
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale patterning of polymers on DNA origami†
Nico
Alleva
,
Pia
Winterwerber
,
Colette J.
Whitfield
 ,
David Y. W.
Ng
* and
Tanja
Weil
*
,
David Y. W.
Ng
* and
Tanja
Weil
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: david.ng@mpip-mainz.mpg.de; weil@mpip-mainz.mpg.de
First published on 8th June 2022
Abstract
The combination of DNA–origami and synthetic polymers paves the way to a new class of structurally precise biohybrid nanomaterials for diverse applications. Herein, we introduce the grafting to method with high conversions (70–90%) under ambient conditions to generate DNA–polymer conjugates, which can hybridized precisely to DNA–origami architectures. We generated homo and block copolymers from three different polymer families (acrylates, methacrylates and acrylamides), coupled them to single stranded DNA (ssDNA) and pattern different DNA–origami architectures to demonstrate the formation of precise surface nanopatterns.
 David Y. W. Ng | David received his BSc in Chemistry with first class honours in 2009 at the National University of Singapore. In 2010, his PhD studies on dendrimer–protein transporters was funded by the Max Planck Institute for Polymer Research (MPIP) to work under the supervision of Prof. Tanja Weil in Ulm. David graduated with summa cum laude in 2014 and led a junior group until 2016. He then moved to the MPIP and formed the synthetic life-like systems group focusing on constructing dynamic bioactive structures in living cells. In 2021, he is appointed to concurrently lead the BioCore facility of the MPIP. |
Importantly, the unique facet of DNA–polymer conjugates lies in a versatile platform to create architectures of higher complexity where DNA nanotechnology can be used to guide polymers with high precision in the nanoscale.8 This strategy entails the use of DNA–origami as a template, where a long single-stranded (ss) DNA is folded by short oligonucleotides, so called staple strands, into complex 3-D DNA origami architectures like rectangles, tubes, stars or many other architectures.9 As each grid position on DNA–origami can be independently functionalized, radical initiators10 and/or photocatalysts11 have been patterned to promote polymerization at the designated locations. However, conducting polymerization reactions directly on DNA–origami requires stringent conditions such as high ionic strength buffers with divalent cations Mg2+/Ca2+, low reaction volumes typically below 100 μl and mild reaction conditions during polymerization. This significantly limits both the monomer scope and polymerization technique. Additionally, it is often difficult to characterize the polymers (i.e. molecular weights, dispersity) grafted from the DNA origami due to its very low quantities. Herein, we circumvent these limitations by adapting the grafting to approach, where polymers are synthesized in organic solvents (Fig. 1) before binding to DNA–origami for creating precise 3-D polymeric nanopatterns. This approach is already used in literature for the creation of functional DNA–polymer conjugates for e.g. fluorescent polythiophenes to form patterned DNA–origami in a high precise way.8,12
