 °C are shown in Fig. 6b and the corresponding one-dimensional line cuts are shown in Fig. S17 (ESI†). P3 exhibits a pronounced (100) reflection at qxy = 0.30 Å−1 and a (010) reflection at qz = 1.78 Å−1, corresponding to the lamellar stacking distance (dl) of 20.93 Å and π–π stacking distance (dπ) of 3.53 Å. Upon thermal annealing at 180 °C, the diffraction patterns are dramatically intensified, a result in agreement with the AFM analysis and enhanced OTFT electron mobilities.
Finally, we note that the molecular design of P3 and P4, and the position of their LUMO energies, suggests potential in bulk-heterojunction solar cells. Thus, we screened blends based on two donor polymers, P3HT and PTB7; however the OSC power conversion efficiencies were very low (Fig. S15b, c and Table S6, ESI†) and at the best approaching ∼0.02%. The poor performance reflects the low solubility of P4 and the poor energy matching (high HOMO, low bandgap) of these molecules with common donor polymers, preventing efficient charge separation (see details in the ESI†). Furthermore, the frontier molecular orbital topologies of P3 and P4, and particularly the LUMO, are mainly localized on the pentalene core, which differs considerably from that of high performance NFAs (e.g., Y6) where the LUMO delocalizes on whole molecule enabling good electron transport.
3. Conclusions
A series of new antiaromatic compounds with pentalene antiaromatic cores, substituted with π-extended electron-donating and electron-accepting groups, were synthesized and characterized by a variety of methods. From optical and CV measurements, the bandgap and HOMO and LUMO energy levels can be efficiently tuned by introducing electron-donating and electron-withdrawing units, with the latter resulting in strong, red-shifted absorption and deeper energy levels. Single crystal diffraction data and DFT calculations demonstrate that introduction of strong electron-donating units enhances the antiaromatic character of the pentalene central core, while the introduction of electron-withdrawing units diminishes it. Charge transport in OFETs was also investigated, demonstrating that introduction of strong electron-donating substituent in P2 and electron-withdrawing units in P3 results in p-type and n-type charge transport, respectively, in accord with common trends for aromatic π-conjugated cores. Thus, these results indicate that the design and exploration of new antiaromatic molecular cores is promising for further expanding the scope of organic semiconductors in opto-electronics technologies.4. Experimental
4,8-Di(4-pentylphenyl)-2,6-dithia-dicyclopenta[a,e]-pentalene (P1)
