
Molecularly imprinted polymer nanogels targeting the HAV motif in cadherins inhibit cell–cell adhesion and migration†
Paulina X.
Medina Rangel‡
*a,
Alejandra
Mier
a,
Elena
Moroni
a,
Franck
Merlier
a,
Levi A.
Gheber
b,
Razi
Vago
b,
Irene
Maffucci
 a,
Bernadette
Tse Sum Bui
*a and
Karsten
Haupt
*a
a,
Bernadette
Tse Sum Bui
*a and
Karsten
Haupt
*a
aCNRS Enzyme and Cell Engineering Laboratory, Université de Technologie de Compiègne, Rue du Docteur Schweitzer, CS 60319, 60203 Compiègne Cedex, France. E-mail: paulina.medinarangel@yale.edu; jeanne.tse-sum-bui@utc.fr; karsten.haupt@utc.fr
bThe Avram and Stella Goldstein-Goren Department of Biotechnology Engineering, Ben-Gurion University of the Negev, P.O. Box 653, Beer-Sheva 84105, Israel
First published on 9th May 2022
Abstract
Cadherins are cell–surface proteins that mediate cell–cell adhesion. By regulating their grip formation and strength, cadherins play a pivotal role during normal tissue morphogenesis and homeostasis of multicellular organisms. However, their dysfunction is associated with cell migration and proliferation, cancer progression and metastasis. The conserved amino acid sequence His-Ala-Val (HAV) in the extracellular domain of cadherins is implicated in cadherin-mediated adhesion and migration. Antagonists of cadherin adhesion such as monoclonal antibodies and small molecule inhibitors based on HAV peptides, are of high therapeutic value in cancer treatment. However, antibodies are not stable outside their natural environment and are expensive to produce, while peptides have certain limitations as a drug as they are prone to proteolysis. Herein, we propose as alternative, a synthetic antibody based on molecularly imprinted polymer nanogels (MIP-NGs) to target the HAV domain. The MIP-NGs are biocompatible, have high affinity for N-cadherin and inhibit cell adhesion and migration of human cervical adenocarcinoma (HeLa) cells, as demonstrated by cell aggregation and Matrigel invasion assays, respectively. The emergence of MIPs as therapeutics for fighting cancer is still in its infancy and this novel demonstration reinforces the fact that they have a rightful place in cancer treatment.
Introduction
Cadherins are a large family of cell-surface transmembrane glycoproteins that mediate Ca2+-dependent cell-cell adhesion.1 Cadherin-mediated adhesion plays a central role in tissue morphogenesis during embryonic development, maintenance of tissue and organ architecture, cell motility and gene expression.2 However, the dysfunction of these molecules due to various pathological conditions is implicated in uncontrolled cell migration and proliferation, tumour and metastastatic cancer progression.3–6 Type I classical cadherins which comprise E (epithelial), N (neural) and P (placental) cadherins are characterized by their five ectodomains (EC1-EC5), a conserved tryptophan residue localized at the second position (Trp2) at the N-terminal and a conserved His-Ala-Val (HAV) sequence in the EC1 ectodomain2,7 (Fig. 1A). Cadherin-mediated cell-cell adhesion occurs via the reciprocal insertion of Trp2 from one cadherin into the hydrophobic pocket, lined by the conserved HAV domain of an identical cadherin from an adjacent cell.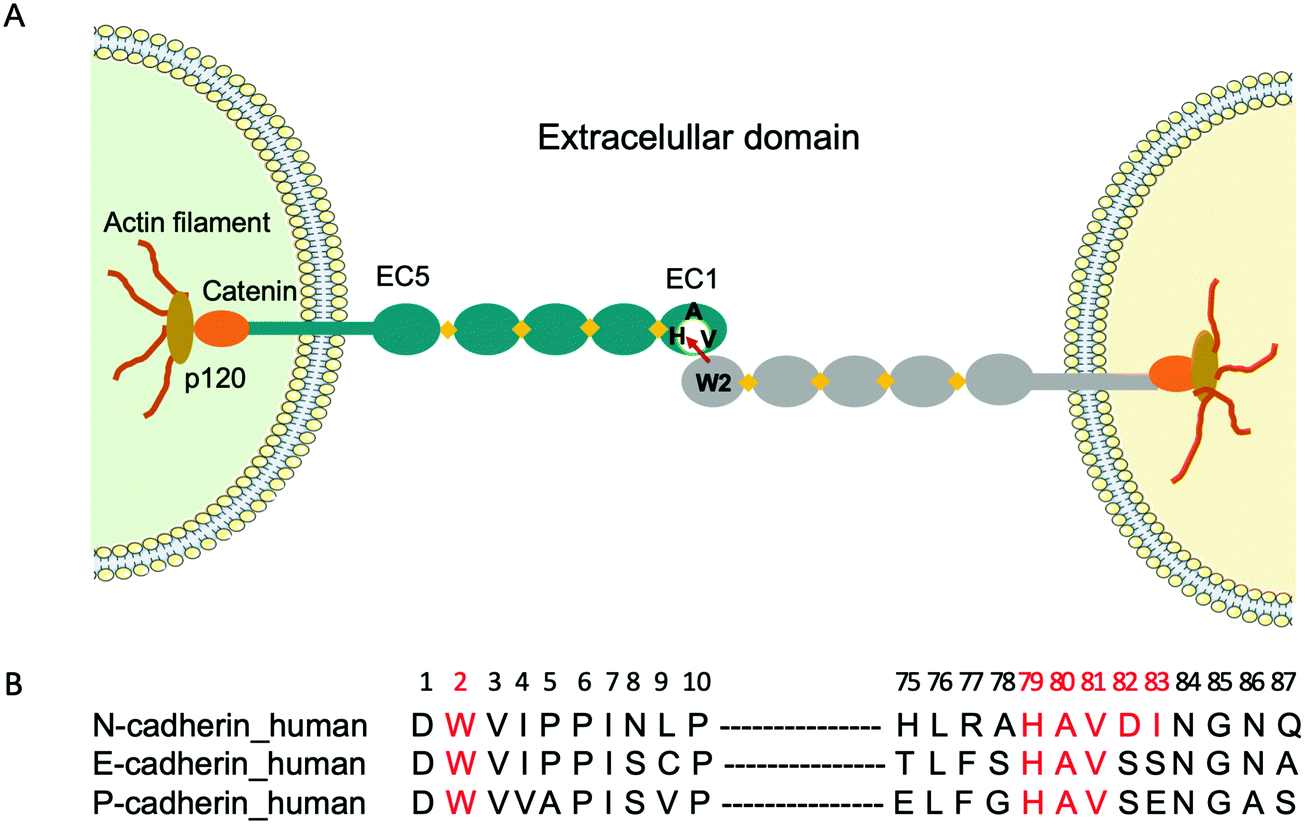 | ||
| Fig. 1 (A) Schematic representation of trans dimerization of cadherins. The five ectodomains (EC1 to EC5) are represented as grey and green ovals, and Ca2+ ions are shown as yellow rhombi. Adhesion occurs by the insertion of W2 from one cadherin into the hydrophobic pocket, lined by HAV located in the EC1 of the adjacent cadherin. (B) Multiple sequence alignment showing the amino acids (red) in human E-, N- and P-cadherins, involved in cell adhesion. Alignment performed with EMBL-EBI. | ||