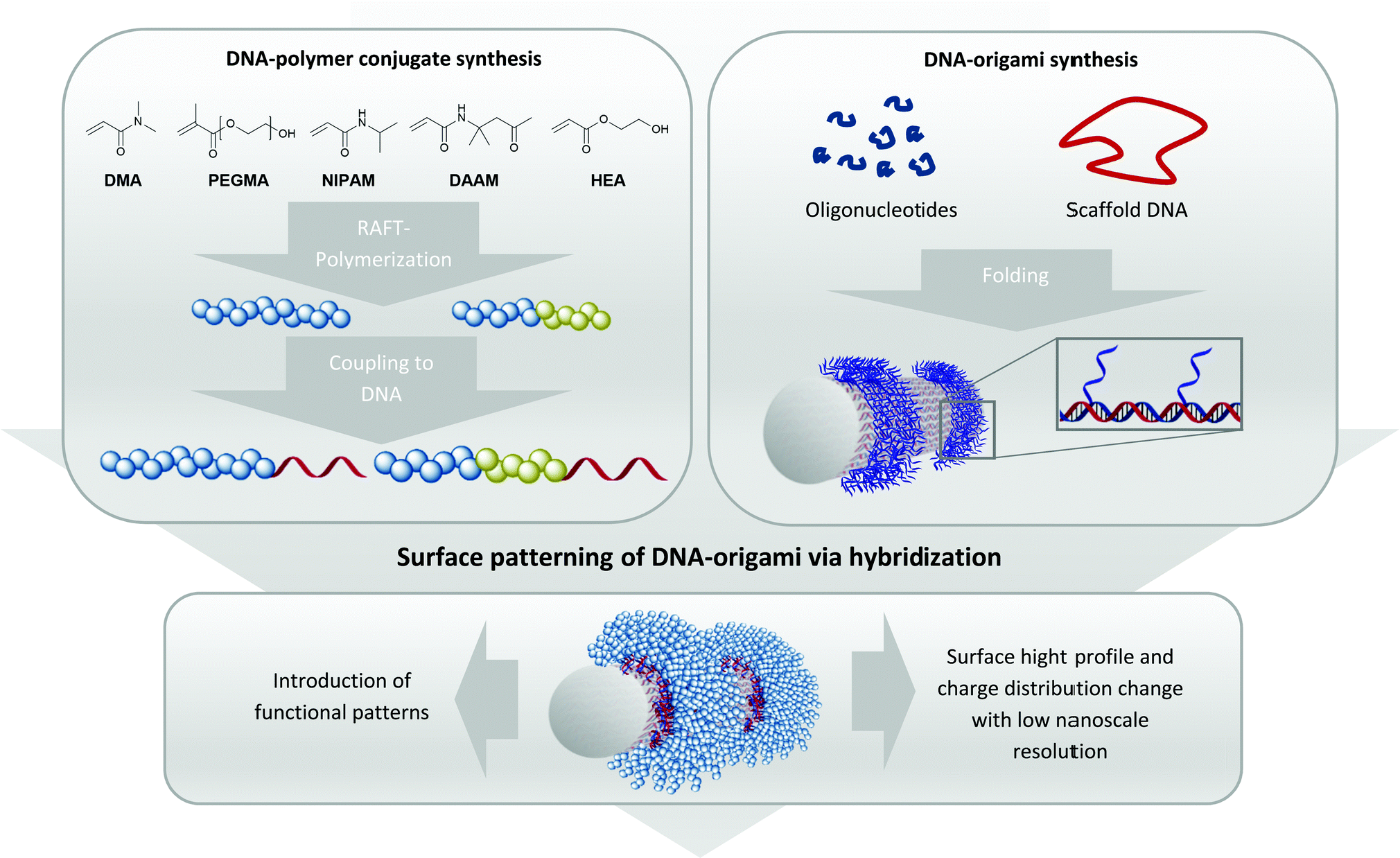 | ||
| Fig. 1 Concept of the combination of the grafting to method for generating DNA–polymer conjugates and the surface coating of DNA–origami due to protruding single stranded sticky sequences on the surface. The selected monomers form polymers with different features to diversify the inherent properties of DNA–origami nano-objects. The monomers were polymerized via RAFT polymerization and then coupled to single stranded DNA via NHS–amine conjugation chemistry under mild and ambient conditions. This versatile method generates a wide range of DNA–polymer conjugates for precise surface patterning of DNA–origami nanostructures. | ||
In order to position polymer chains onto DNA–origami, staple strands corresponding to the designated positions have to be elongated by a sticky DNA sequence. These extensions protrude as a ssDNA from the DNA–origami surface, allowing complementary sequences appended onto the polymer chains to recognize.13 Hence, polymer chains with different sets of sticky DNA sequences can self-assemble onto their corresponding sites to afford a customizable architecture. As such, this technique grants access towards achieving precise geometric shapes that cannot be constructed via conventional polymer synthesis methods.14,15 Three different kinds of monomer backbone (acrylates, methacrylates and acrylamides) were selected to underline the versatility of the approach. Technically, the production of 1–1 DNA–polymer conjugates can be performed via the grafting from or the grafting to method. In the grafting from method, the DNA block contains an initiator molecule and polymerization of the monomer proceeds in situ to form the respective DNA–polymer conjugate.16 However, the polymerization reaction has to be performed under conditions that accommodate the monomer, polymer and DNA components and there is a general loss of controllability and dispersity of the resultant polymers. On the contrary, the grafting to method synthesizes the polymer independently, which also facilitates characterization and easy scalability. After successful synthesis, bioconjugation to the DNA strand furnishes the target conjugate. As the polymer is synthesized prior the bioconjugation, the polymer block can be tailored with high flexibility even if the polymer chain is hydrophobic.17 Due to these advantages, the grafting to method opens access to various DNA–polymer conjugates containing homo and block copolymers with hydrophilic and hydrophobic monomer units.18
Dimethyl acrylamide (DMA), oligoethylene glycol acrylate (OEGMA), N-isopropyl acrylamide (NIPAM), hydroxyethyl acrylate (HEA) and diacetone acrylamide (DAAM) were polymerized via reversible–addition–fragmentation chain-transfer polymerization (RAFT) to form the homo polymers and block copolymers (Fig. 2a). These monomers constitute widely used classes in the polymer community with DMA and OEGMA promoting aqueous solubility19 whereas NIPAM, HEA and DAAM are common functional monomers in self-assembly systems.16,20 As with RAFT polymerization, the control over dispersity and chain length can be accomplished along with a wide selection of monomers