
Bioinspired nanocatalytic tumor therapy by simultaneous reactive oxygen species generation enhancement and glutamine pathway-mediated glutathione depletion†
Huijia
Mao‡
a,
Yangyang
Wen‡
b,
Yonghui
Yu
a,
Hongyan
Li
 *a,
Jing
Wang
*a and
Baoguo
Sun
a
*a,
Jing
Wang
*a and
Baoguo
Sun
a
aChina-Canada Joint Lab of Food Nutrition and Health (Beijing), School of Food and Health, Beijing Technology and Business University (BTBU), 11 Fucheng Road, Beijing 100048, China. E-mail: hongyan.li@btbu.edu.cn; wangjing@th.btbu.edu.cn
bCollege of Chemistry and Materials Engineering, Beijing Technology and Business University (BTBU), 11 Fucheng Road, Beijing 100048, China
First published on 21st November 2022
Abstract
An insufficient intracellular H2O2 level and overexpressed glutathione (GSH) are still the major challenges for effective chemodynamic therapy (CDT). Inspired by the unique glutamine metabolism pathway in cancer cells, herein, intelligent nanocatalytic theranostics is used to enhance intracellular reactive oxygen species (ROS) accumulation via the production of H2O2 by a biomimetic nanozyme, and simultaneously reduce ROS consumption via the depression of GSH synthesis by the glutamine metabolic inhibitor. In this reactor, nano-sized Au and Fe3O4 coloaded dendritic mesoporous silica nanoparticles (DMSN-Au-Fe3O4) serve as the bifunctional nanozyme, where intracellular glucose is catalyzed into H2O2 by the glucose oxidase-mimicking Au nanoparticles and then immediately transformed into ˙OH by the peroxidase-like Fe3O4 nanoparticles. Then, CB839, the glutaminase (GLS) inhibitor, is grafted on the nanozyme, blocking the glutamine pathway and GSH biosynthesis. As a result, the as-designed nanoplatform with a three-pronged integration of Au-mediated H2O2 self-supply, Fe3O4-triggered Fenton-like reaction, and glutamine pathway-mediated GSH depletion significantly boosts the CDT efficacy, achieving remarkable and specific antitumor properties both in vitro and in vivo. This work not only paves a new way for rationally designing multi-functional nanozymes for achieving high therapeutic efficacy, but also provides new insights into the construction of bioinspired synergetic therapy by combining CDT and a key anticancer pathway.
1. Introduction
Reactive oxygen species (ROS) are a group of highly reactive ions or free radicals, including singlet oxygen (1O2), hydroxyl radicals (˙OH), superoxide anions (O2˙−), and peroxides (O22−), which are pivotal signal indicators associated with numerous physiological processes.1,2 High levels of ROS could oxidize unsaturated fatty acids and amino acids, resulting in irreversible damage to organelles, and ultimately inducing cell apoptosis or necrosis.3 In this respect, utilizing ROS cytotoxicity as a tumor therapy strategy has received great attention in recent years.4–7 Chemodynamic therapy (CDT) is one of the most promising modalities that adopts ROS for cancer treatment.8–12 By employing advanced inorganic or organic nanozymes, which process Fenton or Fenton-like reaction activity to catalyze the conversion of hydrogen peroxide (H2O2) into highly toxic ROS species (˙OH), CDT has achieved the remarkable effect of inducing cell apoptosis.13Given the features of acidity, hypoxia, and H2O2 overexpression in the tumor microenvironment (TME) due to its abnormal metabolism pathways, ROS are produced in tumor regions rather than the normal tissues, endowing CDT with high therapeutic specificity and low invasiveness.11,14,15 Nevertheless, the intratumoral H2O2 level (100 μM–1 mM) is still inadequate to generate enough ROS for achieving satisfactory CDT efficacy.16–18 Besides, tumor cells also feature a high GSH concentration (up to 10 mM), which could largely scavenge as-produced ROS groups and weaken the CDT efficacy.19,20 For this reason, the design of a multi-functionalized nanozyme with both H2O2-supplying and GSH-depleting performance is highly desirable for generating synergistic anticancer effects.
As is known, considerable efforts have been devoted to preparing nanozymes with the “synergetic effect” of both H2O2-supplying functionality and GSH-depleting performance. For instance, Fu et al. fabricated an intelligent nanocatalyst, glucose oxidase and doxorubicin coloaded copper-doped calcium phosphate nanoparticles, which possessed both H2O2 self-supply and GSH-elimination properties, and significantly enhanced the CDT efficacy.13 Sang et al. reported a H2O2 homeostasis disruptor, the 3-amino-1,2,4-triazole modified zeolitic imidazole framework-67 nanozymes, which perturbed the H2O2 balance and gave rise to the accumulation of H2O2 in cancer cells, and ultimately amplified the CDT efficiency.21 Although both the H2O2 self-supply and GSH-elimination increase the ROS level to some extent, the previously reported works could not deplete GSH continuously.
Glutamine metabolism has recently been widely recognized as an essential pathway to tumor cell proliferation. Glutamine is the abundant circulating amino acid involved in the generation of tricarboxylic acid (TCA) cycle intermediates and amino acids, and maintaining redox homoeostasis by synthesizing GSH and NADPH.22 Therefore, targeting glutamine metabolism is an appealing therapeutic option in many malignant tumor cell subtypes.23–26 The glutaminase (GLS) inhibitor strategy, the binding of inhibitors and GLS causes drastic conformational changes of the key ring near the catalytic site, thereby inhibiting GLS activity and suppressing GSH synthesis, has been successfully applied to enhance the anticancer efficacy because of the synergetic effect of both glutamine starvation and GSH depletion.27 CB839 (telaglenastat), as a highly selective allosteric GLS1 inhibitor with high anticancer activity, has already been approved by the U.S. Food and Drug Administration for cancer clinical trials (Stage II).28–30 However, like other drug therapeutics, the drawbacks of CB839, e.g., non-targeted drug delivery, non-selective distribution, and limited objective response as a monotherapy, greatly restrict its therapeutic efficacy.31,32 In this regard, it is of significance to explore the multifunctional targeted delivery platform to enhance the specificity and efficacy of CB839 drugs.
Herein, for the first time, we have designed and constructed an intelligent nanocatalytic platform that possesses both glutamine and glucose starving effects by incorporating the CB839 mediated GSH elimination and a biomimetic dual nanozyme-triggered TME-responsive cascade catalytic reaction together for the enhancement of CDT efficacy (Scheme 1). In this platform, a biomimetic nanozyme DMSN-Au-Fe3O4 NPs, the unique glucose oxidase (GOx)-mimicking catalytic activity of Au nanoparticles (NPs) and peroxidase (POD)-mimicking catalytic activity of Fe3O4 NPs that were co-decorated on the dendritic mesoporous silica nanoparticles, was firstly adopted to catalyze the TME-responsive cascade reaction for facilitating the production of H2O2 and ROS. Followed by loading the glutamine metabolic inhibitor (CB839) and further modification with polyethylene glycol (PEG), the resultant system, on one hand, could convert glucose into H2O2 by the catalysis of Au NPs and generate ˙OH via the catalysis of Fe3O4 NPs via Fenton-like reactions; on the other hand, the scavenging of H2O2 and ROS by GSH could be interrupted by inhibiting the biosynthesis of GSH via blocking the glutamine metabolism pathway. Taken together, the as-prepared DMSN-Au-Fe3O4-CB839 NPs exhibited a superior tumor-suppression rate and tumor inhibition effect in both in vitro and in vivo experiments, proposing a new way for the rational design of cancer therapy nanozymes and strategies.