To fight cancer development and metastasis, E-, N- and P-cadherins have served as viable therapeutic targets for numerous antibodies5,8,9 and small molecule inhibitors.10 The therapeutic antibodies were directed towards various epitopes on the extracellular domain and a few have been tested in preclinical studies.11,12 Regarding small molecule inhibitors, peptides containing the cell adhesion recognition sequence HAV have been used principally as N-cadherin antagonists.10,13 For instance, the cyclic peptide N-Ac-CHAVC-NH2, known as ADH-1 and developed by Adherex Technologies Inc., has successfully completed a number of phase I and II clinical trials, for its ability to inhibit tumour growth when used alone14 or in combination with anti-cancer drugs.15 Importantly, the amino acids that flank the HAV domain determine both the specificity and the activity of the peptides. Consequently, several peptides that act as N-cadherin inhibitors have been synthesized by incorporating flanking amino acids specific to N-cadherin to the HAV peptide. One of the first reported peptides with this purpose is the linear decapeptide N-Ac-LRAHAVDING-NH2, that inhibits human N-cadherin in neurite outgrowth.16 More recently, a shorter cyclic peptide containing the HAVDI sequence13,17 was identified as a highly potent inhibitor of N-cadherin cell adhesion and cell differentiation. It was also shown that when the HAV motif is flanked by a single serine, which mimics the natural HAVS sequence of E-cadherin, it loses its ability to inhibit the N-cadherin response, underlining the high specificity of interaction. However, peptides have certain limitations as a drug, as they are prone to proteolysis. As an alternative, we propose to use molecularly imprinted polymer nanogels (MIP-NGs) as synthetic antibodies to target the HAVDI domain, for blocking N-cadherin adhesion.
Molecularly imprinted polymers (MIPs) are tailor-made synthetic antibodies possessing specific cavities designed for a target molecule.18–20 They are obtained by polymerizing functional and crosslinking monomers around the molecule of interest which acts as a template. After polymerization, the template is removed, resulting in a polymer with binding sites that are complementary in size, shape, and position of the functional groups to the template. The MIP can subsequently recognize and bind the molecule of interest with high affinity and specificity similar to that of an antibody with its antigen. Recently, we showed that MIP-NGs targeting the conserved N-terminal epitope Asp1-Trp2-Val3-Ile4-Pro5-Pro6-Ile7 (DWVIPPI) (Fig. 1B) had high selectivity and affinity for E- and N-cadherins. Consequently, they could inhibit cell-cell adhesion in cell aggregation assays, disrupt three-dimensional tumour spheroids and prevent the invasion of human cervical adenocarcinoma (HeLa) cells in Matrigel invasion assays.21 Thus MIP-NGs are very promising to function as therapeutic drugs,22,23 especially they seem to be stronger inhibitors than small molecule inhibitors, probably because they can cover large surface areas at the protein-protein interfaces. While both E- and N-cadherins can be involved in cancer development depending on the cell and tissue type, we wanted to explore the possibility of targeting preferentially one of the subtypes. Since the DWVIPPI epitope is common to E- and N-cadherins, we opted for the second epitope involved in the cadherin-mediated interaction, containing the HAV sequence, which shows some subtype specificity (Fig. 1B). Our present results show that the MIP-NGs targeting the HAVDI domain have higher affinities for N-cadherin than the small HAV-based peptide antagonists reported in the literature, and that they can specifically inhibit N-cadherin cell adhesion and invasion, as evaluated in cell aggregation and Matrigel invasion assays, respectively.
Results and discussion
Solid-phase synthesis of MIP-NGs