and flexible end group modifications.16,21 Polymerizations of DMA, NIPAM, HEA and DAAM were performed with 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid N-hydroxysuccinimide ester (NHS-DDMAT) acting as the chain transfer agent (CTA) in dioxane or DMF. Block co-polymerization consisting of hydrophobic P(DAAM) and hydrophilic P(DMA) were synthesized to demonstrate the robustness of the functionalization reaction and subsequent patterning. To prevent side reactions during the subsequent bioconjugation reaction with oligonucleotides, the CTA group of the NHS-polymers was removed post polymerization with an excess of azobisisobutyronitrile (AIBN) in dioxane at 75 °C. The obtained NHS–polymers revealed narrow molecular weight distributions (Đ = 1.08–1.27, Table S3, ESI†) and a wide range of polymers with different molecular weights were synthesized (9.6–48.6 kDa, analysed by GPC (Fig. 2(b)). To perform the bioconjugation reaction of the NHS-functionalized polymers with the 5′amino oligonucleotide (complementary sticky A (StAc; 5′-NH2-TTTTCTCTACCACCTACTA-3′)13 or complementary sticky E (StEc; 5′-NH2-CAGTCAGTCAGTCAGTCAGT-3′)15) (Fig. 2c), the solvent has a high impact on the conversion. The accessibility of the reactive functionalities drives the reaction efficiency and therefore requires a good solvent that prevents aggregation of the polymer and the oligonucleotide chains. Optimization on the solvent conditions was performed in acetonitrile (ACN), dimethylformamide (DMF), water and mixtures thereof using P2 as a model polymer, monitored by native polyacrylamide gel electrophoresis (PAGE) (Fig. S2 and S3, ESI†). In comparison, DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (3:1) was the best reaction solvent to afford a conversion between 70–90% (Fig. 2d), quantified by integrating the band intensity of the PAGE gels (Fig. S1 and Table S2, ESI†). The PAGE revealed that the conversion depends on the chain length of the respective polymer, which is exemplarily shown for the DMA polymers (P1–P3) with increasing intensity of the unreacted 5′amino oligonucleotide. Comparing across polymer families, P(NIPAM-b-DMA) (P6) showed higher conversions than P(DMA) (P2–P3) and even the functionalization of P(OEGMA) (P4) proceeds well despite its brush like structure. Importantly, the reaction conditions were also robust for amphiphilic type block copolymer P(DAAM-b-DMA) P8, with an estimated conversion of ∼90%.
:water (3:1) was the best reaction solvent to afford a conversion between 70–90% (Fig. 2d), quantified by integrating the band intensity of the PAGE gels (Fig. S1 and Table S2, ESI†). The PAGE revealed that the conversion depends on the chain length of the respective polymer, which is exemplarily shown for the DMA polymers (P1–P3) with increasing intensity of the unreacted 5′amino oligonucleotide. Comparing across polymer families, P(NIPAM-b-DMA) (P6) showed higher conversions than P(DMA) (P2–P3) and even the functionalization of P(OEGMA) (P4) proceeds well despite its brush like structure. Importantly, the reaction conditions were also robust for amphiphilic type block copolymer P(DAAM-b-DMA) P8, with an estimated conversion of ∼90%.