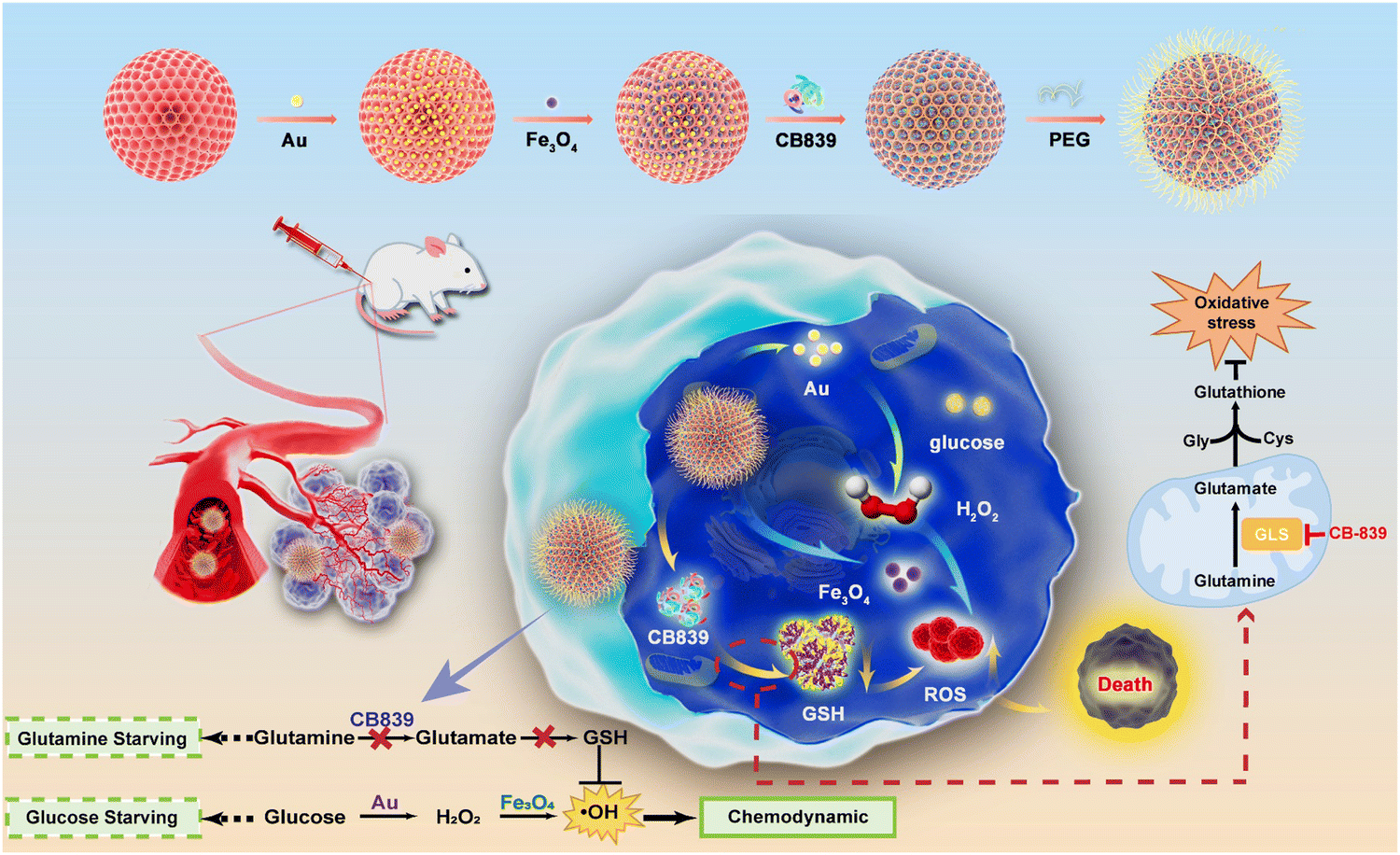 | ||
| Scheme 1 Schematic representation of the synthesis of DMSN-Au-Fe3O4-CB839 nanoparticles and the use of DMSN-Au-Fe3O4-CB839 to induce intensive CDT therapy. | ||
2. Experimental section
2.1 Synthesis of DMSN-Au-Fe3O4
2.2 Material characterization
Scanning electron microscopy (SEM) images were recorded on a field-emission JSM7401 microscope (Tokyo, Japan). Transmission electron microscopy (TEM) images and corresponding elemental mapping analysis were measured on a JEOL-2100F transmission electron microscope (Tokyo, Japan). X-ray powder diffraction (XRD) pattern was determined on a D2 Phaser diffractometer (Bruker, Germany) with a Cu Kα irradiation source. Specific surface area and pore size distribution were evaluated on a Micromeritics Tristar 3000 system, calculated by N2 sorption isotherms using a Brunauer–Emmett–Teller (BET) method. The zeta potential test was performed on Zetasizer Nano-ZS90 (Malven, England). Fourier-transform infrared (FT-IR) spectra were recorded on a FTIR spectrometer (Nicolet iS10, Thermo Fisher Scientific Co. Ltd, Waltham, MA, USA). UV-vis absorption spectra were recorded on a Cary 100 UV/vis spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Inductively coupled plasma mass spectrometry (iCAP Q, Thermo, Waltham, USA) was conducted for quantitative elemental analysis.2.3 Catalytic activity of DMSN-Au-Fe3O4
TMB was employed to study the catalytic activity of DMSN-Au-Fe3O4 in Hac-NaAc buffer solution (pH = 6.5, 7.4). The steady-state kinetic catalytic activities of DMSN-Au-Fe3O4 NPs were investigated by monitoring the absorbance variation at 652 nm on a UV–vis spectrophotometer, which was conducted in a reaction system consisted of DMSN-Au-Fe3O4 NPs, TMB, and H2O2 of varied concentrations (2.5, 5, 10, 20, 30, 40, and 50 × 10−3 M) in Hac-NaAc buffer solution (pH 6.5). According to the absorbance-changing rate, obtained from Beer–Lambert law, A = εlc (A is the absorbance, ε is the molar absorbance coefficient, l is the path length, and c is the molar concentration) with l = 10 mm and ε of 39 000 M−1 cm−1, the concentration-changing rate (v) was calculated of oxTMB of each concentration of H2O2. The Michaelis–Menten kinetics model was applied to analyze the kinetic catalytic behaviors of DMSN-Au-Fe3O4 NPs. Glucose, instead of H2O2, was used to further evaluate the self-organized enzymatic cascade reaction of the DMSN-Au-Fe3O4 NPs (pH = 6.5). The DMSN-Au-Fe3O4 NPs, glucose and TMB were incubated for 1 h. The absorbance at 370 nm and 650 nm were detected.2.4 Drug loading and release
Loading efficiency = 100% × (mtotal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) CB839 − munloaded CB839)/mtotalCB839 CB839 − munloaded CB839)/mtotalCB839 | (1) |
| Loading capacity = 100% × (mtotalCB839 − munloadedCB839)/mtotalDMSN | (2) |
2.5 In vitro cytotoxicity assays
4T1 cells were seeded in 96-well plates at a density of 1 × 104 cells per well. After 24 h incubation, the media were removed. Fresh medium (pH= 7.4 or 6.5) containing nanoparticles (DMSN-CB839, DMSN-Au-Fe3O4, or DMSN-Au-Fe3O4-CB839) at varied concentrations (6.25, 12.5, 25, 50, 100, 200 mg mL−1) were inoculated into 96-well plates and co-cultured for 24 h. The sample-free tissue culture plate was used as the control experiment. Cell viability was assessed by the standard 3-(4,5 dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazollium bromide (MTT) assay.2.6 Cell apoptosis analysis
4T1 cells were seeded in 6-well plates at a density of 1 × 105 cells mL−1 and adhered for 24 h. Then, 4T1 cells were incubated in DMSN-CB839 (200 μg mL−1), DMSN-Au-Fe3O4 (200 μg mL−1), and DMSN-Au-Fe3O4-CB839 (200 μg mL−1) solutions for 24 h, separately. After trypsinization and washing, cells were stained with 5 μL Annexin V-FITC for 10 min, followed by 5 μL PI for 5 min in the dark at room temperature. Afterwards, the cells were evaluated immediately by flow cytometry (Cytoflex, Beckman Coulter, USA).2.7 Intracellular ROS detection
Cells were seeded in 6-well plates and incubated for 24 h. After incubation with nanoparticles (200 μg mL−1) for 4 h, fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCF-DA) was employed for the determination of ROS levels within 4T1 cells and confocal laser scanning microscopy (FV3000, Olympus company, Japan) was performed to monitor the fluorescence intensity of cells.2.8 Intracellular endocytosis observation
4T1 cells were first planted in 6-well plates and incubated for 24 h. Then, the media were replaced with fresh DMEM containing FITC modified DMSN-CB839, DMSN-Au-Fe3O4, or DMSN-Au-Fe3O4-CB839 nanoparticles (100 μg mL−1). After 4 h incubation, the cells were washed with PBS for 3 times and fixed with 4% paraformaldehyde solution for 15 min. 4′,6-Diamidino-2-phenylindole (DAPI) was used to stain cellular nuclei for 10 min. After washing with PBS thoroughly, the fluorescence images of cells were observed by the confocal laser scanning microscopy.2.9 LIVE/DEAD® staining
4T1 cells with a density of 1 × 105 cells mL−1 were cultured in a 6-well plate for 24 h to allow the attachment of cells. Then, the cells were incubated with DMSN-CB839 (200 μg mL−1), DMSN-Au-Fe3O4 (200 μg mL−1), and DMSN-Au-Fe3O4-CB839 (200 μg mL−1) for 24 h. Later, calcein acetoxymethyl ester (calcein AM) and propidium iodide (PI) were used to confirm the visualized viability of cells. Fluorescence images were collected on Olympus FV3000 optical system.2.10 Cellular GSH detection
4T1 cells were randomly seeded in 6-well culture dishes and allowed to adhere for 24 h. Then the cells were incubated with nanoparticles (200 μg mL−1) for 24 h. The GSH Assay Kit (Beyotime Biotechnology, Shanghai) was employed for the detection of cellular GSH concentration.2.11 In vivo anti-tumor treatment