As reported above, the strongest inhibitor of N-cadherin adhesion, based on the HAV sequence is a cyclic peptide inhibitor composed of HAVDI.13 Herein, the main challenge is to obtain a synthetic antibody able to recognize the HAVDI sequence (Fig. 1B and 2A), which is an internal peptide. We selected the sequence AHAVDING as epitope, complemented with two cysteine residues at both ends for cyclisation, so as to closely mimic the conformation of the HAVDI peptide (Fig. 2B).24 The final cyclic template peptide, CAHAVDINGC-K(N3) (C-C) contains a modified lysine residue bearing an azide moiety for the immobilization of the template via click chemistry.25 The 3D structure of the free template epitope, flanked by 2 Cys (Fig. 2C, violet) was predicted with PEP-FOLD2 and superposed onto the native protein with PyMol. PEP-FOLD2 indicates a progressive increase in an extended secondary structure preference up to alanine in position 4 (0.8) followed by a prevalence of an unstructured conformation, which is coherent with the conformation in the native protein.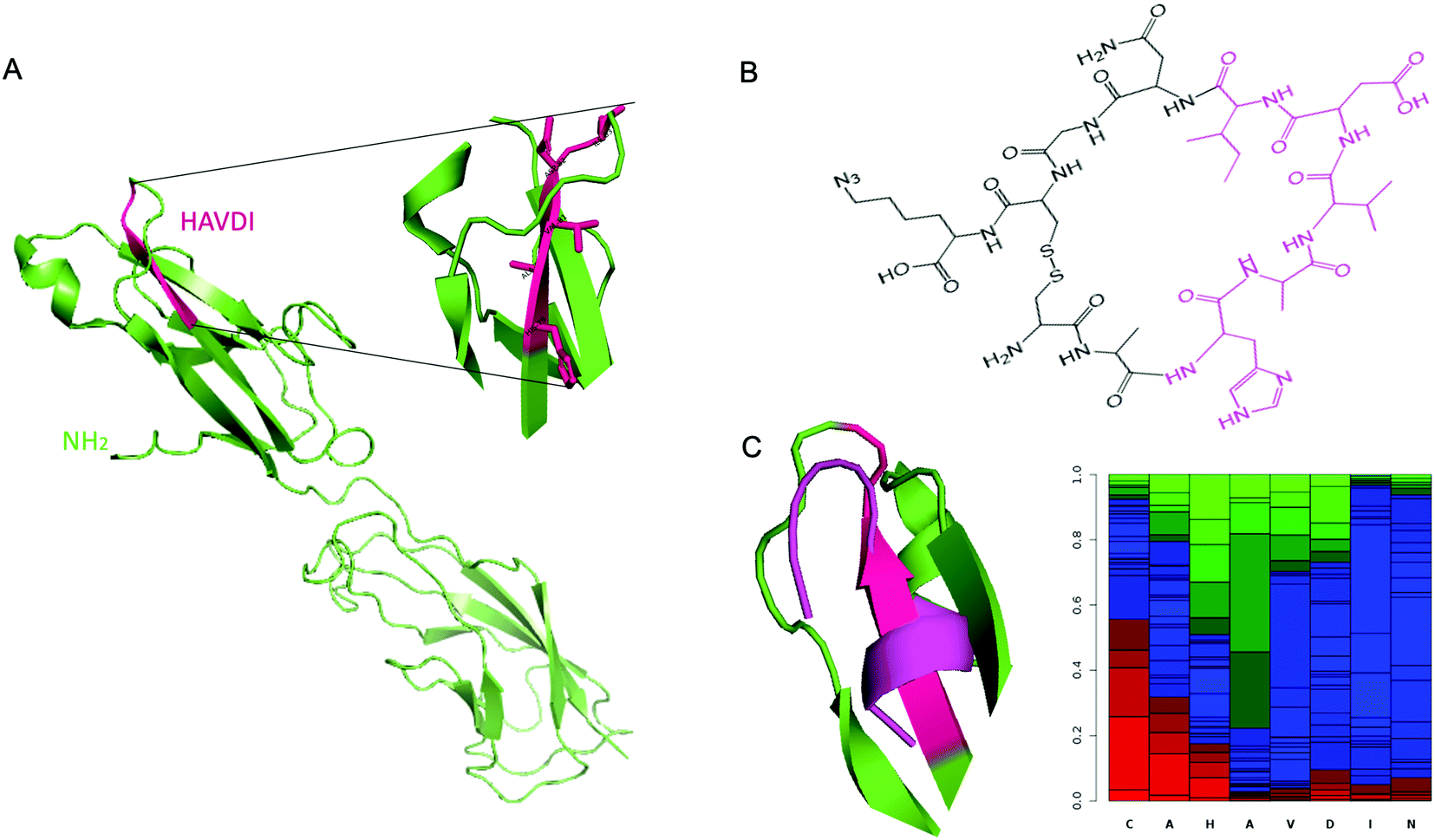 | ||
| Fig. 2 (A) Crystal structure of murine N-cadherin domains EC 1–2 (PDB accession code 2QVI). Murine N-cadherin has 96% identity with human N-cadherin.30 The sequence HAVDI is highlighted in fuchsia (B) Chemical structure of cyclic template peptide NH2-CAHAVDINGCK(N3)-COOH. (C) (Left) Superposition of the predicted 3D structure of the template peptide (flanked by 2 Cys) (violet), onto the native protein epitope. (Right) PEP-FOLD2 prediction of secondary structures probabilities associated to the cyclic template amino acids residues. Green: extended, blue: coil and red: helix. | ||
MIPs were produced by a solid-phase synthesis approach,26–28 where the template molecule is covalently immobilized on glass beads (GBs) as solid support (Fig. 3). This configuration allows an oriented immobilization of the epitope upon which polymer nanogels are synthesized. The binding sites of the resulting MIPs all have the same orientation, thus MIPs synthesized by the solid-phase approach can be considered analogous to monoclonal antibodies. Prior to MIP synthesis, GBs were first activated by boiling in NaOH to introduce -OH groups. After activation, they were functionalized with O-(propargyloxy)-N-(triethoxysilylpropyl)urethane to introduce terminal alkyne groups.29 The template peptide, functionalized with an azide moiety was immobilized on the alkyne-derivatized GBs through a copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC). Click chemistry offers the advantage that the peptide is immobilized without compromising the amino acid side chains that play a crucial role during the imprinting process.
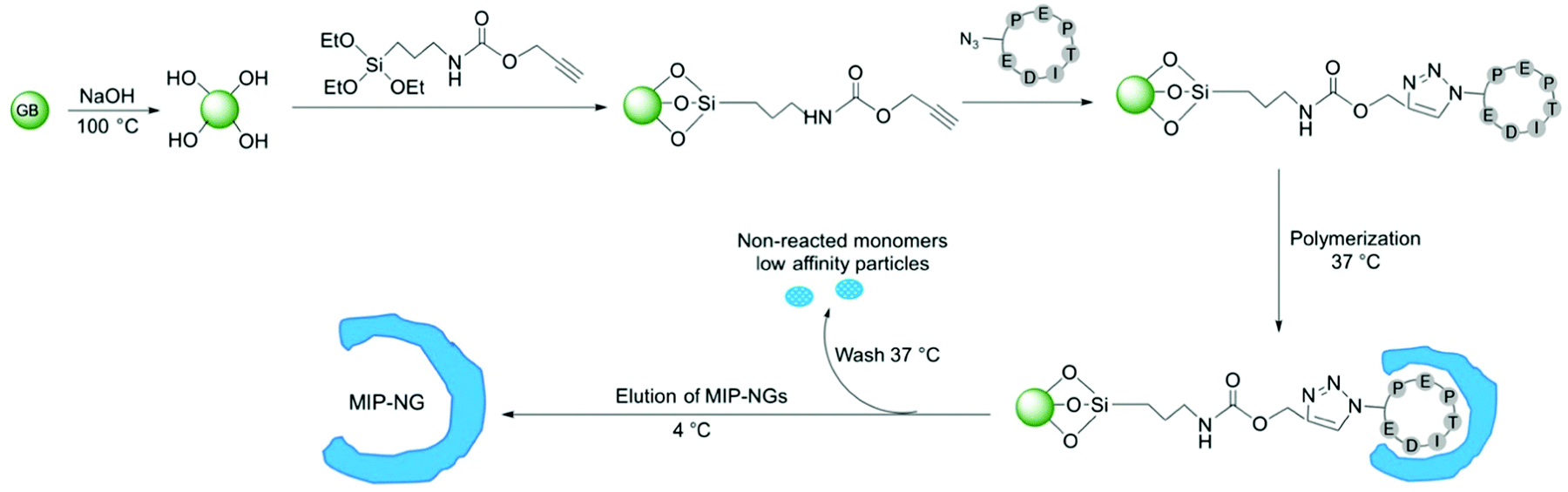 | ||
| Fig. 3 Schematic representation of the solid-phase synthesis of MIP. The cyclic peptide template bearing an azide moiety is immobilized on alkyne-GBs via click chemistry, followed by polymerization at 37 °C to form thermoresponsive MIP-NGs. After washing away the non-reacted monomers and low affinity polymers, the MIP-NGs are released from the GBs by decreasing the temperature to 4 °C. | ||
To synthesize the MIP-NGs, a mixture of monomers capable of interacting with the amino acid side chains of the peptide, was employed. N-Isopropylacrylamide (NIPAM) constituted the major monomer to yield thermoresponsive polymers that can be easily released from the support by changing the temperature.25–27N-tert-butyl acrylamide (TBAM) was used to target the hydrophobic amino acids. A benzamidine-based monomer, 4-acrylamidophenyl(amino)-methanimium acetate (AB) was employed to form a salt bridge with the carboxylic group of aspartic acid. N,N′-methylenebis(acrylamide) (BIS) was used as cross-linker and free radical polymerization was carried out at 37 °C in sodium phosphate buffer pH 7.0. At this temperature, the polymers are formed in the collapsed state, encapsulating the immobilized peptide. After washing away the low-affinity nanogels and the unreacted monomers, the polymers that presented high affinity to the peptide were eluted at 4 °C, below the lower critical solution temperature of p(NIPAM) (LCST, ∼32 °C), where the NGs swell and are detached from the column. The yield of polymerization of the MIP-NGs was 0.25 ± 0.03 mg g−1 GBs (n = 3), comparable to previous reports.25,27,29