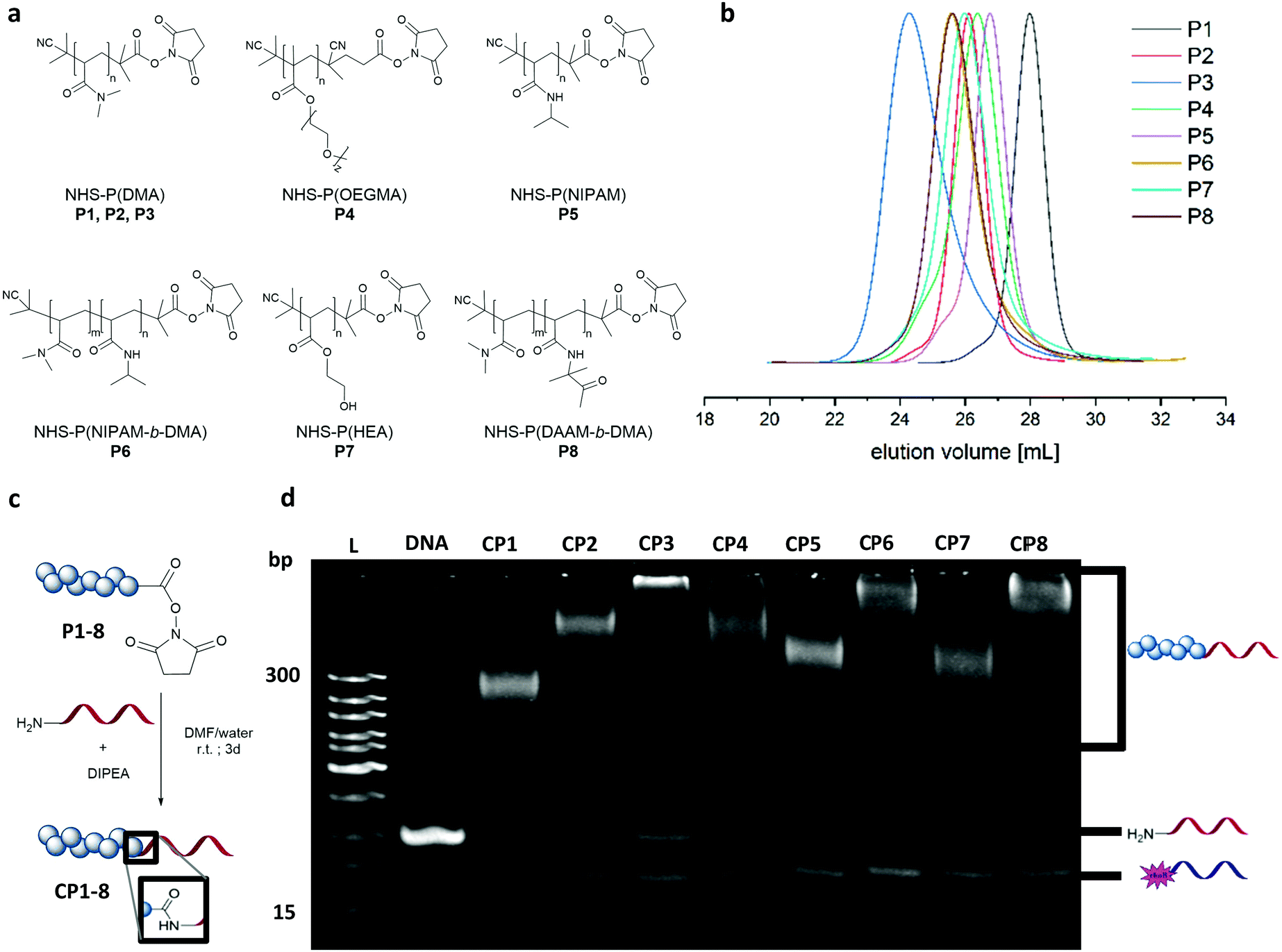 | ||
| Fig. 2 (a) Obtained polymers after CTA group removal, containing the NHS group for bioconjugation to DNA. (B) GPC traces of the NHS polymers P1–P8 as measured by DMF GPC using polymethylmethacrylate (PMMA) as calibration standard. (c) Schematic representation of the coupling reaction of the generated NHS polymers with the 5′ amino oligonucleotide using DIPEA as the auxiliary base. (D) DNA and DNA–polymer conjugation reaction solutions analysed by 15% PAGE, stained with SYBR Gold. Complementary rhodamine DNA was used for hybridization to obtain better staining. L: DNA ladder; DNA: used 5′amino oligonucleotide; CP1–CP8: coupling reaction solutions of P1–P8 with 5′amino oligonucleotide (StA). | ||
Next, the DNA–polymer conjugates were patterned onto the surface of DNA origami nanotubes. First, the DNA–DMA conjugates of three different polymer chain length (P1: 9.6 kDa; P2: 22.1 kDa; P3: 48.6 kDa), were purified via spin filtration to remove unreacted amino oligonucleotides. Subsequently, the conjugates were hybridized to the DNA–origami tube containing patterned StA sequences (Fig. 3a). The attachment was performed in origami buffer (1 mM Na2EDTA, 5 mM NaCl, 5 mM Tris, 12 mM MgCl2 pH 8) at 37 °C for 1 h. The resulting architectures (O1, O2, O3) were monitored via atomic force microscopy (AFM) to determine the height profile of the DMA coated DNA–origami against uncoated DNA–origami (O0) (Fig. 3b). The increase in height as a function of polymer weight demonstrated successful hybridization of the polymer chains and showed a nonlinear dependence between polymer length and height change. The trend is expected due to the increase in chain collapse as the molecular weight of the polymer chains increases. As such, the z-axis contribution per monomer is weighted less as the polymer grows larger. The attachment of the polymers was independently characterized using agarose gel, with shifts in molecular weight to charge ratios corresponding to the size of the polymers used (Fig. S4, ESI†).