All animal work was performed in accordance with Animal Ethics (P2021041) approved by Charles River Institutional Animal Care and Use Committee. Balb/c mice (4 weeks, female, ∼15 g) were purchased from Charles River (Beijing, China). To develop the animal tumor model, ∼1 × 106 4T1 cells were subcutaneously injected into the right flank of each female Balb/c mice (16–18 g). The tumor volume was calculated according to the following formula: volume = length × (width)2/2. Once tumors reached ∼50 mm3, the mice were divided randomly into four groups consisting of 8 mice in each group for in vivo therapy experiments. DMSN-CB839, DMSN-Au-Fe3O4, DMSN-Au-Fe3O4-CB839 (20 mg kg−1) were intratumorally injected into the mice in three groups respectively, while 100 μL of saline solution was injected into the other group as the control group. All mice received one injection at day 1, 5 and 9. During the treatment, tumor sizes and body weights of the mice were recorded every other day during the 14 day period. Representative tumors and the main organs (heart, liver, spleen, lung, and kidney) were collected on the 14th day post injection and the H&E staining was performed to evaluate the pathologic change.2.12 Statistical analysis
Quantitative data were expressed as mean results ± standard deviation (s.d.). Statistical evaluation was conducted by using unpaired Student's two-sided t test analysis. Asterisk corresponds to significant differences (*P < 0.05, **P < 0.01, ***P < 0.001).3. Results and discussion
3.1 Structure, catalytic activity and loading performance of DMSN-Au-Fe3O4-CB839
To verify our protocol, the dual-functioned nanozyme, DMSN-Au-Fe3O4, was first prepared. The fabrication procedures were schematically illustrated in Scheme 1, with detailed experimental methods in the ESI.† Firstly, dendritic mesoporous silica nanoparticles (DMSN) were chosen as the desirable nanoplatforms for the decoration of small Au and Fe3O4 NPs. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) observations disclosed that DMSN possessed a highly uniform particle size of ∼130 nm, exhibiting an unique central-radial porous structure, as shown in Fig. 1(A), (B) and Fig. S1 (ESI†). The corresponding homogeneous Si, O elemental distributions from high-angle annular dark-field (HADDF) TEM confirmed the successful synthesis of DMSN (Fig. S2, ESI†). The nitrogen sorption isotherms in Fig. S3 (ESI†) unveiled the mesoporous nature of DMSN, with a high specific surface area of 333.9 m2 g−1 and an average pore diameter of 12.7 nm. Thus, the large mesopores with highly accessible surface areas are extremely favorable for the follow-up deposition of Au and Fe3O4 NPs, as well as the drug (CB839 inhibitor) loading.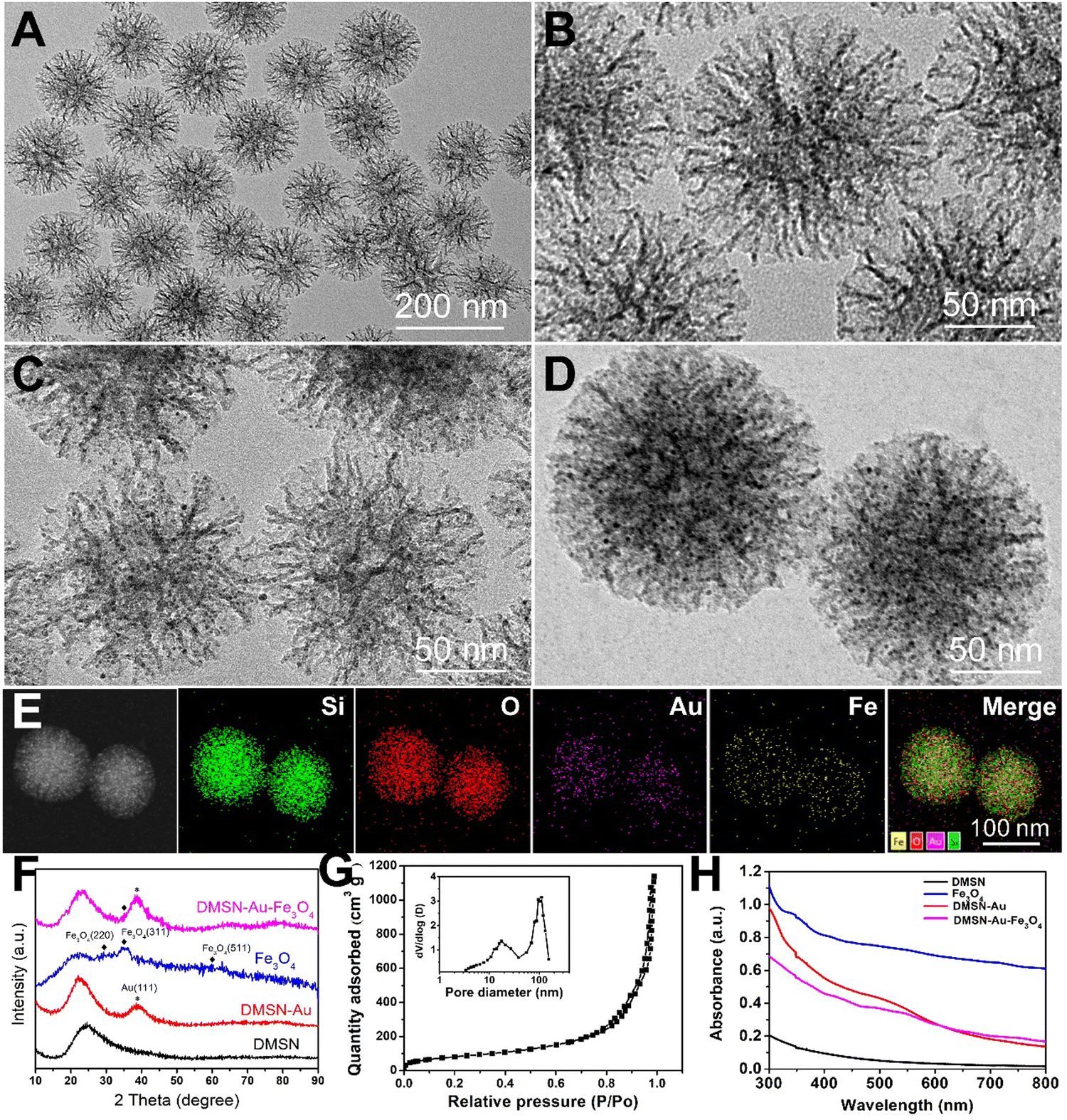 | ||
| Fig. 1 TEM images of DMSN (A) and (B), DMSN-Au (C) and DMSN-Au-Fe3O4 NPs (D). (E) Dark-field STEM image of DMSN-Au-Fe3O4 NPs and corresponding element mappings for elements of O, Si, Au, Fe and merge, respectively. (F) XRD patterns of DMSN, DMSN-Au, Fe3O4, and DMSN-Au-Fe3O4 NPs. (G) N2 sorption isotherms and pore size distributions (insert) of DMSN-Au-Fe3O4 NPs. (H) UV-vis spectra of DMSN, Fe3O4, DMSN-Au, and DMSN-Au-Fe3O4 NPs. | ||
The as-obtained DMSN NPs were initially negatively charged with a zeta potential of −11 mV due to the presence of a large number of silanol terminations on the surface (Fig. S4, ESI†). For the in situ deposition of Au NPs, DMSN were functionalized with amine groups by aminopropyltriethoxysilane (APTES), producing positively charged DMSN-NH2 NPs with a zeta potential of +10.91 mV (Fig. S4, ESI†). These amine groups absorbed the AuCl4− ions quickly and then grew Au NPs with the aid of a reducing agent-NaBH4 (labeled as DMSN-Au). The TEM images in Fig. 1€ and Fig. S5 (ESI†) suggested the ultrasmall Au NPs with a diameter of only 2–4 nm, and the corresponding Au elemental mapping verified that Au NPs were homogeneously dispersed in the mesopores of DMSN. Subsequently, the pre-synthesized 1 nm-sized Fe3O4 NPs (as presented in Fig. S6, ESI†) were firmly confined within the mesopores of DMSN-Au NPs due to the hydrophobic nature of Fe3O4,35 forming the DMSN-Au-Fe3O4 NPs. The SEM observation showed that DMSN-Au-Fe3O4 NPs retained the same porous structure and particle size (∼130 nm, Fig. S7, ESI†) as pristine DMSN, revealing its unchanged particle morphology after the nanocomposite preparation. Fig. 1(D) was the high-resolution TEM image of DMSN-Au-Fe3O4, depicting the dendritic structure uniformly decorated with numerous ultrasmall nanoparticles inside the mesopores. Then, HADDF TEM image and corresponding elemental mappings were conducted (Fig. 1(E)), disclosing the homogeneous distribution of O, Si, Au, and Fe elements in the whole nanoparticles, and thus revealing the uniformly dispersed Au and Fe3O4 NPs in DMSN. The inductively coupled plasma mass spectrometry (ICP-MS) measurement confirmed that the atomic ratio of Si, Au, and Fe elements were 1:0.75:0.42.
XRD analysis was applied to identify the formation of metallic Au and crystallized Fe3O4 in the nanocomposites. As shown in Fig. 1(F), both DMSN-Au and DMSN-Au-Fe3O4 displayed strong diffraction peaks at 38.2°, corresponding to the (111) lattice plane for metallic Au (JCPDS no. 04-0784). Similarly, the as-prepared Fe3O4 NPs presented small humps at 31.2°, 36.8° and 59.3° that indexed to the (220), (311), and (511) planes for the Fe3O4 (JCPDS no. 26-1136), respectively. However, the diffraction intensity of Fe3O4 peaks decreased significantly in the DMSN-Au-Fe3O4 NPs, possibly resulted from an overlap by strong diffractions of the Au and DMSN matrix.33Fig. 1(G) was the N2 sorption analysis of DMSN-Au-Fe3O4 NPs, which revealed a slightly decreased specific surface area of 299.7 m2 g−1 and larger portion of macropores (>40 nm), possibly caused by the deposition of Au and Fe3O4 NPs. Moreover, the UV-vis absorption spectra further verified the formation of DMSN-Au and DMSN-Au-Fe3O4 nanocomposites, showing a characteristic plasma resonance peak of Au NPs at 505 nm, as shown in Fig. 1(G). Here, it is noteworthy that the typical absorption peak in DMSN-Au-Fe3O4 NPs slightly shifted rightward in comparison to DMSN-Au NPs, which might be related with the interaction effect between Au NPs and Fe3O4 NPs.