Physicochemical characterization and binding properties of MIP-NGs
The hydrodynamic size of MIP-NGs was analyzed by dynamic light scattering (DLS) and found to be 47.7 ± 2.5 nm, PDI 0.32 ± 0.05 and a zeta potential of +9.8 mV (Fig. S1A and B, ESI†). Transmission electron microscope (TEM) images showed that the MIP-NGs were monodisperse (Fig. S1C, ESI†) and confirmed the size determined by DLS. The recognition of the peptide epitope by the MIP was evaluated at 37 °C by equilibrium binding assays with the fluorescently labelled template (coupled to fluorescein dibenzocyclooctyne, FAM DBCO) via a copper-free click chemistry reaction (Fig. S2A, ESI†). This fluorophore was chosen for its bulky DBCO, to favor the accomodation of only the peptide in the MIP binding cavity. The fluorescent peptide was purified by preparative HPLC and its purity analyzed by electrospray ionization high-resolution mass spectrometry (Fig. S2B, ESI†). The concentration of the pure fluorescent peptide was found to be 22.4 ± 3.5 μM (Fig. S3, ESI†). Equilibrium binding experiments showed that the binding of the MIP-NGs to the fluorescent peptide was specific as no binding was observed with the control polymer, a MIP with the same chemical composition synthesized with the peptide DWVIPPI (Fig. 4). These results confirmed the presence of specific imprinted sites at the surface of the nanogels.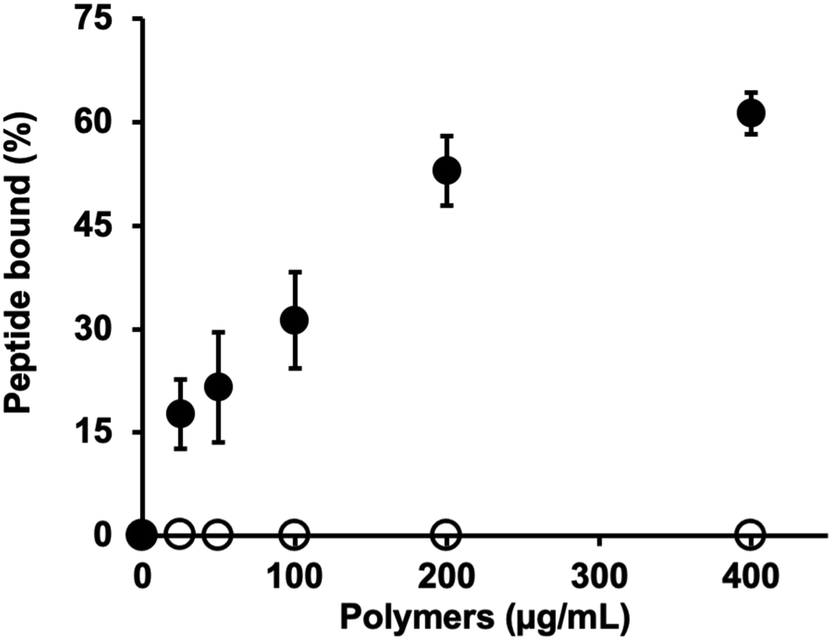 | ||
| Fig. 4 Equilibrium binding isotherms of 10 nM of fluorescent template peptide to MIP-NGs (full circles) and control polymer (empty circles) at 37 °C in sodium phosphate buffer pH 7.0 (n = 3, mean ± s.e.m.). | ||
Selectivity was evaluated by competitive binding assays in the presence of the native template peptide CAHAVDINGC-K(N3), a shorter cyclic peptide CHAVDINC-K(N3) containing the same motif, linear peptides HAVDI and DWVIPPI, as well as recombinant E-, N- and P-cadherins (Fig. S4, ESI†). Increasing concentrations of the peptides or proteins competed with a fixed amount of the fluorescently labelled peptide for binding to the MIP-NGs. The IC50 values (concentration of inhibitor required to displace 50% of the fluorescent peptide from the MIP) are shown in Table 1. IC50 values of 300 nM and 25 nM were found for the template peptide and N-cadherin respectively, indicating the excellent recognition of the MIP-NGs for their target. The higher affinity for the protein, as compared to the peptide epitope, may be due to the contribution of the protein surface in contact with the MIP-NGs (displacement of solvent molecules, conformational rearrangements of main and side chain atoms), which leads to improved complementarity and stability. MIP-NGs’ binding to the shorter version of the cyclic peptide was negligible, probably due to the fact that the different lengths of the peptides lead to different curvatures and flexibility. Thus, the shorter peptide has less contact area with the cavity in the polymer. It is interesting to note that the MIP-NGs have almost no affinity for the linear HAVDI peptide. A reduced interaction has been previously observed between a MIP directed against a cyclic epitope, and the linear version of the epitope.31 No interaction with the peptide DWVIPPI was observed. E-cadherin, containing the sequence SHAVSSNG and P-cadherin, with the sequence GHAVSENG, presented IC50 values of 41 and 200 nM, respectively, implying that the MIP-NGs also recognized E-cadherin and P-cadherin to a certain extent, but less well as compared to the target N-cadherin. Globally, the MIP-NGs appear to bind the proteins somewhat more strongly than the template epitope peptide, a phenomenon that we have often observed with other examples (unpublished data), and that can be attributed to the induced-fit and surface effects mentioned above.