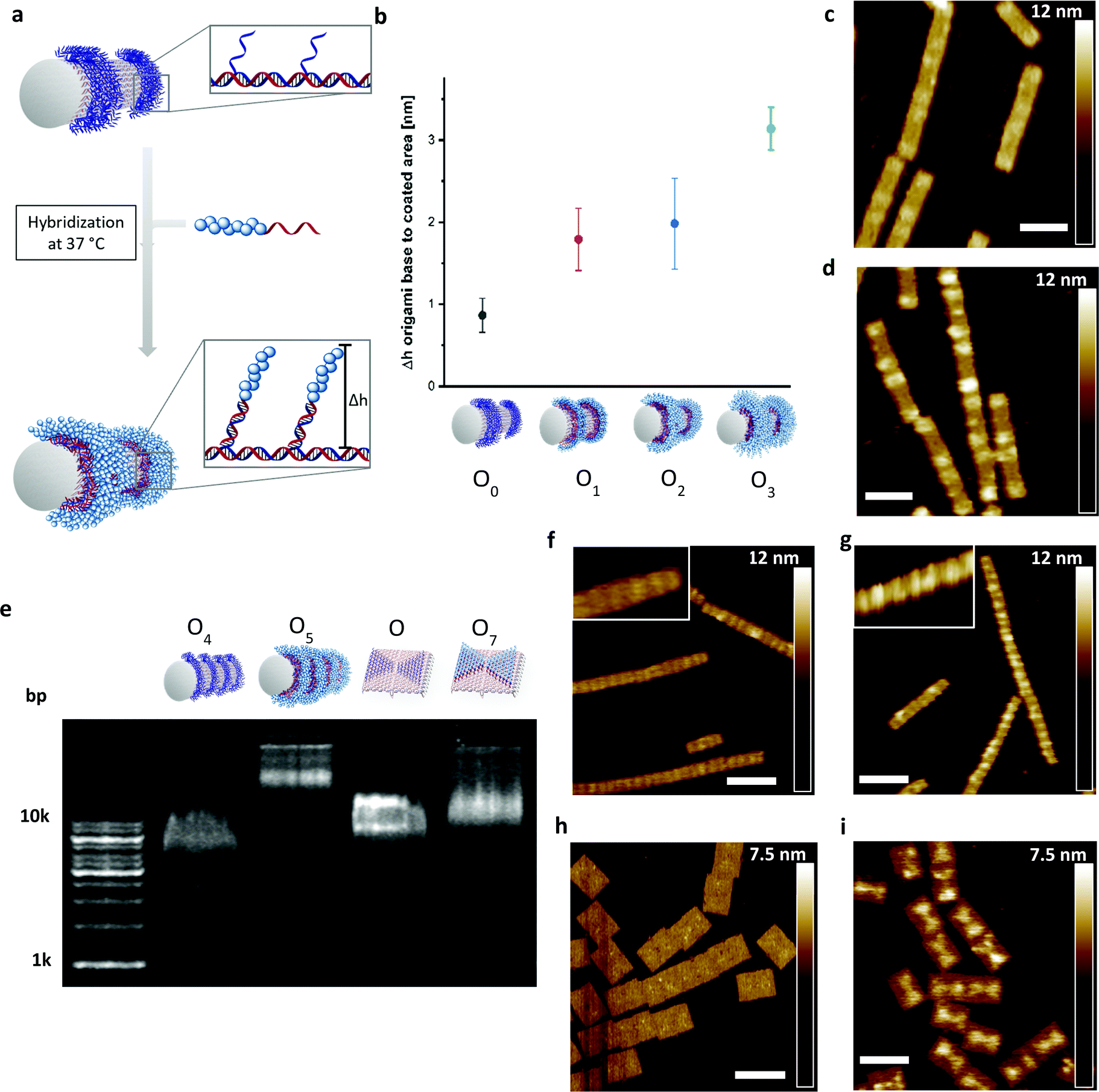 | ||
| Fig. 3 (a) Schematic representation of the DNA–origami coating with DNA–polymer conjugates by hybridization at 37 °C. (b) Height changes of the DNA origami surface obtained via AFM, where the respective uncoated and the coated sections on the origami nanostructures were measured to obtain the height difference (n = 10 times for each coating). Heights from Fig. S5–S8 and Tables S4–S7. (c) Uncoated DNA–origami imaged by AFM. Scale bar = 80 nm. (d) DNA–origami coated with CP3 imaged by AFM. Scale bar = 80 nm. (e) Monitoring of the coated and uncoated DNA–origami tube and rectangle containing StA by 1% agarose gel, stained with SYBR Gold; L: DNA ladder; O4: uncoated DNA–origami; O5: coated DNA–origami with CP3; O6: uncoated DNA–origami; O7: coated DNA–origami with CP2. (f–i) Respective DNA–origami architectures uncoated and coated with the respective DNA–polymer conjugate; tube: CP3; rectangle: CP2. Scale bar = 120 nm. | ||
To show the flexibility to accommodate patterns of varying shapes and of different origami templates, a DNA–origami tube containing four StA rings (O4) and a DNA–origami tile with two StA triangles (O6) were hybridized with CP2 or CP3 and imaged via AFM (Fig. 3f–i). The topological height change observed in the AFM image and the band shift in the agarose gel (Fig. 3e) indicate that the patterning of the polymeric architectures was successful, demonstrating the nanoscale resolution of the coating, even for larger (∼50 kDa) DNA–polymer conjugates. Further customization can be achieved by using different sticky sequences to assemble different polymers onto a single nano-object. A