The successful fabrication of the nanozyme encouraged us to systematically investigate its catalytic functions. The DMSN-Au-Fe3O4 NPs possess two catalytic activities: Au-induced glucose oxidase (GOx) mimicking activity that catalyzes glucose in the presence of oxygen into gluconic acid and H2O2, and Fe3O4-induced peroxidase (POD) mimicking activity that converts the produced H2O2 into ˙OH via Fenton reaction. The GOx-mimicking activity of DMSN-Au-Fe3O4 NPs was first confirmed by the generation of gluconic acid from glucose in the presence of the nanozyme, resulting in the major absorbance peak at 505 nm after the addition of hydroxylamine and Fe3+ (Fig. S8, ESI†), accompanied by the production of H2O2. Then, the POD-like activity of the nanozyme was detected by a typical colorimetric method based on the indicator of 3,3′,5,5′-tetramethylbenzidine (TMB).35 As illustrated in Fig. 2(A), the nanozyme can catalyze H2O2 into H2O and ˙OH species, which could oxidize the uncolored TMB into blue-colored oxTMB immediately and thus display a strong absorption peak at 652 nm. The UV-vis absorption spectra in Fig. 2(B) showed that no absorbance peak was observed in TMB and the mixed TMB + H2O2 system. However, once added the DMSN-Au-Fe3O4 nanozyme, strong absorbance signals peaked at 370 and 652 nm were detected, indicative of the formation of ˙OH species and strong POD-like activity of DMSN-Au-Fe3O4 NPs. To validate the pH-dependent POD-like activity of DMSN-Au-Fe3O4 NPs, catalytic performances at different pH conditions were also implemented. As shown in Fig. S9 (ESI†), no peaks were detected at a neutral condition (pH = 7.4), whereas strong absorbance peaks at 370 and 652 nm were obtained at a weak acid condition (pH = 6.5), implying the selective POD-like activity of DMSN-Au-Fe3O4 NPs that only activated in the weak-acidic intratumoral circumstances, rather than in the normal cells with physiological pH of 7.4.
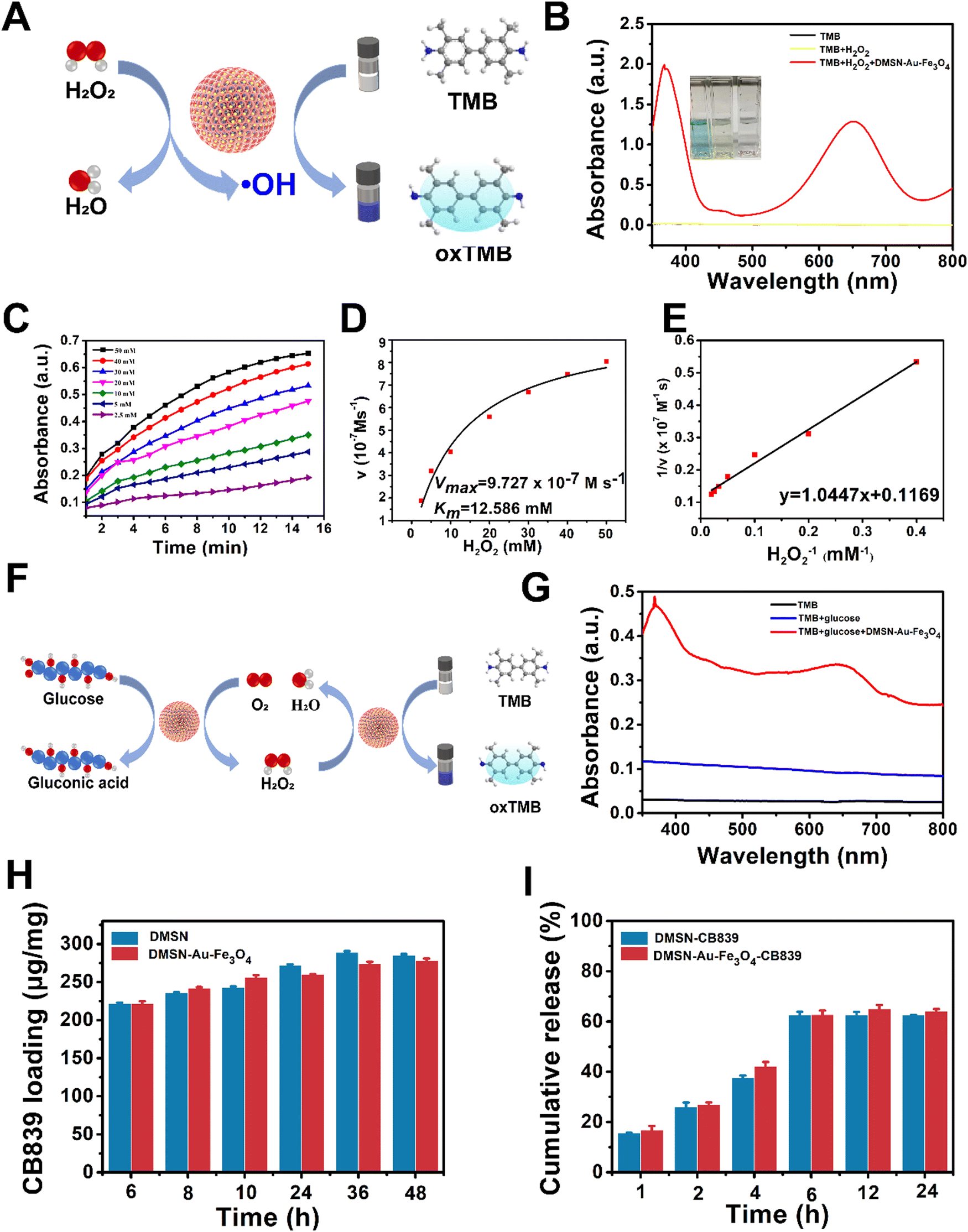 | ||
| Fig. 2 (A) Schematic illustration of the POD-mimic catalytic process of DMSN-Au-Fe3O4 NPs. (B) UV-vis absorption spectra of the catalyzed oxidation of TMB (oxTMB) in the different reaction system (pH = 6.5). (C) Time-dependent absorbance changes at 652 nm as a result of the catalyzed oxidation of TMB at different H2O2 concentrations (2.5, 5, 10, 20, 30, 40, 50 × 10−3 M). (D) Michaelis–Menten kinetic analysis and (E) Lineweaver–Burk plotting for DMSN-Au-Fe3O4 with H2O2 as substrate. The steady-state catalytic rate (v) was calculated from the initial slopes of absorbance versus time plots in panel (C). (F) Schematic illustration of DMSN-Au-Fe3O4 nanoplatform-catalyzed glucose oxidation reaction. (G) UV-vis absorption spectra of the catalyzed oxidation of TMB (oxTMB) in the different reaction system (pH = 6.5). (H) CB839 loading capacities of DMSN and DMSN-Au-Fe3O4 NPs. (I) CB839 release property of DMSN-CB839 and DMSN-Au-Fe3O4-CB839 in PBS solution (1 mg mL−1, pH 6.5). | ||
The catalytic activity of DMSN-Au-Fe3O4 NPs was then studied using steady-state kinetics with H2O2 substrates. To obtain the kinetic data, time-dependent absorbance changes with different H2O2 concentrations were conducted, as depicted in Fig. 2(C). The initial reaction rates were plotted against corresponding H2O2 concentrations, and the Michaelis–Menten curves were fitted (Fig. 2(D)) according to eqn (3):36
| v = Vmax [S]/(Km + [S]) | (3) |
| 1/v = (Km/Vmax) × (1/[S]) + 1/Vmax | (4) |
where v, Vmax, Km, and [S] represent the initial velocity, the maximal reaction velocity, Michaelis constant, and the substrate concentration, respectively.
Subsequently, the double reciprocal of the Michaelis–Menten equation (eqn (4)) was conducted to determine Km and Vmax for the catalytic reaction by DMSN-Au-Fe3O4 NPs (Fig. 2(E)). The calculated Km and Vmax for DMSN-Au-Fe3O4 NPs were 12.58 mM and 9.72 × 10−7 M s−1, respectively, revealing a good binding affinity with H2O2 for the enzyme.35
Subsequently, the self-organized enzymatic cascade reaction of DMSN-Au-Fe3O4 NPs was further evaluated. As demonstrated in Fig. 2(F), the Au NPs firstly oxidized glucose into H2O2, and then the Fe3O4 NPs catalyzed the as-produced H2O2 into highly toxic ˙OH species. Afterwards, colorless TMB substrate was oxidized into oxTMB (blue color) by ˙OH. Fig. 2(G) was the detected UV-vis spectra after the above-mentioned cascade reaction, proving that DMSN-Au-Fe3O4 NPs produced the ˙OH directly without any H2O2, exhibiting both GOx and POD mimicking activities.