| Competitor | Molecular weighta (Da) | Isoelectric pointa | IC50 (nM) |
|---|---|---|---|
| a Theoretical molecular weight and isoelectric point. | |||
| Template peptide CAHAVDINGC-K(N3) | 1153.49 | 4.98 | 300 |
| Short cyclic peptide CHAVDINC-K(N3) | 1025.44 | 4.98 | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
| HAVDI linear peptide | 553.29 | 4.98 | >100000 |
| DWVIPPI linear peptide | 838.46 | 3.11 | No binding |
| N-cadherin | 62910 |
4.7 | 25 |
| E-cadherin | 60586 |
4.28 | 41 |
| P-cadherin | 87400 |
4.55 | 200 |
Despite the MIP having 60% cross-reactivity with E-cadherin, this synthetic receptor represents a superior antagonist of N-cadherin as compared to the different peptides reported in the literature.13 The simplest cyclic peptide N-Ac-CHAVC-NH2 inhibited N-cadherin with an IC50 of 0.323 ± 0.020 mM. When the cyclic HAV peptide was turned into an N-cadherin peptide by incorporating flanking amino acids specific to N-cadherin, the antagonistic properties of the peptides were increased. For instance, the cyclic peptides N-Ac-CHAVDC-NH2, N-Ac-CHAVDIC-NH2 and N-Ac-CAHAVDIC-NH2 presented IC50 values of 0.114 ± 0.006 mM, 0.065 ± 0.005 mM and 0.210 mM respectively. Interestingly, when the HAVDI peptide was tested as a linear peptide (N-Ac-HAVDI-NH2), a 7-fold reduction in the activity was observed (IC50 of 0.44 ± 0.014 mM) as compared to the cyclic analogue. These results imply that the MIP is a better antagonist than the peptides by almost three orders of magnitude.
Recognition of cadherins on cells
Since the MIP-NGs seem to recognize free N-cadherin (and to a lesser extent E-cadherin) protein in solution, the next step was to verify whether they could recognize these proteins at the cell surface. Fluorescent MIP-NGs were synthesized by incorporating polymerizable rhodamine B in the pre-polymerization mixture, as previously reported.21,29 Their hydrodynamic diameter was measured by DLS and found to be 44 ± 2.1 nm, with a polydispersity index of 0.29 ± 0.04 (n = 3). The MIP-NGs were then applied to two human cancer cell lines with different cadherin expression profiles: HeLa (expressing N-cadherin) and MCF-7 (breast cancer expressing E- and P-cadherins). The cadherin profile in the two cell lines was prior verified by staining with anti E-, N- and P-cadherin antibodies (Fig. 5A and B). MIP-NGs were found to bind to the surface of HeLa cells, indicating that their preferential target is N-cadherin. While in free solution, the MIP-NGs seemed to bind (to a lesser extent also E-cadherin), no evidence of staining by rhodamine MIP-NGs was observed on fixed MCF-7 cells. This result indicates that the selected epitope CAHAVDINGCK(N3) generated a MIP-NG selective for N-cadherin, although we have no explanation for this improved specificity over the solution experiment; the only difference being that in solution we have used recombinant proteins. No staining on L929 mouse fibroblasts, an additional control cell line, which does not express endogeneous cadherins was observed either (Fig. S5, ESI†).32 No staining was obtained with the non-imprinted control nanogels (NIP-NGs).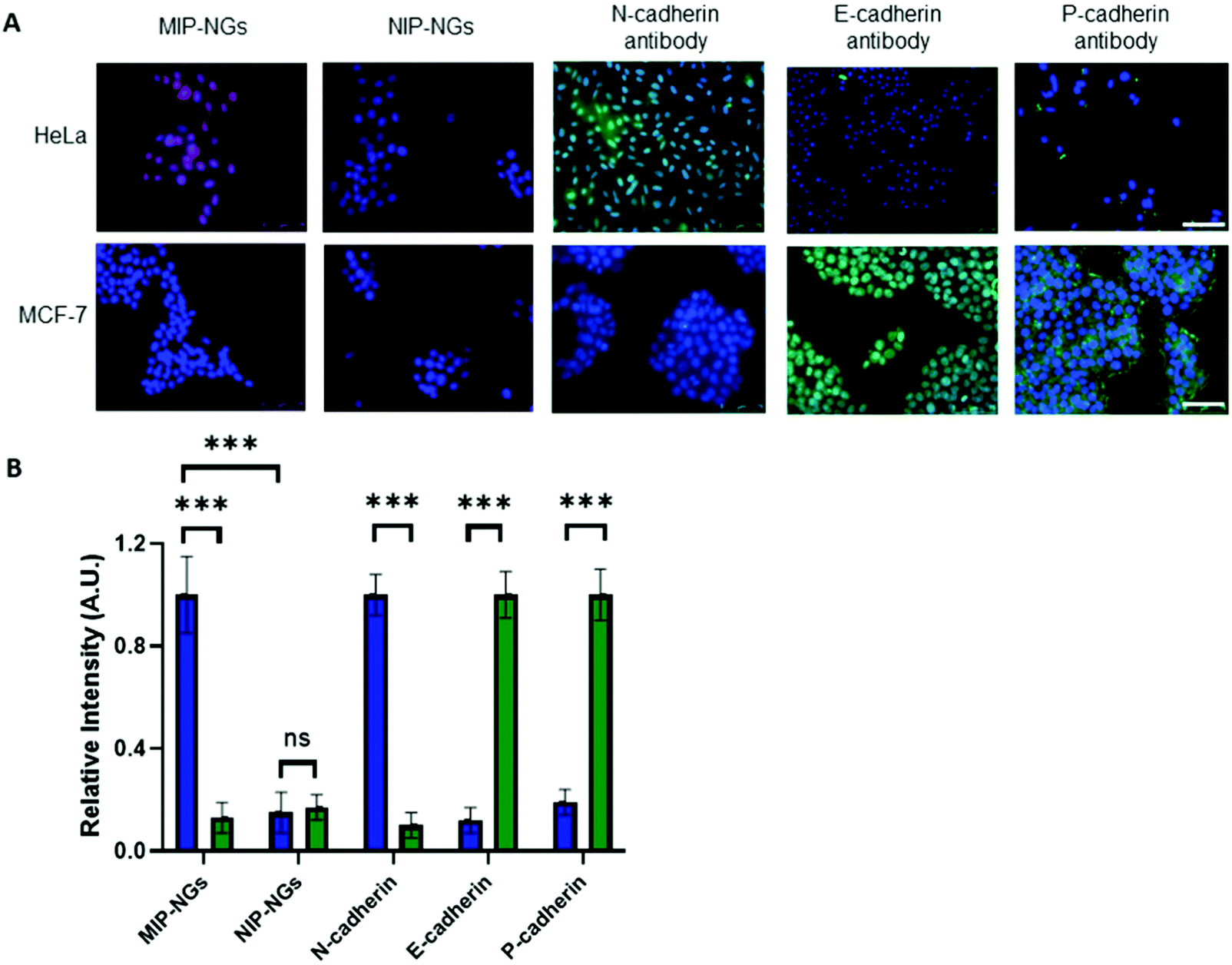 | ||
| Fig. 5 (A) Epifluorescence micrographs of HeLa (overexpressing N-cadherin) and MCF-7 cells (overexpressing E- and P-cadherin), stained with rhodamine MIP-NGs (red) or rhodamine NIP-NGs (red) or anti N-, E- and P-cadherin antibodies (green). Nuclei stained with Hoechst (blue). Scale bar: 75 μm. (B) Corresponding relative fluorescence intensity of HeLa (blue) and MCF-7 (green) cells after imaging with MIP-NGs or NIP-NGs or immunostaining with N-, E- or P-cadherin antibodies. Data are expressed as mean ± SEM, statistical analysis was performed using Student's test (***p < 0.001), ns: not significant. | ||
