
Exciton funneling amplified photoluminescence anisotropy in organic radical-doped microcrystals†
Zhonghao
Zhou
 ,
Chan
Qiao
,
Jiannian
Yao
,
Yongli
Yan
* and
Yong Sheng
Zhao
*
,
Chan
Qiao
,
Jiannian
Yao
,
Yongli
Yan
* and
Yong Sheng
Zhao
*
CAS Key Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: ylyan@iccas.ac.cn; yszhao@iccas.ac.cn
First published on 4th August 2021
Abstract
We demonstrate a controllable photoluminescence anisotropy amplification in organic luminescent radical-doped microcrystals via exciton funneling. The widely tunable doping ratio resulting from very similar molecular structures between hosts and guests leads to a freely tailorable exciton funneling process, which paves an avenue for the construction of high-performance polarizing optical elements.
Introduction
Polarization, which is a fundamental characteristic of light, plays an increasingly important role in various fields such as biosensing, quantum communication and 3D displays.1–3 Traditionally, polarized light is obtained using bulky linear polarizers, which not only limits the conversion efficiency due to the greatly reduced vertical component of light, but also hinders the development of miniaturized polarized optical devices.4,5 Utilizing polarization-sensitive materials that can directly generate polarized light is a promising route to manipulate photons at the micro/nanoscale.6–12 Benefiting from tailorable excited-state processes, abundant intermolecular interactions and high optical orientation,13–15 organic microcrystals provide an ideal platform to precisely control the polarization of photoluminescence (PL).16–19 Nevertheless, the emitted light from these materials generally suffers from a limited degree of polarization (DOP) owing to the relatively small projection of the transition dipole moment (μ) at the optical excitation plane, which impedes their practical applications in modern optoelectronics.A promising approach to improve the DOP is to concentrate polarized excitons by constructing light-harvesting systems composed of donor–acceptor pairs,20 which has been widely utilized to enhance acceptor emission through exciton funnelling.21–24 In general, aggregation of organic chromophores enables the formation of lower-energy excimer states surrounded by monomers with high-lying energy levels,25–28 which can serve as artificial light-harvesting antennas.29–31 Excitation light energy can be harvested by the antennas as linearly polarized excitons and subsequently concentrated in acceptors via exciton funneling (Fig. 1a),32,33 possibly triggering an amplification of PL anisotropy. However, the general differences between the molecular structures of guest and host compounds result in a limited doping concentration,34 which impedes the formation and modulation of energy transfer processes in crystalline states. Recently, it was reported that excimers with lower-energy were formed in organic radical precursor crystals doped with the corresponding luminescent radicals.35 The very similar molecular structures of the radical and corresponding precursor lead to a widely tunable doping ratio for free tailoring of excimer states, which is promising for the amplification and in situ modulation of PL anisotropy.
 | ||
| Fig. 1 (a) Diagram of exciton funneling in light-harvesting systems. (b and c) Molecular structures and unit cells of (b) PyBTM (CCDC† number: 1006484)36 and (c) αH-PyBTM (CCDC† number: 2093989). (d) SEM image of a typical PyBTM doped αH-PyBTM microplate. Scale bar is 10 μm. (e) Transmission electron microscopy image and SAED pattern of a typical PyBTM doped αH-PyBTM microplate. Scale bar is 1 μm. (f) SAED and (g) XRD patterns of pure and PyBTM doped αH-PyBTM microcrystals, with the doping ratio indicated. | ||
Herein, we propose a strategy to amplify PL anisotropy in organic radical-doped microcrystals, in which exciton funneling was exploited to modulate the DOP of excimer emission. Radical precursor microcrystals doped with the corresponding luminescent radicals were synthesized through a controllable liquid-phase self-assembly, where interactions between monomers and excimers were well-modulated by the doping concentration of the radical guests. In these composites, the radical monomers acted as antennas to efficiently capture the excitation energy, which was subsequently delivered to the excimers. Benefiting from the efficient coupling between exciton funneling and PL anisotropy transfer, the concentration of polarized excitons in the excimer states resulted in a distinct amplification of PL polarization and the DOP was successfully modulated by varying the temperature. These results not only offer a comprehensive understanding of exciton funneling in organic radical excimers but also provide guidance for the rational design of high-performance micro/nanoscale polarizing optical elements with specific functionalities.