Encouraged by the excellent ˙OH-generating activity of DMSN-Au-Fe3O4, the CB839 loading capacity of DMSN-Au-Fe3O4 NPs was also determined for pristine DMSN as a control experiment. As presented in Fig. S4 (ESI†), the zeta-potential results between DMSN, DMSN-NH2, DMSN-Au, DMSN-Au-Fe3O4, CB839, and DMSN-Au-Fe3O4-CB839 NPs unveiled the charging variations in each synthesis step. Apparently, the introduction of amine groups by APTES treatment of DMSN NPs turned the negative (−11 mV) zeta potential into positive (+10.91 mV). After the loading of Au and Fe3O4 NPs, the eventual DMSN-Au-Fe3O4 NPs exhibited the negative (−7.54 mV) potential, indicating an excellent capacity for the anchoring of positively charged CB839 (+9.4 mV) via strong electrostatic interactions. As displayed in Fig. 2(H), DMSN-Au-Fe3O4 exhibited a high loading capacity of up to 278.25 ± 2.56 μg mg−1 for CB839, almost close to the loading efficiency of the pristine DMSN NPs (285.95 ± 1.25 μg mg−1). To obtain high biocompatibility and physiological stability, DMSN-Au-Fe3O4-CB839 NPs were further functionalized by polyethylene glycol (PEG) molecules, which were validated by the Fourier transform infrared spectroscopy (FTIR) in Fig. S10 (ESI†). The peaks at 2890 and 2740 cm−1 were the stretching vibration of CH2 and CH3,37 respectively, and the peaks at 1060, 1110, 1150, 1250, and 1280 cm−1 corresponded to the stretching vibration band of C–O–C,38 suggesting the successful modification of mPEG-SH molecule on the DMSN-Au-Fe3O4-CB839 NPs. Besides, the corresponding drug releasing studies of DMSN-Au-Fe3O4-CB839 and DMSN-CB839 were also performed in PBS solution (pH = 6.5). As shown in Fig. 2(I), a burst release of ∼40% was observed at the first 4 h, followed by a cumulative release increasing to 60% at 6 h. After 6 h, no significant increment of the releasing percent was observed. It is presumable that the small CB839 molecules were adsorbed in the mesoporous silica pore structure that far from the center, giving rise to a relatively rapid release kinetics within 6 h. The fast release of CB839 can create a GSH-deficient tumor environment by inhibiting glutamine metabolism, which further boosts the accumulation of ROS generated by the self-organized enzymatic cascade reaction of DMSN-Au-Fe3O4 NPs in tumor cells.
3.2 In vitro tumor chemodynamic therapy
Initially, the intracellular uptake of fluorescein isothiocyanate (FITC)-labeled DMSN NPs was evaluated by confocal laser scanning microscopy (CLSM), showing that 4T1 tumor cells treated with FITC-labeled DMSN NPs exhibited the obvious green fluorescence. This indicated the effective intracellular uptake after incubation for only 2 hours (Fig. 3) and guaranteed the FITC-functionalized DMSNs can be effectively internalized in 4TI cells. Then, the cytotoxicity of DMSN-Au-Fe3O4-CB839 NPs was tested on two normal cell lines, exhibiting little cytotoxicity toward L6 and HUVEC cells at concentrations ranging from 0 to 200 μg mL−1 within 24 h (Fig. S11, ESI†), suggesting its favorable biocompatibility and high biosafety for the further in vitro and in vivo anticancer therapy.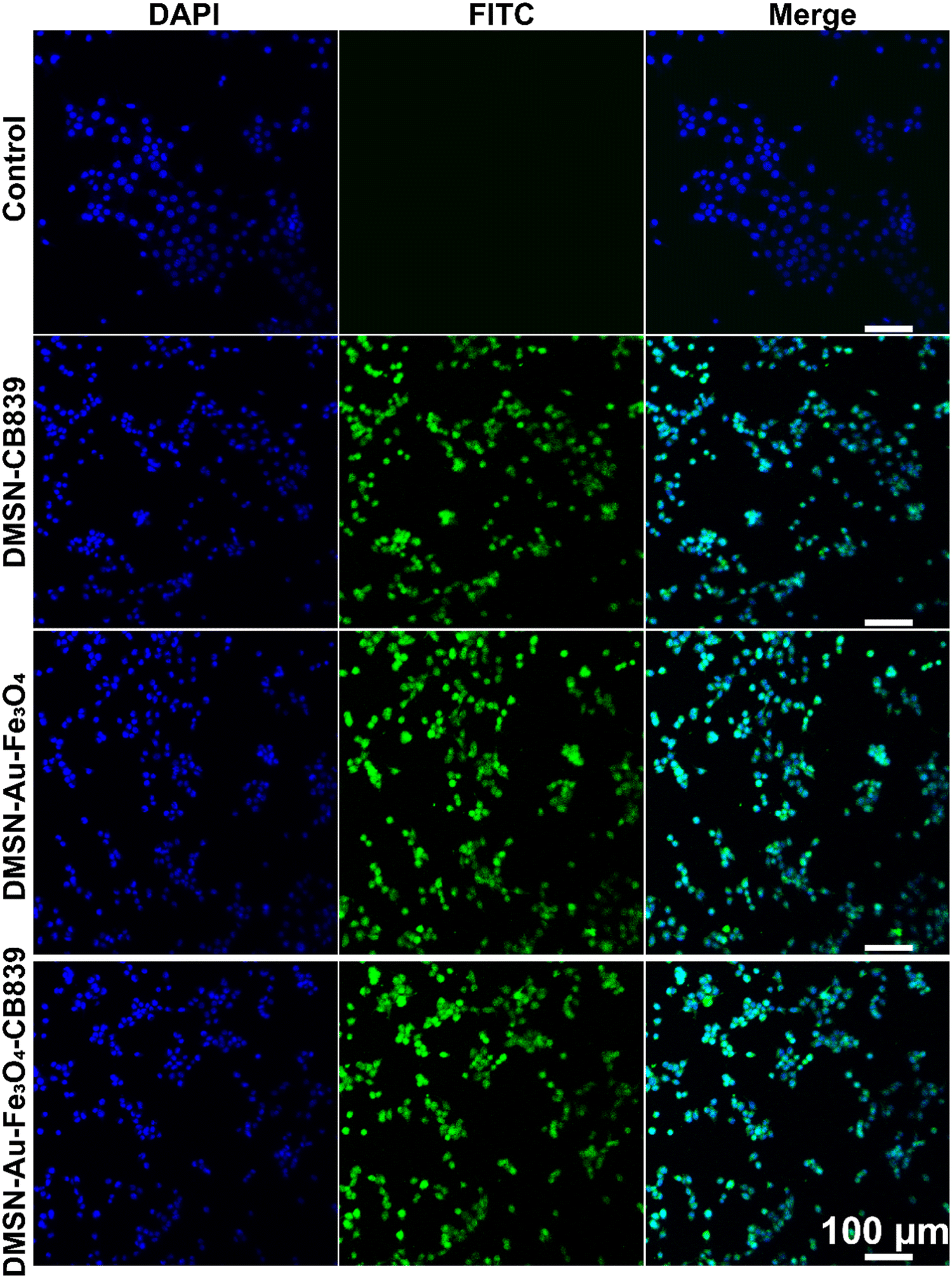 | ||
| Fig. 3 CLSM images of 4T1 cells after coincubation with FITC-labeled-DMSN-CB839, FITC-labeled-DMSN-Au-Fe3O4, and FITC-labeled-DMSN-Au-Fe3O4-CB839 for 2 h. Left column (blue): DAPI, middle column (green): FITC, right column: Merge. | ||
Furthermore, the specificity and selectivity towards tumor cells of DMSN-Au-Fe3O4 nanozyme were evaluated on 4T1 cells. The in vitro cytotoxicity of DMSN-Au-Fe3O4 nanozyme at different pH conditions was conducted by the standard cell-counting kit 8 (CCK-8) assay. As seen in Fig. 4(A), DMSN-Au-Fe3O4 NPs showed a much lower cytotoxicity at pH = 7.4, with cell viability higher than 80% even at the concentration up to 200 μg mL−1. However, the cell viability of 4T1 tumor cell incubated at pH = 6.5 decreased significantly as DMSN-Au-Fe3O4 NPs concentration increased, showing a dose-dependent manner and reached to the cell viability of higher than 60% at 200 μg mL−1, keeping consistent with the catalytic activity characterization in Fig. S8 (ESI†). We also compared relative cell viabilities after being incubated with the DMSN-Au NPs and DMSN-Fe3O4 NPs under the same experimental conditions. As shown in Fig. S12 (ESI†), notably, both DMSN-Au and DMSN-Fe3O4 NPs had no effect on the growth of 4T1 cancer cells proliferation even at the high NPs concentration of 200 μg mL−1, further validating that the high cytotoxicity of DMSN-Au-Fe3O4 NPs was attributed from the cascade catalytic reactions by incorporating Au mimicking GOx-like activity and Fe3O4 mimicking POD activity, rather than a single component NPs.