A mixture of 2 (3.6 g, 10.8 mmol), hydroquinone (2.38 g, 21.6 mmol), Cs2CO3 (7.04 g, 21.6 mmol), CsF (3.28 g, 21.6 mmol), Pd2(dba)3 (197.8 mg, 0.216 mmol), and [(t-Bu)3PH]BF4 (188 mg, 0.65 mmol) in anhydrous 1,4-dioxane (80 mL) was heated at 140 °C under Ar for 48 h. After cooling to room temperature, the mixture was filtered and the collected solid washed thoroughly with chloroform. The filtrate was then collected and evaporated to dryness. The residue was purified by chromatography on silica gel to give the titled product as a brown solid (0.40 g, 14.6%).1H NMR (500 MHz, CDCl3, δ): 7.53 (d, J = 8.0 Hz, 4H, ArH), 7.24 (d, J = 8.0 Hz, 4H, ArH), 6.81 (d, J = 4.8 Hz, 2H, ArH), 6.70 (d, J = 4.8 Hz, 2H, ArH), 2.64 (t, J = 8.0 Hz, 4H, CH2), 1.68–1.61 (m, 4H, CH2), 1.59–1.31 (m, 8H, CH2), 0.91 (t, J = 7.6 Hz, 6H, CH3). 13C NMR (125 MHz, CDCl3, δ): 148.16, 144.80, 142.57, 139.04, 137.00, 130.60, 128.83, 127.64, 127.17, 121.25, 35.96, 31.55, 30.97, 22.55, 14.03. MALDI-TOF MS: calcd for C34H35S2 (M + H+): 507.2180, found (M + H+): 507.2177.4,4′-(4,8-Bis(4-pentylphenyl)pentaleno[2,1-b:5,4-b′]dithiophene-2,6-diyl)bis(N,N-dimethylaniline) (P2)
A mixture of 3 (100 mg, 0.15 mmol), N,N-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-aniline (111.5 mg, 0.45 mmol), Pd(PPh3)4 (6.4 mg, 0.006 mmol) and K2CO3 (2 M) (0.4 mL, 0.8 mmol) in a solvent mixture of toluene (10 mL) and water (2 mL) was heated at 100 °C under argon for 24 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was purified by chromatography on silica gel to give the product as a dark green solid (89 mg, 80%).1H NMR (600 MHz, C2D2Cl4, δ): 7.63 (d, J = 7.8 Hz, 4H, ArH), 7.41 (d, J = 7.8 Hz, 4H, ArH), 7.33 (d, J = 7.8 Hz, 4H, ArH), 6.80 (s, 2H, ArH), 6.75 (d, J = 7.8 Hz, 4H, ArH), 3.02 (s, 12H, CH3), 2.74 (t, J = 7.8 Hz, 4H, CH2), 1.82–1.73 (m, 4H, CH2), 1.53–1.42 (m, 8H, CH2), 1.00 (t, J = 7.2 Hz, CH3). 13C NMR (150 MHz, C2D2Cl4, δ):150.4, 144.8, 131.0, 128.8, 127.9, 126.2, 123.6, 115.9, 112.8, 40.4, 36.0, 31.6, 30.6, 22.5, 13.9. MALDI-TOF MS: calcd for C50H52N2S2 (M+): 744.3572, found (M+): 744.3568.2,2′-((2Z,2′Z)-(((4,8-Bis(4-pentylphenyl)pentaleno[2,1-b:5,4-b′]dithiophene-2,6-diyl)bis(2-((2-octyldodecyl)oxy)-4,1-phenylene))bis(methanylylidene))bis(3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (P3)
Pyridine (0.1 mL) was added to a solution of 8 (100 mg, 0.076 mmol) and IC (118 mg, 0.62 mmol) in dry CHCl3 (10 mL) under argon. The mixture was heated to reflux for 8 h and then allowed to cool to room temperature. After addition of 20 mL methanol a solid formed which was collected by filtration. The crude product was purified by recrystallization with mixture of chloroform and methanol to obtain the pure compound as a dark green solid (107 mg, 85%).1H NMR (600 MHz, C2D2Cl4, δ): 9.10 (s, 2H, CH), 8.75 (d, J = 7.8 Hz, 2H, ArH), 8.50 (d, J = 8.4 Hz, 2H, ArH), 7.97 (d, J = 7.2 Hz, 2H, ArH), 7.85 (t, J = 7.8 Hz, 2H, ArH), 7.80 (t, J = 7.2 Hz, 2H, ArH), 7.69 (d, J = 7.8 Hz, 4H, ArH), 7.42 (d, J = 7.8 Hz, 4H, ArH), 7.27 (d, J = 8.4 Hz, 2H, ArH), 7.18 (s, 2H, ArH), 7.06 (s, 2H, ArH), 4.10 (d, J = 6.0 Hz, 4H, CH2), 2.78 (t, J = 7.8 Hz, 4H, CH2), 2.02–1.97 (m, 2H, CH), 1.83–1.77 (m, 4H, CH2), 1.60–1.54 (m, 4H, CH2), 1.54–1.44 (m, 16H, CH2), 1.41–1.26 (m, 48H, CH2), 1.05–0.92 (m, 18H, CH3). 