Results and discussion
(3,5-Dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl (PyBTM, Fig. 1b),36 a typical luminescent organic radical,37,38 was selected as the guest compound and the corresponding precursor αH-PyBTM (Fig. 1c and Table S1, ESI†) was chosen as the host matrix to fabricate the light-harvesting microcrystals for the following reasons. (i) The molecular structures of PyBTM and αH-PyBTM are very similar, facilitating the free doping of PyBTM into αH-PyBTM without a fixed stoichiometric proportion. The widely tunable doping concentration of PyBTM molecules in the host crystals is critical to control the aggregation of PyBTM for tailorable interaction between the monomer states and excimer states.35,39 (ii) The rigid environment provided by αH-PyBTM crystals would improve the emission stability of PyBTM (Fig. S1, ESI†).40 (iii) The large spectroscopic overlap between the emission spectrum of αH-PyBTM and the absorption spectrum of PyBTM enables energy transfer from the host crystals to guest molecules (Fig. S2, ESI†). (iv) The excitation energy harvested by the monomers would concentrate on the lower-energy excimer states via exciton funneling, which might be beneficial for the amplification of PL anisotropy.αH-PyBTM microcrystals doped with controllable concentrations of PyBTM were synthesized through a liquid-phase self-assembly method (see the ESI† for details). The doping concentration of PyBTM was determined by measuring the absorption spectra of the corresponding samples.35 As shown in the scanning electron microscopy (SEM) image (Fig. S3, ESI†), the pure αH-PyBTM microcrystal has a well-defined plate-like morphology with smooth and flat facets. The doped αH-PyBTM microcrystal shares the same morphology (Fig. 1d), indicating that the introduction of PyBTM molecules does not affect the growth of the αH-PyBTM microcrystals. In Fig. 1e and f, all the αH-PyBTM microplates doped with different concentrations of PyBTM are high-quality crystals growing along the [100] direction. These microcrystals exhibit similar selected-area electron diffraction (SAED) patterns and X-ray diffraction (XRD) peaks (Fig. 1f and g), which indicates that the αH-PyBTM microcrystals retain their original crystal structure and relatively high crystallinity after being doped with PyBTM.41,42 As demonstrated in the confocal microscopy images (Fig. S4, ESI†), the PyBTM molecules are uniformly dispersed in the αH-PyBTM matrices, further verifying the excellent material compatibility between the guests and hosts.