DNA–origami tube patterned with StE and StA sequences (O8) was hybridized with StEc-P7 conjugate and CP2 (Fig. 4). The recognition between each set of sticky sequences and their complementary binding partners was characterized stepwise. Using O8, hybridization was performed with StEc-P7 to form O9. The attachment of StEc-P7 was observed via AFM only on one end of the DNA–origami tube resulting in a height difference of ∼3–4 nm (Fig. 4e), thus underlining the specificity of the hybridization reaction. The influence of the attached polymers in terms of molecular weight and charge change of the DNA–origami were determined by agarose gel electrophoresis (Fig. 4d). With both StEc-P7 and CP2 present, the designated two-ring structure was formed.
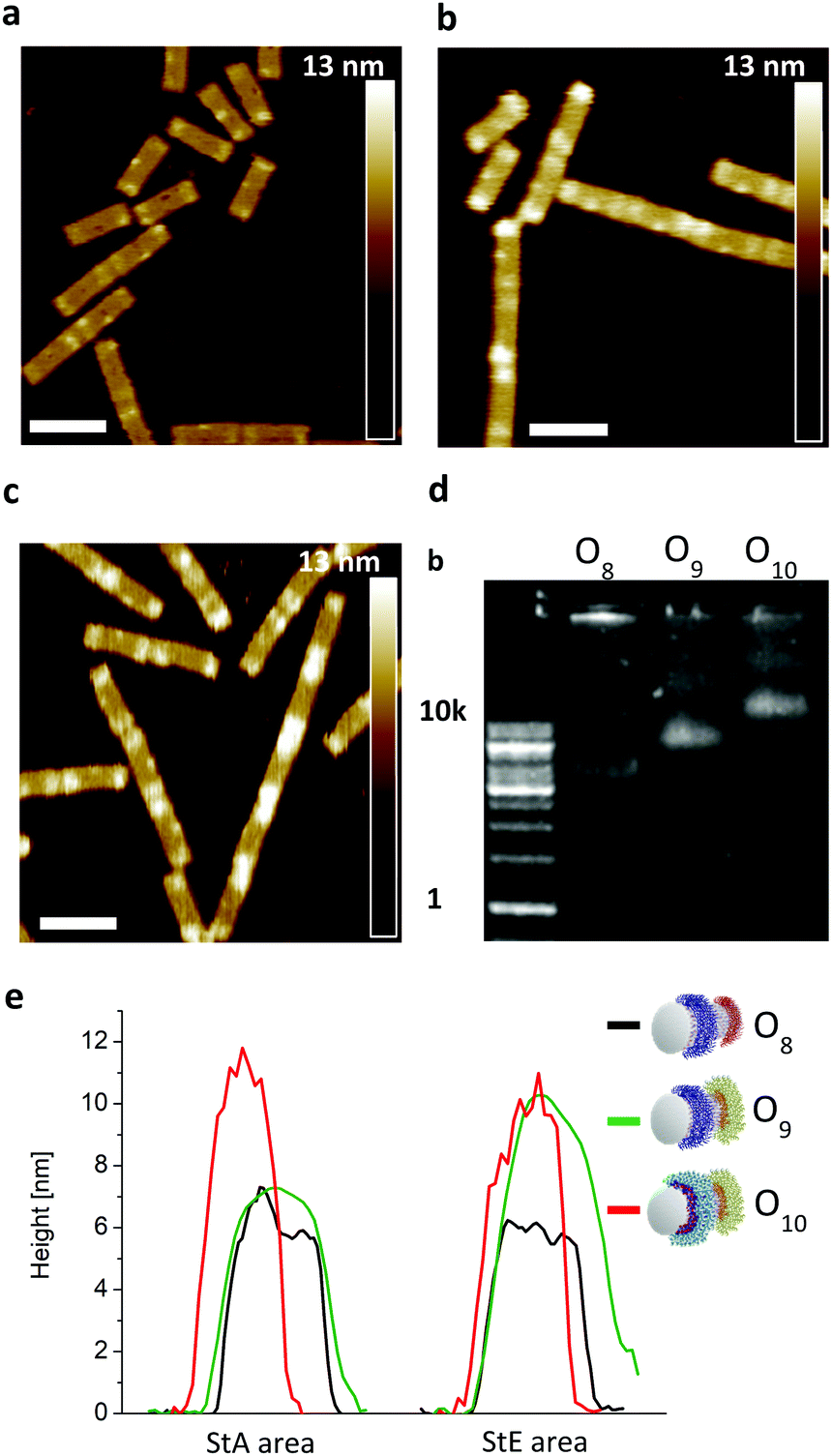 | ||
| Fig. 4 (a) Uncoated DNA–origami (O8) monitored via AFM. Scale bar = 100 nm (b) DNA–origami (O9) with StEc-P7 monitored via AFM. Scale bar = 100 nm (c) DNA–origami (O10) coated with StEc-P7 and CP2. Scale bar = 100 nm (d) Coated and uncoated DNA–origami monitored with 1% agarose gel, stained with SYBR Gold. O8 is uncoated, O9 is coated with StEc-P7 and O10 is coated with CP2 and StEc-P7. (e) AFM topographic images reveal a significant increase in height of the respective coating area. | ||
Collectively, we showed that the grafting to strategy present several advantages over the grafting from strategy. First, the polymers can be fully characterized prior to patterning. This allows us to attribute material characteristics to each polymer scaffold and composition. In addition, the methodology is more modular, where polymer combinations one origami can be easily achieved. Furthermore, the grafting to strategy showed better sustainability and cost effectiveness evaluated by Eco Scale,22 a green metric factor (see ESI†).