13C NMR (150 MHz, C2D2Cl4, δ): 180.0, 161.0, 150.5, 150.4, 146.5, 144.0, 142.4, 141.3, 140.0, 139.5, 138.5, 137.8, 129.2, 128.2, 128.0, 124.2, 122.0, 121.8, 119.4, 119.0, 116.6, 114.0, 108.1, 73.1, 71.7, 36.1, 31.9, 31.8, 30.6, 30.0, 29.7, 29.3, 27.1, 22.6, 22.5, 14.0. MALDI-TOF MS: calcd for C112H130N4O4S2 (M+): 1658.9534, found (M+): 1658.9534.2,2′-((2Z,2′Z)-(((4,8-Bis(4-pentylphenyl)pentaleno[2,1-b:5,4-b′]dithiophene-2,6-diyl)bis(2-((2-octyldodecyl)oxy)-4,1-phenylene))bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (P4)
P4 was prepared as a dark solid with a yield of 90% using the same procedure used for the synthesis of P3, but with IC2F rather than IC as the reactant. 1H NMR (600 MHz, C2D2Cl4, δ): 9.12 (s, 2H, CH), 8.62–8.56 (m, 2H, ArH), 8.50 (d, J = 8.4 Hz, 2H, ArH), 7.72 (t, J = 7.8 Hz, 2H, ArH), 7.68 (d, J = 8.4 Hz, 2H, ArH), 7.42 (d, J = 7.8 Hz, 4H, ArH), 7.26 (d, J = 8.4 Hz, 2H, ArH), 7.18 (s, 2H, ArH), 7.06 (s, 2H, ArH), 4.10 (d, J = 5.4 Hz, 4H, CH2), 2.79 (t, J = 7.8 Hz, 4H, CH2), 2.03–1.98 (m, 2H, CH), 1.83–1.77 (m, 4H, CH2), 1.60–1.42 (m, 16H, CH2), 1.40–1.29 (m, 48H, CH2), 1.02 (t, J = 7.2 Hz, 6H, CH3), 0.96–0.92 (m,12H, CH3). 13C NMR (150 MHz, C2D2Cl4, δ): 184.1, 161.4, 150.9, 146.4, 146.0, 144.0, 142.5, 139.5, 138.3, 135.5, 130.1, 129.2, 128.0, 127.0, 121.6, 119.6, 116.8, 114.2, 113.5, 107.6, 73.2, 72.2, 36.1, 31.9, 31.8, 31.6, 30.6, 30.0, 29.6, 29.3, 27.1, 22.6, 22.5, 14.0, 13.9. MALDI-TOF MS: calcd for C112H126F4N4O4S2 (M+): 1730.9157, found (M+): 1730.9139.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support for this work from the National Natural Science Foundation of China (project no. 21875148) (Y. H., Z. L., J. W., J. L.). T. M., A. F., and Y. C., J. W. are grateful to the Center for Light Energy Activated Redox Processes (LEAP), an Energy Frontier Research Center funded by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under award DE-SC0001059 (J. W.: fabricating device and other characterization, Y. C. and G. Li: materials synthesis, A. F. and T. J. M.: project advising), AFOSR grant FA9550-18-1-0320, the Flexterra Corporation, and the Office of Naval Research Contract N00014-20-1-2116. The use of the Advanced Photon Source, an Office of Science User Facility operated by the US DOE Office of Science by Argonne National Laboratory, was supported by the US DOE under contract DE-AC02-06CH11357. J. W. acknowledges the joint-PhD program supported by the China Scholarship Council (No. 201906240142) for a fellowship.Notes and references
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- J. K. Pagano, J. Xie, K. A. Erickson, S. K. Cope, B. L. Scott, R. Wu, R. Waterman, D. E. Morris, P. Yang, L. Gagliardi and J. L. Kiplinger, Nature, 2020, 578, 563–567 CrossRef.
- J. Gomes and R. Mallion, Chem. Rev., 2001, 101, 1349–1384 CrossRef PubMed.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- M. J. Cook, A. R. Katritzky and P. Linda, Adv. Heterocycl. Chem., 1974, 17, 255–356 CrossRef.