The concentration of PyBTM in the host crystals is tunable over a wide range, enabling precise modulation of the aggregation of the radical molecules. As the PyBTM concentration increases, the emission colors of the doped crystals distinctly changed from yellow to red with decreased PL quantum yields (Fig. 2a and Fig. S5, ESI†), suggesting the formation of a new excited state at high concentrations. When the doping concentration is 1.1 wt%, the PL spectrum of the microcrystals was dominated by the monomer emission located at ∼570 nm (Fig. 2b). With increasing doping concentration from 1.9 wt% to 8.3 wt%, a broadband emission with a peak wavelength at approximately 680 nm appeared, while, in comparison, no obvious change was found in their absorption spectra (Fig. S6, ESI†). These results imply the formation of organic excimers in the PyBTM doped αH-PyBTM microcrystals.43
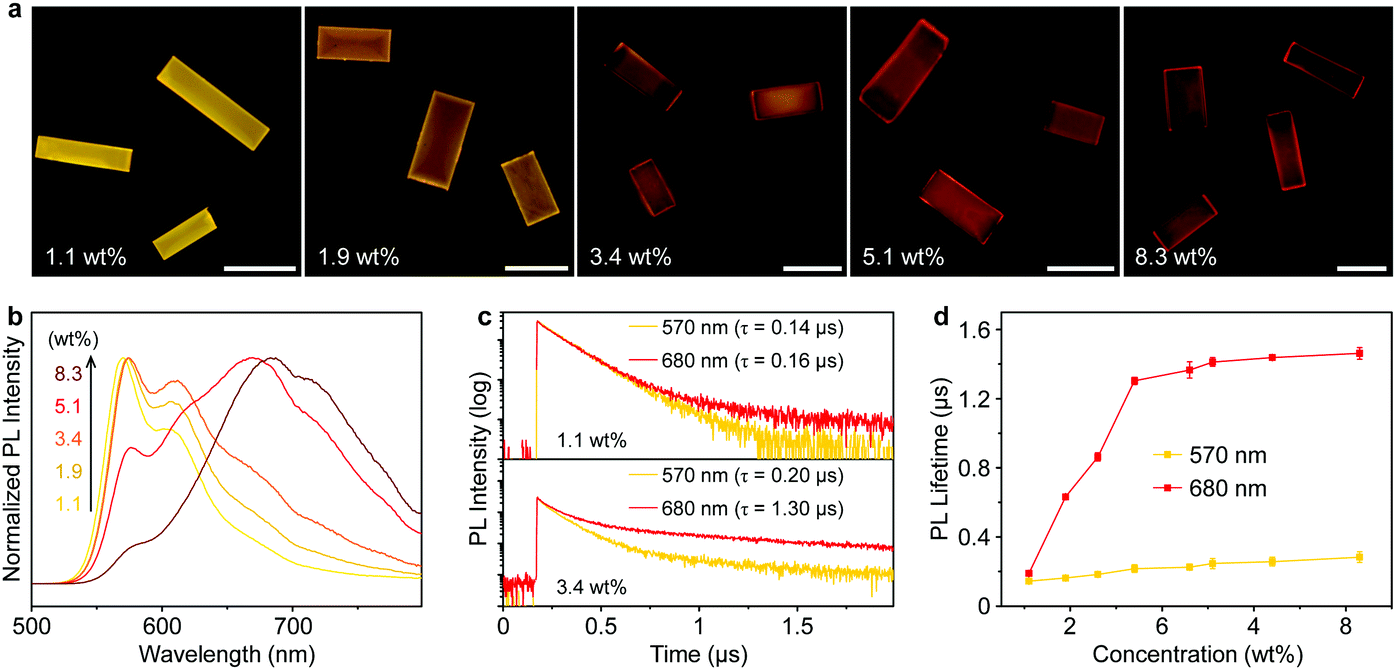 | ||
| Fig. 2 (a) PL images and (b) PL spectra of PyBTM doped αH-PyBTM microcrystals with different doping concentrations. Scale bars are 20 μm. (c) PL decay profiles of the PyBTM at the monomer emission band (570 nm, yellow) and the excimer emission band (680 nm, red), from the PyBTM doped αH-PyBTM microcrystals with concentrations of 1.1 wt% (top) and 8.3 wt% (bottom), respectively. (d) Concentration-dependent profiles of the average PL decay lifetime of PyBTM monitored at 570 nm (yellow) and 680 nm (red). Error bars represent three individual measurements. | ||
The excimer state can be distinguished from the monomer state by its relatively long lifetime.44 At a low concentration (1.1 wt%), the intermolecular distance among adjacent PyBTM molecules was large enough to prevent the formation of excimers and no obvious difference was observed between the PL lifetimes measured at 570 and 680 nm (top panel of Fig. 2c). When the doping concentration increased to 3.4 wt%, the decay time at 680 nm was distinctly longer than that at 570 nm (bottom panel of Fig. 2c), which clearly demonstrates the formation of PyBTM excimers. The prolonged lifetime at 680 nm could be attributed to the partial spin-forbidden characters in the excimer emission rather than thermally activated delayed fluorescence or triplet–triplet annihilation mechanisms. Fig. 2d summarizes the PL decay lifetimes of different PyBTM doped αH-PyBTM microcrystals monitored at 570 nm (yellow) and 680 nm (red). There was no obvious difference between the PL decay times at the two wavelengths when the doping ratio is 1.1 wt%, implying that the emission originates from an identical excited species. A dramatic increase in the PL lifetime at 680 nm was observed in the 1.9 wt% doped crystals, while that at 570 nm was almost unchanged. The decreasing short-lived component of the lifetime measured at 570 nm with concentration points to a migration of excitation energy from the wide-bandgap monomers to the lower-energy excimer states (Fig. S7, ESI†).45,46 The efficiency of energy transfer from monomers to excimers in the doped microcrystals was estimated to be ∼78% (Table S2, ESI†), which ensures an efficient exciton funneling to the excimer states in such light-harvesting microcrystals.