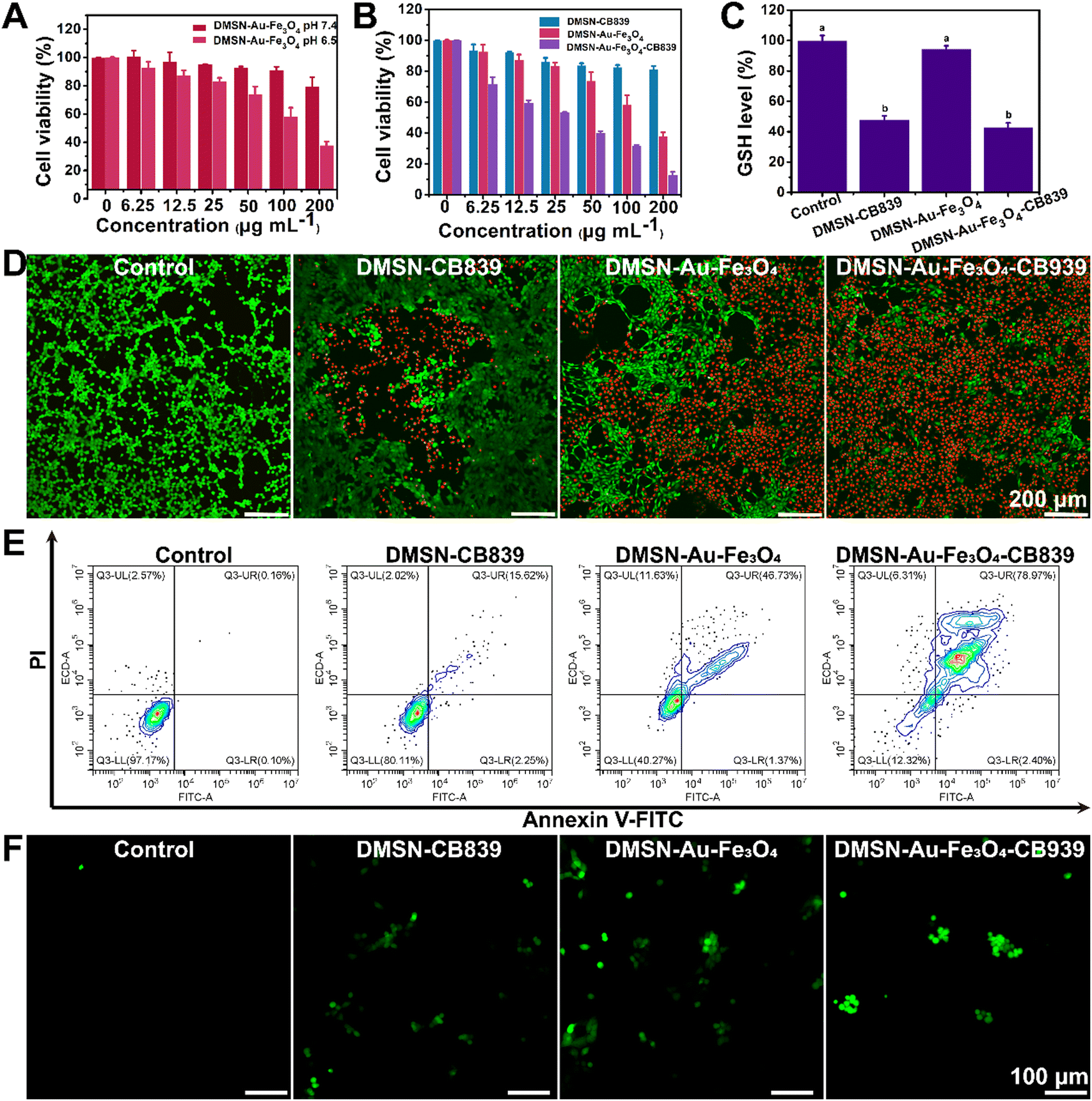 | ||
| Fig. 4 (A) Relative viabilities of 4T1 cells after being incubated with DMSN-Au-Fe3O4 NPs at varied concentrations (0, 6.25, 12.5, 25, 50, 100, and 200 μg mL−1) for 24 h at pH 7.4 and pH 6.5. (B) Relative viabilities of 4T1 cells with different treatments. (C) GSH levels in 4T1 cells harvested from mice after treatment with DMSN-CB839, DMSN-Au-Fe3O4, and DMSN-Au-Fe3O4-CB839, respectively. (D) Live/dead staining of 4T1 cells incubated with different gr€s. (E) Fluorescein-annexin V and PI staining assays of 4T1 cells handled with different formulations. (F) Fluorescence images of DCFH-DA stained 4T1 cells after incubation with fresh medium, DMSN-CB839, DMSN-Au-Fe3O4, and DMSN-Au-Fe3O4-CB839 for 4 h. | ||
Afterwards, the in vitro anti-tumor effect of DMSN-Au-Fe3O4, DMSN-CB839 and DMSN-Au-Fe3O4-CB839 NPs was evaluated at pH 6.5. As expected, DMSN-Au-Fe3O4-CB839 NPs exhibited the highest cytotoxicity between the three NPs (Fig. 4(B)), displaying the synergistic effect of CDT and chemotherapy. As shown in Fig. S13 (ESI†), glutamate, transformed via glutamine pathway under the catalysis of GLS1, is the precursor for both GSH biosynthesis and tricarboxylic acid cycle (TCA). Thus, targeting GLS to interrupt glutamine pathway as the anti-cancer chemotherapy has been intensively applied in the clinical treatment. For instance, CB839, as the most potential allosteric inhibitor of GLS1 with an IC50 of 22 nM for GLS1 inhibition activity,39,40 exhibited favorable antiproliferative activity against HCT116 cells (IC50 = 0.11 μM) and MDA-MB-436 cells (IC50 = 0.43 μM),41 and preferably repressed a triple-negative breast cancer cell line with an IC50 value of 20–30 nM.42 It is proposed that CB839 as the GSL1 inhibitor induces the drastic conformational changes of the key ring near the catalytic site from the binding between the inhibitor and GLS, and consequently suppressing the generation of GSH. Therefore, the effect of DMSN-Au-Fe3O4-CB839 NPs on the intracellular GSH levels was conducted. As presented in Fig. 4(C), GSH levels in 4T1 cells incubated with DMSN-CB839 NPs and DMSN-Au-Fe3O4-CB839 NPs were decreased by 48.34% and 53.65%, respectively, while DMSN-Au-Fe3O4 NPs displayed no apparent influences on GSH levels, exhibiting the great advantages in decreasing GSH levels in comparison of a previous reported 30% reduction of GSH after treating HCT116 cancer cells with CB839 at a concentration of 200 nM.40 This further validated that CB839 could effectively eliminate cellular GSH levels and thereby contributing to the synergistic antitumor effect with DMSN-Au-Fe3O4 NPs.
Similar antiproliferative evidence can be found in the cell live/dead double staining experiments. To analyze the distributions of live and dead cells clearly, 4T1 cells treated with different NPs for 24 h were stained with calcein acetoxymethyl ester (Calcein-AM) and propidium iodide (PI), as presented in Fig. 4(D). Obviously, no cell death was observed in the control group, while the group treated with DMSN-CB839 NPs and DMSN-Au-Fe3O4 NPs showed 19% and 60% of cell death rate, respectively. Impressively, when treated with DMSN-Au-Fe3O4-CB839 NPs, the cell death rate reached as high as 90%. As shown in Fig. 4(E), the typical flow cytometric apoptosis assay exhibited an obvious increasing in apoptosis cell percentage in 4T1 cells with different treatments for 24 h compared with control group. The percent of apoptotic cells treated with DMSN-CB839, DMSN-Au-Fe3O4 and DMSN-Au-Fe3O4-CB839 NPs were significantly reduced to 80.11%, 40.27% and 12.32%, respectively (Fig. 4(E)). Furthermore, a ROS fluorescence probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA) was used to monitor the intracellular ˙OH generation, which can be oxidized by ROS into 2′,7′-dichlorofluorescein (DCF) with strong green fluorescence.43 As presented in Fig. 4(F), the cells without any treatment exhibited weak fluorescence, whereas the most bright and strong green fluorescence was detected in the cells treated with DMSN-Au-Fe3O4-CB839 nanoparticles, suggesting the strongest intracellular ROS production and the occurrence of oxidative stress in these tumor cells. Glutathione peroxidase 4 (GPX4) is a phospholipid peroxidase that catalyzes lipid peroxides and it reduces the accumulation of lipid-based ROS, in the presence of glutathione as an essential cofactor. As shown in Fig. S14 (ESI†), the expression of GPX4 was significantly upregulated in DMSN-Au-Fe3O4-CB839 group, resulting in intracellular ˙OH generation and provoking endogenous negative feedback regulation mechanisms. In addition, the in vitro evaluation of the broad-spectrum CDT efficacy of DMSN-Au-Fe3O4-CB839 NPs was validated, showing the A549 cells were also effectively eliminated by applying the DMSN-Au-Fe3O4-CB839 NPs (Fig. S15, ESI†). All the above results demonstrated that the remarkable upregulation of ROS and cell apoptosis was triggered by the synergistic effects of CDT and chemotherapy, i.e., the generation of H2O2 by GOx-like activity of Au NPs, the accumulation of ˙OH by POD-mimicking activity of Fe3O4 NPs, and the elimination of cellar GSH levels by CB839.