- K. E. Horner and P. B. Karadakov, J. Org. Chem., 2013, 78, 8037–8043 CrossRef.
- K. Najmidin, A. Kerim, P. Abdirishit, H. Kalam and T. Tawar, J. Mol. Model., 2013, 19, 3529–3535 CrossRef.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- H. Usta, D. Kim, R. Ozdemir, Y. Zorlu, S. Kim, M. C. Ruiz Delgado, A. Harbuzaru, S. Kim, G. k. Demirel, J. Hong, Y. Ha, K. Cho, A. Facchetti and M. Kim, Chem. Mater., 2019, 31, 5254–5263 CrossRef.
- Y. Lin, A. Magomedov, Y. Firdaus, D. Kaltsas, A. El-Labban, H. Faber, D. R. Naphade, E. Yengel, X. Zheng, E. Yarali, N. Chaturvedi, K. Loganathan, D. Gkeka, S. H. AlShammari, O. M. Bakr, F. Laquai, L. Tsetseris, V. Getautis and T. D. Anthopoulos, ChemSusChem, 2021, 14, 3569–3978 CrossRef PubMed.
- K. Feng, H. Guo, J. Wang, Y. Shi, Z. Wu, M. Su, X. Zhang, J. H. Son, H. Y. Woo and X. Guo, J. Am. Chem. Soc., 2021, 143, 1539–1552 CrossRef.
- S. Shi, P. Chen, Y. Chen, K. Feng, B. Liu, J. Chen, Q. Liao, B. Tu, J. Luo, M. Su, H. Guo, M.-G. Kim, A. Facchetti and X. Guo, Adv. Mater., 2019, 31, 1905161 CrossRef.
- X. Zhang, L. Qin, J. Yu, Y. Li, Y. Wei, X. Liu, X. Lu, F. Gao and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 12475 CrossRef.
- A. G. A. Sá, A. C. de Meneses, P. H. H. de Araújo and D. de Oliveira, Trends Food Sci. Technol., 2017, 69, 95–105 CrossRef.
- P. Xing and Y. Zhao, Adv. Mater., 2016, 28, 7304–7339 CrossRef.
- R. Breslow, J. Brown and J. J. Gajewski, J. Am. Chem. Soc., 1967, 89, 4383–4390 CrossRef.
- J. L. Marshall, K. Uchida, C. K. Frederickson, C. Schutt, A. M. Zeidell, K. P. Goetz, T. W. Finn, K. Jarolimek, L. N. Zakharov, C. Risko, R. Herges, O. D. Jurchescu and M. M. Haley, Chem. Sci., 2016, 7, 5547–5558 RSC.
- X. Fu, Y. Meng, X. Li, M. Stępień and P. J. Chmielewski, Chem. Commun., 2018, 54, 2510–2513 RSC.
- A. Konishi, Y. Okada, M. Yasuda, A. Konishi, R. Kishi, M. Nakano and M. Yasuda, J. Am. Chem. Soc., 2019, 141, 560–571 CrossRef PubMed.
- Y. Sun, Y. Guo and Y. Liu, Mater. Sci. Eng., R, 2019, 136, 13–26 CrossRef.
- Z. Wen and J. I. C. Wu, Chem. Commun., 2020, 56, 2008–2011 RSC.
- X. Liu, Y. Wei, X. Zhang, L. Qin, Z. Wei and H. Huang, Sci. China: Chem., 2021, 64, 228–231 CrossRef CAS.
- H. U. Kim, J.-H. Kim, H. Suh, J. Kwak, D. Kim, A. C. Grimsdale, S. C. Yoon and D.-H. Hwang, Chem. Commun., 2013, 49, 10950–10952 RSC.
- Z. Zhang, H. Fan and X. Zhu, Org. Chem. Front., 2017, 4, 711–716 RSC.
- Z. Jin, Z. F. Yao, K. P. Barker, J. Pei and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 2034 CrossRef CAS PubMed.