The anisotropic packing of molecules in organic single crystals leads to a distinct optical anisotropy in these materials.47–49 PL anisotropy measurements of the doped αH-PyBTM microplates were carried out on a home-made angle-resolved polarized PL system (Fig. S8, ESI†). In Fig. 3a, the 1.1 wt% doped αH-PyBTM microcrystal shows a strong polarized emission with a DOP of 0.65 at 570 nm according to DOP = (Imax − Imin)/(Imax + Imin),50 where Imax and Imin are defined as the maximum and minimum of the PL intensity, respectively. This relatively large DOP shows an obvious PL anisotropy of the monomer emission in PyBTM doped αH-PyBTM microcrystals. When the concentration of PyBTM in the host crystals increased to 3.4 wt%, excimer states located at ∼680 nm formed, accompanied with a DOP of ∼0.4 (Fig. 3b). This weak PL anisotropy of the excimer states could be attributed to the relatively small projection of the μ of the PyBTM excimers at the optical excitation plane.
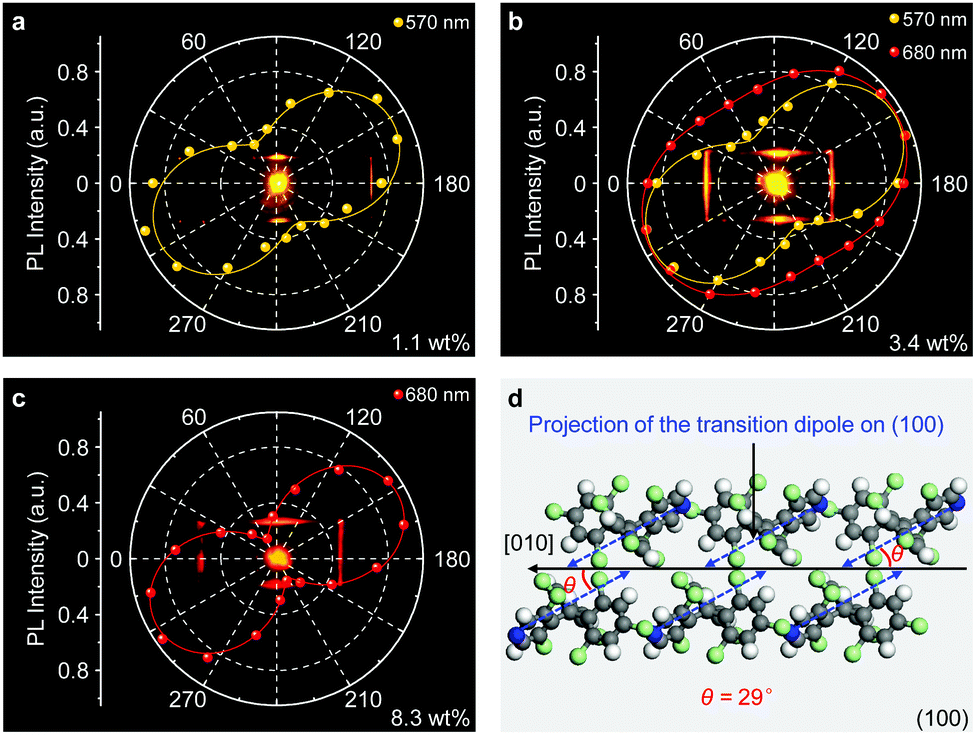 | ||
| Fig. 3 (a–c) Spatial polarization profiles of light emission from the αH-PyBTM microcrystal doped with (a) 1.1 wt%, (b) 3.4 wt%, (c) 8.3 wt% PyBTM. The plotted data were fitted with a sinusoidal function. (d) Schematic depiction of the spatial relationship between the αH-PyBTM molecular transition dipole moment (blue arrows) and the growth direction (black arrow) on the photoexcited plane of the microplate. | ||
Although the emission intensity of the doped microcrystals is partially quenched at higher doping concentrations (Fig. S5, ESI†), the DOP of the excimer emission at 680 nm dramatically increases to 0.85 in an 8.3 wt% PyBTM doped microcrystal (Fig. 3c), which is amplified 2-fold with respect to that in the 3.4 wt% one shown in Fig. 3b. This anisotropy amplification results from the efficient light-harvesting energy transfer process, in which the excitation light energy was harvested by the wide-bandgap monomers and transferred to the low-energy excimer states in a polarized form. The strong PL anisotropy in PyBTM doped αH-PyBTM microcrystals is ascribed to the brickwork arrangement of the αH-PyBTM molecules in the crystalline state (Fig. 3d),20 leading to an antiparallel spatial distribution of the μ (blue arrows in Fig. 3d) on the photoexcited (01−1) plane at an angle of ∼29° to the growth direction of the microcrystals. The same polarized orientation between the μ of the αH-PyBTM host molecules and the PL emission from the PyBTM guest molecules indicates that the μ of PyBTM is also aligned parallel to that of the αH-PyBTM molecules in a dense packing (Fig. S9, ESI†).