Inhibition of cell aggregation by MIP-NGs
Cell aggregation assays have become a useful tool to study cadherin-dependent adhesion, given that cell aggregation is the net result of cell-cell adhesion.7 HeLa and MCF-7 cells were suspended in agar-coated microtiter wells to exclude cell-substrate adhesion while allowing cell-cell interactions to occur. Prior to incubation with cells, MIP-NGs were checked for their non-cytotoxicity using the MTT assay (Fig. S6, ESI†). Cells were incubated in the presence of either MIP, NIP (a control polymer synthesized against azide-functionalized GBs) or the cyclic template CAHAVDINGC-K(N3) or the linear HAVDI peptide for 24 h; the formation of cell aggregates was evaluated under an inverted microscope. Cells incubated with only medium were taken as reference, and cells incubated with ethylenediaminetetraacetic acid (EDTA) served as positive control (EDTA chelates Ca2+ ions and cell-cell aggregation is abrogated).As illustrated in Fig. 6, HeLa and MCF-7 cells incubated in only growth medium showed one tight aggregate. On the contrary, no cell cluster was observed in the presence of EDTA, suggesting that cell aggregation was disrupted. For HeLa cells, addition of MIP and the cyclic peptide CAHAVDINGC resulted in numerous dispersed aggregates, indicating their ability to prevent cell-cell adhesion. Inhibition of cell aggregation by MIP-NGs was concentration-dependent (Fig. S7, ESI†). Slightly smaller aggregates were observed with the MIP than with the peptide, indicating the better antagonist properties of the MIP to N-cadherin than the cyclic peptide. On the other hand, the HAVDI linear peptide when compared with cyclic peptide CAHAVDINGC, was not very effective in cell disruption. In the case of MCF-7 cells which lack N-cadherin but which contain E-cadherin, the MIP caused a very slight dispersion in cell aggregation. For both cell lines, the NIP does not really affect cell aggregation, since the main cell cluster was preserved. These observations suggest that MIPs render cells incapable of establishing cell-cell adhesion by binding to the adhesive motif, HAVDI of N-cadherin. The specificity of the inhibition by the MIP was confirmed by the lower effect produced in MCF-7, a cell line lacking N-cadherin, and by the lower impact observed with the NIP.
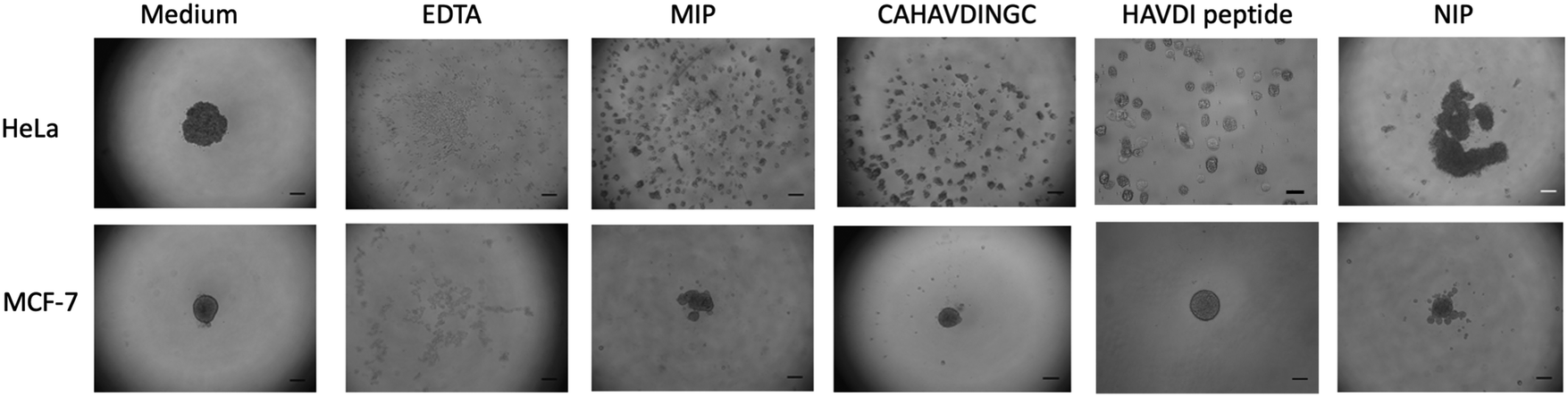 | ||
| Fig. 6 Effect on HeLa and MCF-7 (200000 cells per mL) cell aggregation when incubated with 4 mM EDTA, 20 μg mL−1 MIP, 100 nM template peptide, 100 nM HAVDI linear peptide and 20 μg mL−1 NIP, in growth medium. Scale bar: 250 μm. | ||
Inhibition of cancer cell invasion by MIP-NGs
Matrigel® invasion assay33 was employed to assess the invasion propensity of HeLa cells in the presence of MIP-NGs. Matrigel acts as a reconstituted basement membrane and prevents non-invasive cells from passing across, whereas invasive cells enzymatically degrade the Matrigel membrane and invade through its pores. HeLa cells (25000) were seeded onto a Matrigel-coated insert with or without MIP-NGs (30 μg mL−1) and incubated for 24 h. Cells that migrated to the lower side of the membrane were stained and evaluated by epifluorescence microscopy. Fig. 7 shows that cells seeded with MIP-NGs showed a significant decrease in their invasive capacity as compared to non-treated cells and NIPs. The MIP-NGs synthesized with CAHAVDINGC (IC50: 25 nM) was as efficient as the MIP-NGs synthesized with DWVIPPI21 (IC50: 20 nM) for blocking cancer invasion in vitro.