3.3 In vivo tumor chemodynamic therapy
Stimulated by the superior cancer killing effect of DMSN-Au-Fe3O4-CB839 in vitro, the assessment of their catalytic-therapeutic efficacy in vivo was further investigated. 4T1-tumorbearing mice were chosen as model and intratumorally (i.t.) injected with 100 μL of the following solutions: (i) phosphate buffered saline (PBS) as the control group; (ii) DMSN-CB839 (20 mg kg−1); (ii) DMSN-Au-Fe3O4 (20 mg kg−1); and (iv) DMSN-Au-Fe3O4-CB839 (20 mg kg−1) to investigate the therapeutic performance. Over a 14 day period, the body weight change of mice in the three therapeutic groups and control group exhibited an obviously increasing trend, indicating that DMSN-CB839, DMSN-Au-Fe3O4, and DMSN-Au-Fe3O4-CB839 had inappreciable side effects in vivo (Fig. 5(C)). The tumor volumes of each group were carefully recorded every two days. As shown in Fig. 5(A)–(D), intratumoral administration presented satisfactory tumor growth suppression effects. Compared with other groups, the tumor size of the mice treated with DMSN-Au-Fe3O4-CB839 NPs was the smallest. According to the variation of tumor size during the treatment, the inhibition rate of the DMSN-Au-Fe3O4-CB839 group was calculated to be 75.8%, much higher than that of the DMSN-CB839 group and DMSN-Au-Fe3O4 group (34.9% and 48.6%), demonstrating the superior therapeutic effect of the DMSN-Au-Fe3O4-CB839 treatment. As shown in Fig. 5(E), a similar tendency was also observed in tumor weights at day 14. The tumor weight of mice groups given with nanoparticles was lower than that of the control group. Concretely, the mice dosed with DMSN-Au-Fe3O4-CB839 displayed the lowest tumor weight, showing a 70.5% reduction of tumor weight in comparison with the control group while 30.0% and 41.0% reduction for the DMSN-CB839 and DMSN NP-treated groups, respectively. These results were consistent with the in vitro antitumor results because DMSN-Au-Fe3O4-CB839 NPs could continuously release the cargos of Au NPs, Fe3O4 NPs, and CB839 after injection, and obtain the synergistic antitumor effect by combining the Au-mediated glucose consumption, Fe3O4-mediated Fenton reaction, and glutamine pathway-mediated GSH depletion.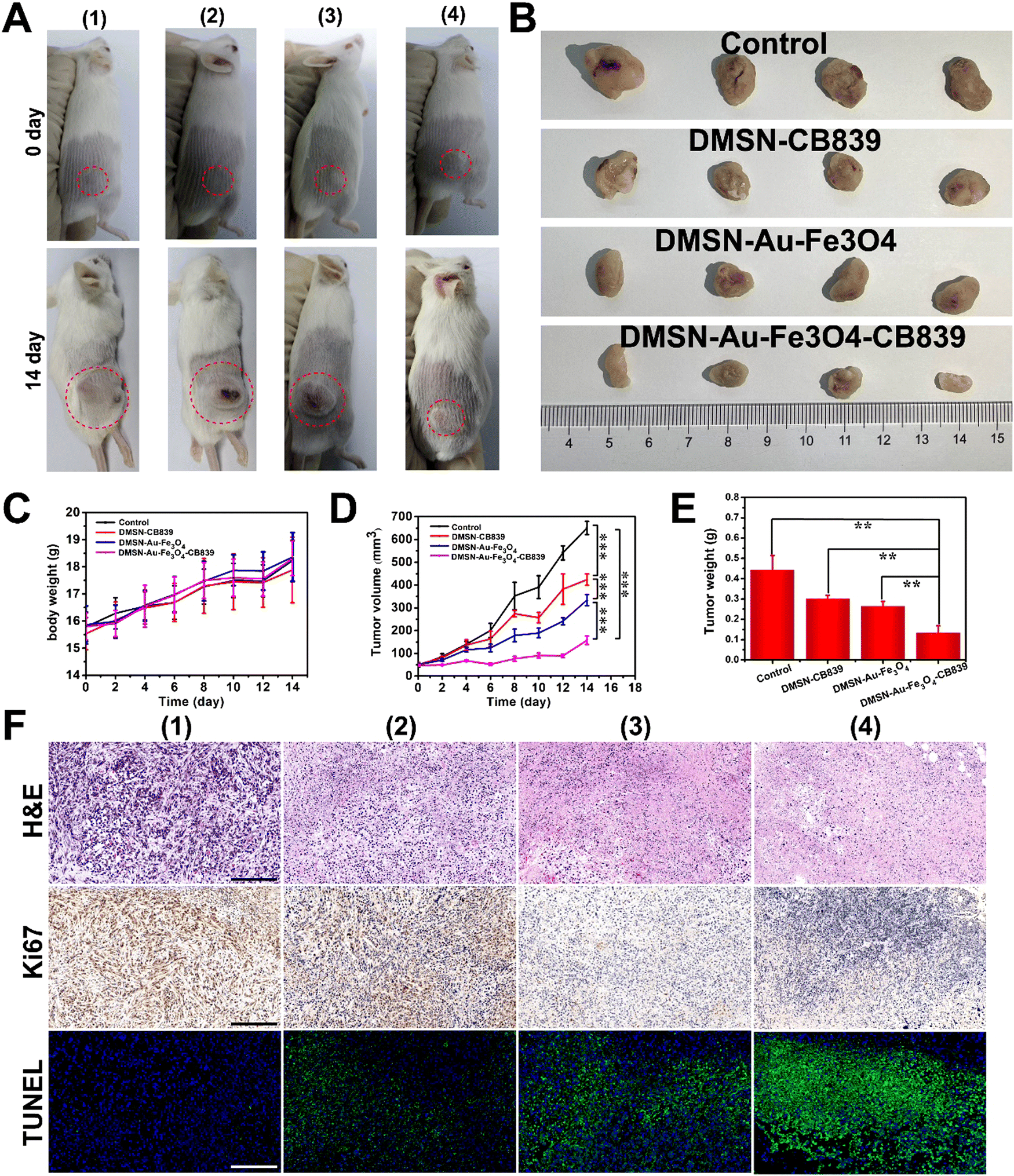 | ||
| Fig. 5 (A) Photos of the tumor-bearing mice before different treatments and after 14 days of treatments. (B) Photographs of the dissected tumors after treatments for 14 days. (C) Changes in body weight after various treatments. (D) Changes in tumor volumes after various treatments. (E) Weight of the tumor dissection after various treatments for 14 days. (F) H&E, Ki67, and TUNEL staining images of the dissected tumor tissues after 14 days of treatment. Scale bar: 200 μm. Groups (1), (2), (3), and (4) represent PBS, DMSN-CB839 (20 mg kg−1), DMSN-Au-Fe3O4 (20 mg kg−1), and DMSN-Au-Fe3O4-CB839 (20 mg kg−1), respectively. | ||
After the treatments, all mice were sacrificed, and their tumors and main organs (heart, liver, spleen, lungs, and kidneys) were dissected and stained with Hematoxylin and Eosin (H&E) to confirm their antitumor effect. As depicted in Fig. 5(F), staining results exhibited partial histological damage in tumor cells in all three therapeutic groups. Compared to the saline group, and tumor slices of DMSN-Au-Fe3O4-CB839 treated mice demonstrated the highest level of cell apoptosis and necrosis than the other groups. Also, no obvious off-target damage and inflammation were detected in the H&E staining images of the heart, liver, spleen, lungs, and kidneys of all the groups after the intervention, indicating the negligible pathological side effects on major organs of mice in therapeutic groups (Fig. S16, ESI†). Furthermore, obvious severe apoptosis and significantly inhibited proliferation capability of tumor cells were achieved in DMSN-Au-Fe3O4-CB839 group from the terminal-deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling (TUNEL) and Ki67 staining, respectively (Fig. 5(F)). Such results suggest the great potential biocompatibility and biosafety of DMSN-Au-Fe3O4-CB839 NPs as an effective antitumor chemodynamic therapy for future clinical application.
4. Conclusions
In summary, bioinspired by the unique glutamine metabolism pathway in tumor cells, we constructed an intelligent nanocatalytic platform to overcome the bottleneck for effective CDT. With high GOx and POD-like activity, Au and Fe3O4 coloaded DMSN nanoparticles catalyzed the TME-responsive cascade reaction, facilitating the production of H2O2 and ROS. Furthermore, CB839, the glutaminase inhibitor, suppressed glutaminase activity and thereby disturbed the glutamine pathway and GSH biosynthesis, reducing the consumption of ROS in tumor sites. The massive generation and accumulation of ROS could induce intracellular oxidative stress for the efficacy of CDT. The reactor exhibited a superior tumor-suppression rate and tumor inhibition effect in both in vitro and in vivo experiments. Overall, our findings provide not only a novel nanocatalytic platform with excellent antitumor efficacy, but also pave a new way for the design of a more effective cancer synergistic therapy for further practical application.Author contributions
Mao Huijia: methodology, validation, formal analysis, investigation, visualization, and writing – original draft. Wen Yangyang: conceptualization, methodology, investigation, formal analysis, and writing – review & editing. Yu Yonghui: investigation. Li Hongyan: writing – review & editing, supervision, and funding acquisition. Wang Jing: conceptualization, funding acquisition, and project administration. Sun baoguo: investigation and visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from the National Natural Science Foundation of China (32102010, 32172236, and 31901729) and the fund of Cultivation Project of Double First-Class Disciplines of Food Science and Engineering, Beijing Technology & Business University (No. BTBUYXTD202206).References
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- Z. Y. Cao, D. D. Li, J. X. Wang and X. Z. Yang, Acta Biomater., 2021, 130, 17–31 CrossRef CAS PubMed.