- G. Dai, J. Chang, W. Zhang, S. Bai, K.-W. Huang, J. Xu and C. Chi, Chem. Commun., 2015, 51, 503–506 RSC.
- G. Dai, J. Chang, X. Shi, W. Zhang, B. Zheng, K. W. Huang and C. Chi, Chem. – Eur. J., 2015, 21, 2019–2028 CrossRef CAS.
- F. Yin, L. Wang, X. Yang, M. Liu, H. Geng, Y. Liao, Q. Liao and H. Fu, New J. Chem., 2020, 44, 17552–17557 RSC.
- H. Oshima, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2017, 56, 3270–3274 CrossRef CAS PubMed.
- A. Konishi, Y. Okada, M. Nakano, K. Sugisaki, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2017, 139, 15284–15287 CrossRef CAS PubMed.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS.
- J. Melidonie, J. Liu, Y. Fu, J. J. Weigand, R. Berger and X. Feng, J. Org. Chem., 2018, 83, 6633–6639 CrossRef CAS PubMed.
- F. G. Klärner, Angew. Chem., Int. Ed., 2001, 40, 3977–3981 CrossRef.
- T. Nishinaga, T. Ohmae, K. Aita, M. Takase, M. Iyoda, T. Arai and Y. Kunugi, Chem. Commun., 2013, 49, 5354–5356 RSC.
- T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura, H. Mori, E. Miyazaki and K. Takimiya, Angew. Chem., Int. Ed., 2010, 49, 7728–7732 CrossRef CAS PubMed.
- C. Liu, S. Xu, W. Zhu, X. Zhu, W. Hu, Z. Li and Z. Wang, Chem. – Eur. J., 2015, 21, 17016–17022 CrossRef CAS.
- M. Nakano, I. Osaka and K. Takimiya, J. Mater. Chem. C, 2015, 3, 283–290 RSC.
- A. D. Allen and T. T. Tidwell, Chem. Rev., 2001, 101, 1333–1348 CrossRef CAS PubMed.
- M. Baranac-Stojanovic, Chem. – Eur. J., 2019, 25, 9747–9757 CrossRef CAS PubMed.
- X.-Y. Wang, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 7668–7671 CrossRef CAS PubMed.
- J. Cao, G. London, O. Dumele, M. von Wantoch Rekowski, N. Trapp, L. Ruhlmann, C. Boudon, A. Stanger and F. Diederich, J. Am. Chem. Soc., 2015, 137, 7178–7188 CrossRef CAS PubMed.
- T. Yoshida, D. Sakamaki, S. Seki and H. Shinokubo, Chem. Commun., 2017, 53, 1112–1115 RSC.
- Z. Jin, Y. C. Teo, S. J. Teat and Y. Xia, J. Am. Chem. Soc., 2017, 139, 15933–15939 CrossRef CAS PubMed.
- A. Konishi, Y. Okada, R. Kishi, M. Nakano and M. Yasuda, J. Am. Chem. Soc., 2018, 141, 560–571 CrossRef.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- J. Wu, D. Yang, Q. Wang, L. Yang, H. Sasabe, T. Sano, J. Kido, Z. Lu and Y. Huang, J. Mater. Chem. A, 2018, 6, 5797–5806 RSC.
- L. Yang, W. Gu, L. Lv, Y. Chen, Y. Yang, P. Ye, J. Wu, L. Hong, A. Peng and H. Huang, Angew. Chem., Int. Ed., 2018, 57, 1096 CrossRef CAS.
- F. S. Melkonyan, W. Zhao, M. Drees, N. D. Eastham, M. J. Leonardi, M. R. Butler, Z. Chen, X. Yu, R. P. Chang, M. A. Ratner, A. F. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 6944–6947 CrossRef CAS.
- T. Hodsden, K. J. Thorley, J. Panidi, A. Basu, A. V. Marsh, H. Dai, A. J. P. White, C. Wang, W. Mitchell, F. Glöcklhofer, T. D. Anthopoulos and M. Heeney, Adv. Funct. Mater., 2020, 30, 2000325 CrossRef CAS.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef PubMed.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- M. He, C. Li, Z. Pang, K. Chen, Y. Tan, Y. Huang and Z. Lu, Chem. – Eur. J., 2020, 26, 12140–12144 CrossRef CAS.