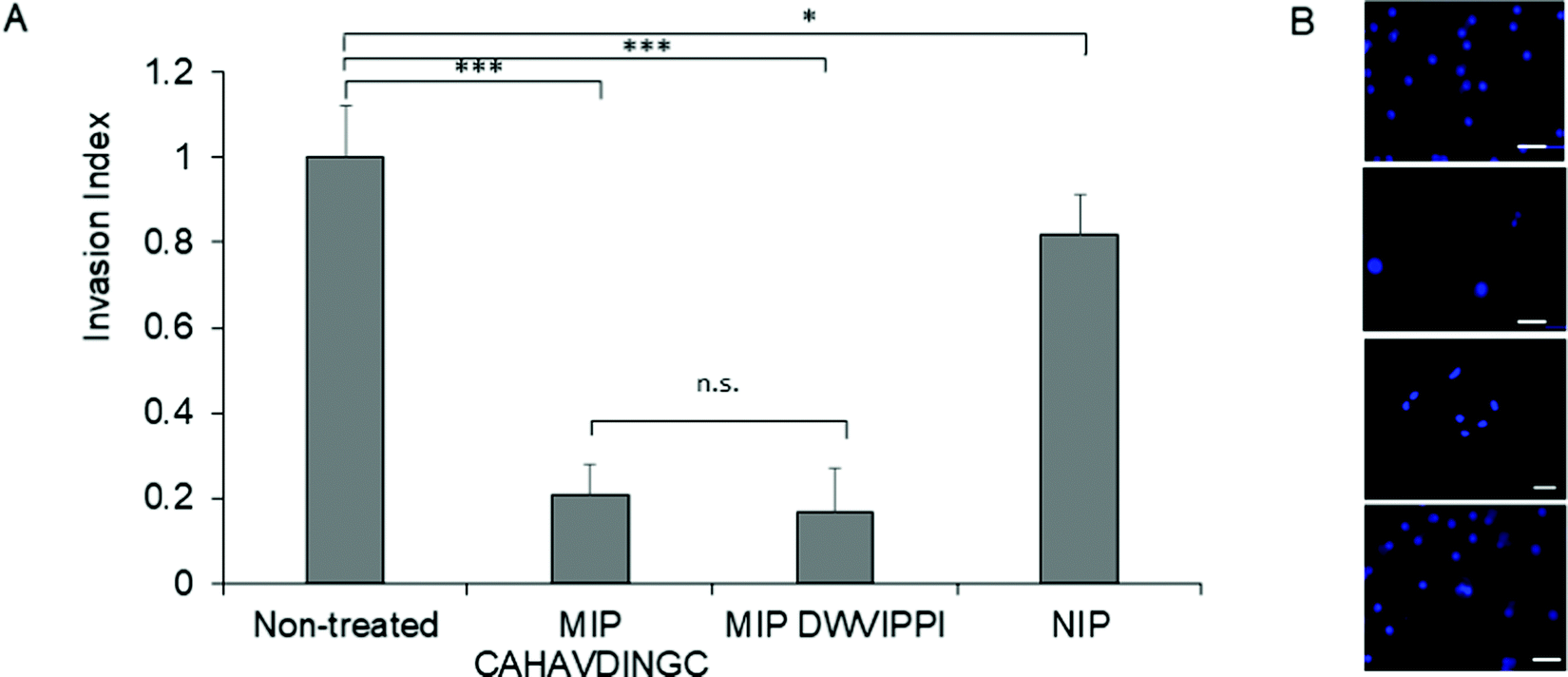 | ||
| Fig. 7 Fig. 7. (A) Invasion index representing the extent of HeLa cells invasion in the presence of MIP or NIP, as studied by Matrigel Invasion assay. As a positive control, MIP targeting DWVIPPI21 was tested in parallel. Mean values are significantly different at 95% confidence (*p < 0.05, ***p < 0.001, n.s, not significant, Student's t-test, mean ± s.e.m.). (B) Epifluorescence micrographs of HeLa cells that invaded to the lower side of the Matrigel membrane. Cells were stained with Hoechst. From top to bottom: untreated cells, cells treated with MIP (CAHAVDINGC), MIP (DWVIPPI) and NIP. Scale bar: 25 μm. | ||
Overall, these results indicate that MIPs can exhibit a biological activity by modulating cadherin activity; this can lead to new approaches to treat diseases associated with dysfunctional cadherin mechanisms.