The above results help us draw a picture of exciton evolution in such a light-harvesting system. As exhibited in Fig. 4a, the pumped energy is first harvested by the wide-bandgap PyBTM monomers and subsequently captured by the lower-energy PyBTM excimer states through an efficient energy transfer process. This energy transfer concentrates the polarized excitons into the excimers, which behaves as exciton funnels to favor the amplification of the polarized PL emission from the excimer states. Moreover, the emission color of the radical-doped microcrystal gradually changes from yellow to red with increasing temperature (Fig. 4b), which implies that the energy transfer processes between the monomer and excimer states have been activated at higher temperatures.51 Therefore, we expect that an increase in temperature will amplify the PL anisotropy in the PyBTM doped αH-PyBTM microcrystals.
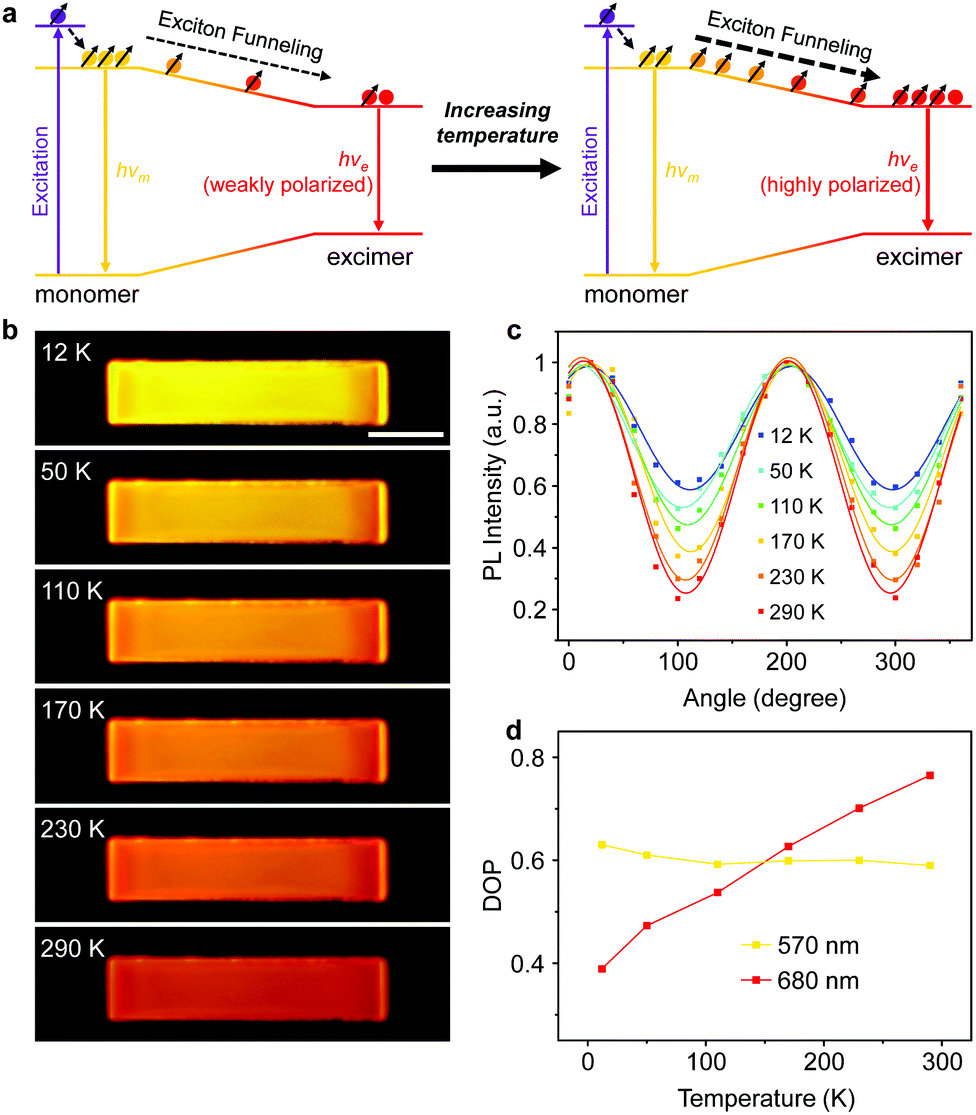 | ||
| Fig. 4 (a) Schematic of the temperature-controlled exciton funneling for the formation and radiative deactivation of excimer excitons in the PyBTM doped αH-PyBTM light-harvesting systems. (b) Temperature-dependent PL images of an identical PyBTM doped αH-PyBTM microcrystal. Scale bar is 10 μm. (c) Plots of PL intensity at 680 nm as a function of emission polarization angle at different temperatures in an identical PyBTM doped αH-PyBTM microplate. (d) Relationship between DOP and temperature at different wavelengths. | ||
Exactly, this is what we observed in the experiments. As shown in Fig. 4c, the Imin of the excimer emission recorded at 680 nm decreases gradually with increasing temperature, clearly indicating an increasing DOP at elevated temperatures (Fig. 4d). This could be attributed to activation of the energy transfer processes from the wide-bandgap monomers to the lower-energy excimer states as temperature rises,51 considering the energy transfer of doublet excitons in organic radicals is a Dexter-like exciton hopping mechanism.52 Due to insufficient sites for hopping at low temperatures, the number of available hopping sites quickly becomes limited during the downhill migration of excitons, which hinders the energy transfer processes from monomers to excimers and results in a low DOP of excimer emission. When the temperature increases, the linearly polarized monomer excitons sufficiently diffuse and funnel to the lower-energy excimer states, leading to sufficient