- L. Shi, Y. Wang, C. Zhang, Y. Zhao, C. Lu, B. Yin, Y. Yang, X. Gong, L. Teng and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 9562–9572 CrossRef CAS PubMed.
- J. Stanicka, E. G. Russell, J. F. Woolley and T. G. Cotter, J. Biol. Chem., 2015, 290, 9348–9361 CrossRef CAS PubMed.
- J. W. Crabb, L. Yan, M. Miyagi, X. Gu, K. A. West, A. M. Kamei, K. Shadrach, M. E. Rayborn, R. G. Salomon and J. G. Hollyfield, FASEB J., 2002, 16, A12–A12 Search PubMed.
- E. Hwang and H. S. Jung, Chem. Commun., 2020, 56, 8332–8341 RSC.
- B. W. Yang, Y. Chen and J. L. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS PubMed.
- Y. Y. Cui, X. Chen, Y. Cheng, X. Y. Lu, J. J. Meng, Z. W. Chen, M. K. Li, C. C. Lin, Y. L. Wang and J. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 22150–22158 CrossRef CAS PubMed.
- Q. W. Tian, F. F. Xue, Y. R. Wang, Y. Y. Cheng, L. An, S. P. Yang, X. Y. Chen and G. Huang, Nano Today, 2021, 39, 101162 CrossRef CAS.
- C. Zhang, W. B. Bu, D. L. Ni, S. J. Zhang, Q. Li, Z. W. Yao, J. W. Zhang, H. L. Yao, Z. Wang and J. L. Shi, Angew. Chem., Int. Ed., 2016, 55, 2101–2106 CrossRef CAS PubMed.
- Z. M. Tang, Y. Y. Liu, M. Y. He and W. B. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- H. J. Mao, Y. Y. Wen, Y. H. Yu, H. Y. Li, J. Wang and B. G. Sun, Mater. Today Bio, 2022, 16, 100436 CrossRef CAS PubMed.
- L. H. Fu, Y. Wan, C. Qi, J. He, C. Li, C. Yang, H. Xu, J. Lin and P. Huang, Adv. Mater., 2021, 33, 2006892 CrossRef CAS PubMed.
- Z. G. Ren, S. C. Sun, R. R. Sun, G. Y. Cui, L. J. Hong, B. C. Rao, A. Li, Z. J. Yu, Q. C. Kan and Z. W. Mao, Adv. Mater., 2020, 32, 1906024 CrossRef CAS PubMed.
- J. T. Xu, R. P. Shi, G. Y. Chen, S. M. Dong, P. P. Yang, Z. Y. Zhang, N. Niu, S. L. Gai, F. He, Y. J. Fu and J. Lin, ACS Nano, 2020, 14, 9613–9625 CrossRef CAS PubMed.
- X. Lin, S. Liu, X. Zhang, R. Zhu, S. Chen, X. Chen, J. Song and H. Yang, Angew. Chem., Int. Ed., 2020, 132, 1699–1705 CrossRef.
- Z. L. Dong, L. Z. Feng, Y. Chao, Y. Hao, M. C. Chen, F. Gong, X. Han, R. Zhang, L. Cheng and Z. Liu, Nano Lett., 2019, 19, 805–815 CrossRef CAS PubMed.
- X. Y. Zhong, X. W. Wang, L. Cheng, Y. A. Tang, G. T. Zhan, F. Gong, R. Zhang, J. Hu, Z. Liu and X. L. Yang, Adv. Funct., 2020, 30, 1907954 CrossRef CAS.
- Y. X. Xiong, C. Xiao, Z. F. Li and X. L. Yang, Chem. Soc. Rev., 2021, 50, 6013–6041 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Y. Sang, F. Cao, W. Li, L. Zhang, Y. You, Q. Deng, K. Dong, J. Ren and X. Qu, J. Am. Chem. Soc., 2020, 142, 5177–5183 CrossRef CAS PubMed.
- L. L. Zhou, Q. Guan, W. Y. Li, Z. Y. Zhang, Y. A. Li and Y. B. Dong, Small, 2021, 17, 2101368 CrossRef CAS PubMed.
- W. Yu, X. Y. Yang, Q. Zhang, L. Sun, S. T. Yuan and Y. J. Xin, Clin. Transl. Oncol., 2021, 23, 2253–2268 CrossRef CAS PubMed.
- C. R. Hoerner, V. J. Chen and A. C. Fan, Kidney Cancer., 2019, 3, 15–29 CAS.
- J. F. Zhang, S. Y. Mao, Y. D. Guo, Y. Wu, X. D. Yao and Y. Huang, Biosci. Rep., 2019, 39, BSR20181826 CrossRef CAS PubMed.
- D. C. Singleton, A. L. Dechaume, P. M. Murray, W. P. Katt, B. C. Baguley and E. Y. Leung, BMC Cancer, 2020, 20, 470 CrossRef CAS PubMed.
- A. C. Boese and S. M. Kang, Redox. Biol., 2021, 42, 101870 CrossRef CAS PubMed.
- M. M. Robinson, S. J. Mcbryant, T. Tsukamoto, C. Rojas, D. V. Ferraris, S. K. Hamilton, J. C. Hansen and N. P. Curthoys, Biochem. J., 2007, 406, 407–414 CrossRef CAS PubMed.
- J.-B. Wang, J. W. Erickson, R. Fuji, S. Ramachandran, P. Gao, R. Dinavahi, K. F. Wilson, A. L. Ambrosio, S. M. Dias and C. V. Dang, Cancer Cell, 2010, 18, 207–219 CrossRef CAS PubMed.
- W. H. Yang, Y. J. Qiu, O. Stamatatos, T. Janowitz and M. J. Lukey, Trends Cancer., 2021, 7, 790–804 CrossRef CAS PubMed.
- B. Giesen, A. C. Nickel, J. Barthel, U. D. Kahlert and C. Janiak, Pharmaceutics, 2021, 13, 295 CrossRef CAS PubMed.
- S. Ghosh, A. Javia, S. Shetty, D. Bardoliwala, K. Maiti, S. Banerjee, A. Khopade, A. Misra, K. Sawant and S. Bhowmick, J. Controlled Release, 2021, 337, 27–58 CrossRef CAS PubMed.
- S. Gao, H. Lin, H. Zhang, H. Yao, Y. Chen and J. Shi, Adv. Sci., 2019, 6, 1801733 CrossRef PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J.-G. Park, T.-Y. Ahn, Y.-W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- M. F. Huo, L. Y. Wang, Y. Chen and J. L. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- L. Wu, G. Wan, S. Shi, Z. He, X. Xu, Y. Tang, C. Hao and G. Wang, New J. Chem., 2019, 43, 5826–5832 RSC.
- J. M. Liu, A. Z. Wang, S. H. Liu, R. Q. Yang, L. W. Wang, F. Gao, H. G. Zhou, X. Yu, J. Liu and C. Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 25328–25338 CrossRef CAS PubMed.
- K. Sun, J. Z. Hu, X. Y. Meng, Y. F. Lei, X. Z. Zhang, Z. X. Lu, L. M. Zhang and Z. F. Wang, Small, 2021, 17, 2101897 CrossRef CAS PubMed.
- K. Koch, R. Hartmann, J. Tsiampali, C. Uhlmann, A.-C. Nickel, X. He, M. A. Kamp, M. Sabel, R. A. Barker, H.-J. Steiger, D. Hanggi, D. Willbold, J. Maciaczyk and U. D. Kahlert, Cell Death Discovery, 2020, 6, 20 CrossRef CAS.
- X. Xu, J. Wang, M. Wang, X. Yuan, L. Li, C. Zhang, H. Huang, T. Jing, C. Wang, C. Tong, L. Zhou, Y. Meng, P. Xu, J. Kou, Z. Qiu, Z. Li and J. Bian, J. Med. Chem., 2021, 64, 4588–4611 CrossRef CAS PubMed.
- V. Ruiz-Rodado, A. Lita, T. Dowdy, O. Celiku, A. C. Saldana, H. Wang, C. Z. Yang, R. Chari, A. Li, W. Zhang, H. Song, M. Zhang, S. Ahn, D. Davis, X. Chen, Z. Zhuang, C. Herold-Mende, K. J. Walters, M. R. Gilbert and M. Larion, Cancer Metab., 2020, 8, 23 CrossRef.
- Z. Chen, D. Li, N. Xu, J. Fang, Y. Yu, W. Hou, H. Ruan, P. Zhu, R. Ma, S. Lu, D. Cao, R. Wu, M. Ni, W. Zhang, W. Su and B. H. Ruan, J. Med. Chem., 2019, 62, 589–603 CrossRef CAS PubMed.
- L. S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang and W. Tang, Angew. Chem., Int. Ed., 2018, 130, 4996–5000 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb02194c |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2023 |