- M. Nakano, I. Osaka, K. Takimiya and T. Koganezawa, J. Mater. Chem. C, 2014, 2, 64–70 RSC.
- D. C. Grenz, M. Schmidt, D. Kratzert and B. Esser, J. Org. Chem., 2018, 83, 656–663 CrossRef CAS.
- R. Ozdemir, S. Park, İ. Deneme, Y. Park, Y. Zorlu, H. A. Alidagi, K. Harmandar, C. Kim and H. Usta, Org. Chem. Front., 2018, 5, 2912–2924 RSC.
- Y. Zeng, R. Duan, Y. Guo, G. Han, Q. Li and Y. Yi, Chin. Chem. Lett., 2019, 30, 211–216 CrossRef CAS.
- D. J. Schipper, L. C. Moh, P. Müller and T. M. Swager, Angew. Chem., Int. Ed., 2014, 53, 5847–5851 CrossRef CAS PubMed.
- T. Bally, S. Chai, M. Neuenschwander and Z. Zhu, J. Am. Chem. Soc., 1997, 119, 1869–1875 CrossRef CAS.
- N. Baird and R. West, J. Am. Chem. Soc., 1971, 93, 3072–3073 CrossRef CAS.
- J. Zhao, K. Oniwa, N. Asao, Y. Yamamoto and T. Jin, J. Am. Chem. Soc., 2013, 135, 10222–10225 CrossRef CAS.
- A. Konishi, Y. Okada, M. Nakano, K. Sugisaki, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2017, 139, 15284–15287 CrossRef CAS PubMed.
- C. Hu and Q. Zhang, Chin. J. Chem., 2013, 31, 1404–1408 CrossRef CAS.
- S.-i. Kato, S. Kuwako, N. Takahashi, T. Kijima and Y. Nakamura, J. Org. Chem., 2016, 81, 7700–7710 CrossRef CAS.
- H. Xi, Z. Zhang, W. Zhang, M. Li, C. Lian, Q. Luo, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2019, 141, 18467–18474 CrossRef CAS PubMed.
- G. London, M. von Wantoch Rekowski, O. Dumele, W. B. Schweizer, J.-P. Gisselbrecht, C. Boudon and F. Diederich, Chem. Sci., 2014, 5, 965–972 RSC.
- S. Bradamante, A. Facchetti and G. A. Pagani, J. Phys. Org. Chem., 1997, 10, 514–524 CrossRef CAS.
- T. J. Aldrich, M. Matta, W. Zhu, S. M. Swick, C. L. Stern, G. C. Schatz, A. Facchetti, F. S. Melkonyan and T. J. Marks, J. Am. Chem. Soc., 2019, 141, 3274–3287 CrossRef CAS.
- J. H. Park, E. H. Jung, J. W. Jung and W. H. Jo, Adv. Mater., 2013, 25, 2583–2588 CrossRef CAS.
- W. Zhu, J. M. Alzola, T. J. Aldrich, K. L. Kohlstedt, D. Zheng, P. E. Hartnett, N. D. Eastham, W. Huang, G. Wang, R. M. Young, G. C. Schatz, M. R. Wasielewski, A. Facchetti, F. S. Melkonyan and T. J. Marks, ACS Energy Lett., 2019, 4, 2695–2702 CAS.
- J. Chen, X. Zhuang, W. Huang, M. Su, L.-w. Feng, S. M. Swick, G. Wang, Y. Chen, J. Yu, X. Guo, T. J. Marks and A. Facchetti, Chem. Mater., 2020, 32, 5317–5326 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, computation, and characterization details. CCDC 2042122 and 2042120. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc03156b |
| This journal is © The Royal Society of Chemistry 2022 |