quenching of the monomer emission and increasing intensity and polarization of the excimer emission (Fig. S10 and S11, ESI†). This tunable PL anisotropy amplification can be further applied in microcrystal-based polarized electroluminescence devices,53 considering this would be beneficial for various applications ranging from optical communications to liquid-crystal displays.54–56
In summary, we report a controlled amplification of PL anisotropy in organic luminescent radical-doped microcrystals via exciton funneling. Organic radical excimers with lower energy were created and surrounded by monomers with high-lying energy levels in dye-doped microcrystals, which serve as exciton funnels to concentrate polarized excitons into the excimer states and therefore results in a PL anisotropy amplification of the excimer emission. With the sensitive responsiveness of the energy transfer process, the anisotropy was well-modulated by temperature for dynamically tunable polarized photonic elements. These results not only provide insight into the exciton funneling process in light-harvesting systems, but also offer a promising route to develop miniaturized optical devices with desired performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Ministry of Science and Technology of China (Grant No. 2017YFA0204502), the National Natural Science Foundation of China (Grant No. 22090023, 21922307 and 21773265) and the Postdoctoral Innovation Talent Support Project (Grant No. BX2021318).Notes and references
- D. M. Jameson and J. A. Ross, Chem. Rev., 2010, 110, 2685–2708 CrossRef PubMed.
- H. Wang, Y.-M. He, T.-H. Chung, H. Hu, Y. Yu, S. Chen, X. Ding, M.-C. Chen, J. Qin, X. Yang, R.-Z. Liu, Z.-C. Duan, J.-P. Li, S. Gerhardt, K. Winkler, J. Jurkat, L.-J. Wang, N. Gregersen, Y.-H. Huo, Q. Dai, S. Yu, S. Höfling, C.-Y. Lu and J.-W. Pan, Nat. Photonics, 2019, 13, 770–775 CrossRef.
- C. Weder, C. Sarwa, A. Montail, C. Bastiaansen and P. Smith, Science, 1998, 279, 835–837 CrossRef PubMed.
- C. Fang, M. Xu, J. Ma, J. Wang, L. Jin, M. Xu and D. Li, Nano Lett., 2020, 20, 2339–2347 CrossRef CAS PubMed.
- X. Wang, Y. Wang, W. Gao, L. Song, C. Ran, Y. Chen and W. Huang, Adv. Mater., 2021, 33, 2003615 CrossRef CAS.
- J. Hu, L.-S. Li, W. Yang, L. Manna, L.-W. Wang and A. P. Alivisatos, Science, 2001, 292, 2060–2063 CrossRef CAS.
- J. Wang, M. S. Gudiksen, X. Duan, Y. Cui and C. M. Lieber, Science, 2001, 293, 1455–1457 CrossRef CAS.