In conclusion, we have introduced a robust grafting to procedure under ambient conditions with high conversions to achieve various DNA–polymer conjugates containing homo and block copolymers. These DNA–polymer conjugates from widely used polymer families (methacrylates acrylates and acrylamides) show conserved DNA-based recognition properties to their complementary sequences. The hybridization process was also not affected by the steric demand of the polymer chain, which was demonstrated by varying the molecular weight from ∼10 kDa to 50 kDa. The attachment of the polymers to the DNA–origami was demonstrated on both tube and tile origamis. Different polymer chains, each equipped with a unique set of sticky sequences, can be attached onto each origami nano-object in a convenient manner also applicable for non-polymer chemists. With the rising importance of precisely engineered interfaces in nanotechnology and biomedicine, hybrid DNA–polymer conjugates remain one of the most accessible strategies to achieve coatings with nanoscale precision.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project No. 364549901 – TRR 234 CataLight (B01) and the Max Planck Society for Open Access Funding.References
- J. M. Gibbs, S.-J. Park, D. R. Anderson, K. J. Watson, C. A. Mirkin and S. T. Nguyen, J. Am. Chem. Soc., 2005, 127, 1170–1178 CrossRef CAS PubMed.
- H. Kang, H. Liu, X. Zhang, J. Yan, Z. Zhu, L. Peng, H. Yang, Y. Kim and W. Tan, Langmuir, 2011, 27, 399–408 CrossRef CAS PubMed.
- (a) F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed; (b) F. Cavalieri, A. Postma, L. Lee and F. Caruso, ACS Nano, 2009, 3, 234–240 CrossRef CAS PubMed.
- T. R. Wilks, J. Bath, J. W. de Vries, J. E. Raymond, A. Herrmann, A. J. Turberfield and R. K. O'Reilly, ACS Nano, 2013, 7, 8561–8572 CrossRef CAS PubMed.
- K. Isoda, N. Kanayama, M. Fujita, T. Takarada and M. Maeda, Chem. – Asian J., 2013, 8, 3079–3084 CrossRef CAS PubMed.
- Z. Zhao, L. Wang, Y. Liu, Z. Yang, Y.-M. He, Z. Li, Q.-H. Fan and D. Liu, Chem. Commun., 2012, 48, 9753–9755 RSC.
- L. Yang, H. Sun, Y. Liu, W. Hou, Y. Yang, R. Cai, C. Cui, P. Zhang, X. Pan, X. Li, L. Li, B. S. Sumerlin and W. Tan, Angew. Chem., Int. Ed., 2018, 130, 17294–17298 CrossRef.
- J. B. Knudsen, L. Liu, A. L. Bank Kodal, M. Madsen, Q. Li, J. Song, J. B. Woehrstein, S. F. J. Wickham, M. T. Strauss, F. Schueder, J. Vinther, A. Krissanaprasit, D. Gudnason, A. A. A. Smith, R. Ogaki, A. N. Zelikin, F. Besenbacher, V. Birkedal, P. Yin, W. M. Shih, R. Jungmann, M. Dong and K. V. Gothelf, Nat. Nanotechnol., 2015, 10, 892–898 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- Y. Tokura, Y. Jiang, A. Welle, M. H. Stenzel, K. M. Krzemien, J. Michaelis, R. Berger, C. Barner-Kowollik, Y. Wu and T. Weil, Angew. Chem., Int. Ed., 2016, 128, 5786–5791 CrossRef.
- P. Winterwerber, S. Harvey, D. Y. W. Ng and T. Weil, Angew. Chem., Int. Ed., 2020, 59, 6144–6149 CrossRef CAS PubMed.
- J. Zessin, F. Fischer, A. Heerwig, A. Kick, S. Boye, M. Stamm, A. Kiriy and M. Mertig, Nano Lett., 2017, 17, 5163–5170 CrossRef CAS PubMed.
- P. Winterwerber, C. J. Whitfield, D. Y. W. Ng and T. Weil, Angew. Chem., Int. Ed., 2021, e202111226 Search PubMed.
- N. P. Agarwal, M. Matthies, F. N. Gür, K. Osada and T. L. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 5460–5464 CrossRef CAS PubMed.
- J. Schill, B. J. H. M. Rosier, B. Gumí Audenis, E. Magdalena Estirado, T. F. A. de Greef and L. Brunsveld, Angew. Chem., Int. Ed., 2021, 60, 7612–7616 CrossRef CAS PubMed.
- T. Lückerath, K. Koynov, S. Loescher, C. J. Whitfield, L. Nuhn, A. Walther, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Angew. Chem., Int. Ed., 2020, 59, 15474–15479 CrossRef PubMed.
- S. Hansson, V. Trouillet, T. Tischer, A. S. Goldmann, A. Carlmark, C. Barner-Kowollik and E. Malmström, Biomacromolecules, 2013, 14, 64–74 CrossRef CAS PubMed.
- M. Safak, F. E. Alemdaroglu, Y. Li, E. Ergen and A. Herrmann, Adv. Mater., 2007, 19, 1499–1505 CrossRef CAS.
- (a) S. Maiez-Tribut, J. P. Pascault, E. R. Soulé, J. Borrajo and R. J. J. Williams, Macromolecules, 2007, 40, 1268–1273 CrossRef CAS; (b) K. G. Neoh and E. T. Kang, Polym. Chem., 2011, 2, 747–759 RSC.
- A. Khan, T. H. Khan, A. M. El-Toni, A. Aldalbahi, J. Alam and T. Ahamad, Mater. Lett., 2019, 235, 197–201 CrossRef CAS.
- S. Goldmann, D. Quémener, P.-E. Millard, T. P. Davis, M. H. Stenzel, C. Barner-Kowollik and A. H. Müller, Polymer, 2008, 49, 2274–2281 CrossRef.
- K. Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00812b |
| This journal is © The Royal Society of Chemistry 2022 |