- Y. Murakami, E. Einarsson, T. Edamura and S. Maruyama, Phys. Rev. Lett., 2005, 94, 087402 CrossRef PubMed.
- S. Kocsis, G. Y. Xiang, T. C. Ralph and G. J. Pryde, Nat. Phys., 2013, 9, 23–28 Search PubMed.
- Y. Gao, L. Zhao, Q. Shang, Y. Zhong, Z. Liu, J. Chen, Z. Zhang, J. Shi, W. Du, Y. Zhang, S. Chen, P. Gao, X. Liu, X. Wang and Q. Zhang, Adv. Mater., 2018, 30, 1801805 CrossRef.
- L. N. Quan, J. Kang, C. Ning and P. Yang, Chem. Rev., 2019, 119, 9153–9169 CrossRef CAS PubMed.
- D. Zhang, Z. Zeng, Q. Tong, Y. Jiang, S. Chen, B. Zheng, J. Qu, F. Li, W. Zheng, F. Jiang, H. Zhao, L. Huang, K. Braun, A. J. Meixner, X. Wang and A. Pan, Adv. Mater., 2020, 32, 1908061 CrossRef CAS.
- W. Zhang, J. Yao and Y. S. Zhao, Acc. Chem. Res., 2016, 49, 1691–1700 CrossRef CAS.
- M.-P. Zhuo, G.-P. He, Y. Yuan, Y.-C. Tao, G.-Q. Wei, X.-D. Wang, S.-T. Lee and L.-S. Liao, CCS Chem., 2020, 3, 413–424 CrossRef.
- Y. Yu, Y.-C. Tao, S.-N. Zou, Z.-Z. Li, C.-C. Yan, M.-P. Zhuo, X.-D. Wang and L.-S. Liao, Sci. China: Chem., 2020, 63, 1477–1482 CrossRef CAS.
- C. Reese and Z. Bao, Adv. Mater., 2007, 19, 4535–4538 CrossRef CAS.
- H.-H. Fang, Q.-D. Chen, J. Yang, H. Xia, Y.-G. Ma, H.-Y. Wang and H.-B. Sun, Opt. Lett., 2010, 35, 441 CrossRef CAS PubMed.
- S. K. Park, S. Varghese, J. H. Kim, S. J. Yoon, O. K. Kwon, B. K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- Z. Li, J. Wu, X. Wang, K. Wang, S. Zhang, W.-F. Xie and L.-S. Liao, Adv. Opt. Mater., 2019, 7, 1900373 CrossRef CAS.
- M.-J. Sun, Y. Liu, W. Zeng, Y. S. Zhao, Y.-W. Zhong and J. Yao, J. Am. Chem. Soc., 2019, 141, 6157–6161 CrossRef CAS PubMed.
- A. Ajayaghosh, V. K. Praveen, S. Srinivasan and R. Varghese, Adv. Mater., 2007, 19, 411–415 CrossRef CAS.
- C.-Y. Sun, X.-L. Wang, X. Zhang, C. Qin, P. Li, Z.-M. Su, D.-X. Zhu, G.-G. Shan, K.-Z. Shao, H. Wu and J. Li, Nat. Commun., 2013, 4, 2717 CrossRef PubMed.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- C. Qiao, C. Zhang, Z. Zhou, J. Yao and Y. S. Zhao, CCS Chem., 2021, 3, 624–632 CrossRef.
- J. B. Birks, Nature, 1967, 214, 1187–1190 CrossRef.
- B. Walker, H. Port and H. C. Wolf, Chem. Phys., 1985, 92, 177–185 CrossRef.
- Z. Zhao, S. Chen, J. W. Y. Lam, Z. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. Ma, H. S. Kwok and B. Z. Tang, J. Mater. Chem., 2011, 21, 7210–7216 RSC.
- K. Wang, H. Zhang, S. Chen, G. Yang, J. Zhang, W. Tian, Z. Su and Y. Wang, Adv. Mater., 2014, 26, 6168–6173 CrossRef PubMed.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352–10356 CrossRef CAS PubMed.
- S. F. Swallen, Z. Zhu, J. S. Moore and P. Kopelman, J. Phys. Chem. B, 2000, 104, 3988–3995 CrossRef.
- T. A. Klar, T. Franzl, A. L. Rogach and J. Feldmann, Adv. Mater., 2005, 17, 769–773 CrossRef.
- K. Wang, Z. Gao, W. Zhang, Y. Yan, H. Song, X. Lin, Z. Zhou, H. Meng, A. Xia, J. Yao and Y. S. Zhao, Sci. Adv., 2019, 5, eaaw2953 CrossRef PubMed.
- P. Yu, Y. Zhen, H. Dong and W. Hu, Chem, 2019, 5, 2814–2853 Search PubMed.
- S. Kimura, T. Kusamoto, S. Kimura, K. Kato, Y. Teki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 12711–12715 CrossRef.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef.
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224–1229 CrossRef.
- K. Kato, S. Kimura, T. Kusamoto, H. Nishihara and Y. Teki, Angew. Chem., Int. Ed., 2019, 58, 2606–2611 CrossRef.
- A. Obolda, X. Ai, M. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35472–35478 CrossRef.
- H. Dong, C. Zhang, Y. Liu, Y. Yan, F. Hu and Y. S. Zhao, Angew. Chem., Int. Ed., 2018, 57, 3108–3112 CrossRef.
- K. Wang, W. Zhang, Z. Gao, Y. Yan, X. Lin, H. Dong, C. Zhang, W. Zhang, J. Yao and Y. S. Zhao, J. Am. Chem. Soc., 2018, 41, 13147–13150 CrossRef.
- C. Wei, M. Gao, F. Hu, J. Yao and Y. S. Zhao, Adv. Opt. Mater., 2016, 4, 1009–1014 CrossRef.
- J. B. Birks and L. G. Cristophorou, Nature, 1962, 194, 442–444 CrossRef.
- C. Zhang, Y. Yan, Y.-Y. Jing, Q. Shi, Y. S. Zhao and J. Yao, Adv. Mater., 2012, 24, 1703–1708 CrossRef.
- A. J. Musser, S. K. Rajendran, K. Georgiou, L. Gai, R. T. Grant, Z. Shen, M. Cavazzini, A. Ruseckas, G. A. Turnbull, I. D. W. Samuel, J. Clark and D. G. Lidzey, J. Mater. Chem. C, 2017, 5, 8380–8389 RSC.
- C. R. L. P. N. Jeukens, P. Jonkheijm, F. J. P. Wijnen, J. C. Gielen, P. C. M. Christianen, A. P. H. J. Schenning, E. W. Meijer and J. C. Maan, J. Am. Chem. Soc., 2005, 127, 8280–8281 CrossRef PubMed.
- Y. Hirai, S. S. Babu, V. K. Praveen, T. Yasuda, A. Ajayaghosh and T. Kato, Adv. Mater., 2009, 21, 4029–4033 CrossRef.
- M.-P. Zhuo, J.-J. Wu, X.-D. Wang, Y.-C. Tao, Y. Yuan and L.-S. Liao, Nat. Commun., 2019, 10, 3839 CrossRef PubMed.
- S. Jursenas, A. Gruodis, G. Kodis, M. Chachisvilis, V. Gulbinas, E. A. Silinsh and L. Valkunas, J. Phys. Chem. B, 1998, 102, 1086–1094 CrossRef.
- O. V. Mikhnenko, P. W. M. Blom and T. Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- J. Han, Y. Jiang, A. Obolda, P. Duan, F. Li and M. Liu, J. Phys. Chem. Lett., 2017, 8, 5865–5870 CrossRef PubMed.
- Z. Qin, H. Gao, J. Liu, K. Zhou, J. Li, Y. Dang, L. Huang, H. Deng, X. Zhang, H. Dong and W. Hu, Adv. Mater., 2019, 31, 1903175 CrossRef.
- T. Komino, H. Kuwae, A. Okada, W. Fu, J. Mizuno, J.-C. Ribierre, Y. Oki and C. Adachi, ACS Appl. Mater. Interfaces, 2017, 9, 27054–27061 CrossRef.
- X. Wu, G. Xie, C. P. Cabry, X. Xu, S. J. Cowling, D. W. Bruce, W. Zhu, E. Baranoff and Y. Wang, J. Mater. Chem. C, 2018, 6, 3298–3309 RSC.
- J. Zhu, J. Wang, H. Lu and L. Qiu, J. Mater. Chem. C, 2020, 8, 9147–9162 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures and additional data. See DOI: 10.1039/d1tc02638k |
| This journal is © The Royal Society of Chemistry 2022 |