Conclusion
In a precedent paper,21 we synthesized a MIP-NG targeting the first seven amino acids of the N-terminal part of E- and N-cadherins, which encompasses Trp2, an amino acid responsible for cell adhesion. The inhibitory potency of this MIP towards cell-cell adhesion in cell aggregation assays proved to be remarkably higher than that of a commercially available monoclonal antibody, known to inhibit cadherin adhesion. Importantly, this MIP could inhibit cancer cells invasion and disrupt three-dimensional tumour spheroids, highlighting the promising therapeutic value of MIPs in cancer treatment. In the present work, we hypothesized that it should also be possible to block the entrance of the acceptor domain of Trp2, a hydrophobic pocket lined by the sequence HAV. Thus a MIP-NG templated with a cyclic peptide CAHAVDINGC was synthesized. It has a high affinity and selectivity for N-cadherin over E and P-cadherins. This MIP-NG was very effective in abrogating cancer cell adhesion and invasion, confirming the high potential of MIPs as a therapeutic drug. These findings are in line with recent literature data where MIPs imprinted against HER2 glycans were shown to inhibit cell proliferation and reduce cancer volume in vivo.34 We believe that MIPs can successfully contribute to the establishment of a novel therapeutic platform in cancer treatment.35–37Experimental section
Materials and methods
All chemicals and solvents were of analytical grade and purchased from VWR International (Fontenay sous Bois, France) or Sigma-Aldrich (St Quentin Fallavier, France), unless otherwise stated. (O-(Propargyloxy)-N-(triethoxysilylpropyl)urethane was purchased from Abcr GmbH, Germany. Peptides (purity >95%) were custom-synthesizsed by GL Biochem (Shanghai, China). Recombinant human E-cadherin (purity ≥90%, expressed in E. coli) was obtained from Sigma-Aldrich. Recombinant human N-cadherin (purity >90%, expressed in Sf9 Baculovirus cells) was bought from Interchim (Montluçon, France). Recombinant human P-cadherin Fc Chimera Protein (purity >90%) was purchased from R&D Systems (Lille, France). FAM DBCO, 6-isomer and tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) were purchased from Interchim. HeLa and MCF-7 were obtained from Cell Lines Service (Eppelheim, Germany). Media, reagents and consumables for cell culture were from Thermo Fisher Scientific (Illkirch, France). Glass beads of diameter 0.1 mm and microscope slides for cell samples were bought from Roth Sochiel E.U.R.L. (Lauterbourg, France). Buffers were prepared with Milli-Q water, purified using a Milli-Q system (Millipore, Molsheim, France). Fluorescence measurements were done on a FluoroLog-3 spectrofluorimeter (Horiba Jobin Yvon, Longjumeau, France). Transmission electron microscopy images were recorded on a a JEOL JEM-2100F instrument (Japan). Cell aggregation was evaluated under a Leica inverted microscope equipped with camera (Nanterre, France).Synthesis of MIP-NGs
:1:5:8 (alkyne peptide:CuSO4:THPTA:sodium ascorbate).29
Solid-phase synthesis of MIP
Solid-phase synthesis was carried out in a glass column equipped with a thermostated jacket (XK 26/20). The solvents were pumped through the column using a peristaltic pump at a flow rate of 2.5 mL min−1. The column was packed with 20 g peptide GBs and equilibrated with 100 mL buffer A. Then, 20 mL of buffer A containing 4-acrylamidophenyl(amino)-methaniminium acetate (AB, benzamidine-based monomer) (12.4 mg, 0.05 mmol, 5 mol%) was percolated through the column. The flow-through was collected and passed through the column again for 1 h to favour the AB interaction with the -COOH moieties of the peptide. The polymerization mixture was prepared by mixing the rest of the monomers, N-isopropylacrylamide (NIPAM) (92.8 mg, 0.82 mmol, 82 mol%), N-tert-butylacrylamide (TBAM) (10.2 mg dissolved in 1 mL ethanol, 0.08 mmol, 8 mol%) and N,N′-methylenebis(acrylamide) (BIS) (7.7 mg, 0.05 mmol, 5 mol%) in 25 mL with buffer A so that the total monomer concentration is 0.5% (w/w). The solution was purged with nitrogen for 30 min. Afterwards, the initiation couple composed of potassium persulfate (KPS) (8.5 mg in 500 μL buffer A) and N,N,N,N′-tetramethylethylendiamine (TEMED) (63 μL of 10 μL of TEMED in 990 μL buffer A) was added to the reaction mixture. The amount of KPS was 3% (mol/mol) with respect to polymerizable double bonds, and 7.5/1 was the molar ratio for KPS/TEMED. The polymerization mixture was percolated through the reactor, and the temperature was set at 37 °C for 17 h. After polymerization, the column was washed with 50 mL buffer A at 37 °C, and the column was cooled down at 4 °C for 1 h. MIP was eluted by fractions of 6 mL (twice) with water and constituted the working stock solution. The hydrodynamic diameter of MIPs, analyzed by dynamic light scattering (DLS), and their zeta potential were determined by using a Malvern Zetasizer Nano ZS. The concentration of the MIPs was calculated with a 3 mL aliquot of MIP, which was lyophilized and weighed.Evaluation of the binding properties of MIP-NGs
000g for 1 h at 40 °C and the fluorescence intensity of a 700 μL aliquot was measured (λex: 490 nm/λem: 515 nm, slit 4 nm) to determine the amount of free peptide. The amount of peptide bound to the polymer was calculated by subtracting the unbound analyte from the total peptide added to the mixture determined from the blank samples.
Application of MIP-NGs to cells
000 cells per well). After 24 h, cells were incubated with MIP-NGs (5–250 μg mL−1) for 24 h in cell culture medium. Dissolution of the crystals of MTT was achieved by DMSO and Sorensen's buffer. Cell viability was determined by dividing the absorbance obtained for treated cells by that of the untreated controls. Cell treated with 1% Triton X-100 served as positive control of cytotoxicity.
Cell fixation and bioimaging
:400 in 1% BSA in PBS for 30 min. For P-cadherin staining, the cell membrane was permeabilized and the cells incubated with primary antibody in 1% BSA in PBS for 3 h, followed by incubation with Alexa Fluor 488 anti-Mouse IgG secondary antibody at a dilution of 1:400 in 1% BSA in PBS for 30 min. For N-cadherin staining, Alexa Fluor 488 anti-Mouse IgG was also used as secondary antibody and for all cells, the nuclei were stained with Hoechst at a dilution of 1:2000 in PBS at room temperature for 10 min. Cells were washed with PBS, mounted on a microscope slide with 5 μL mounting medium (0.5 mL water, 0.5 mL 1 M Tris-HCl buffer pH 8.0 and 9 mL glycerol) and analyzed with a Leica DMI 6000B epifluorescence microscope. Each sample was prepared in duplicate.
:2000 at room temperature for 10 min. Cells were washed with PBS, mounted on a microscope slide with 5 μL mounting medium (0.5 mL water, 0.5 mL 1 M Tris–HCl buffer pH 8.0 and 9 mL glycerol) and analyzed by fluorescence microscopy. Epifluorescence microscopy images were recorded with a Leica DMI 6000B (Germany) microscope. Each sample was prepared in triplicate and the fluorescence intensities were measured with ImageJ. Three images per sample with 40x magnification were analyzed, by measuring the relative fluorescence of 10 cells per image.
000 cells per mL) was added to the agar-coated wells, followed by the addition of 100 μL of the test product in full medium. The polymers were tested at 20 μg mL−1, EDTA at 4 mM, the cyclic peptide CAHAVDINGC-K(N3) at 100 nM. Cells incubated with only medium were taken as control. The well-plates were incubated at 37 °C, 5% CO2 and 100% humidity under horizontal stirring. Cell aggregation was evaluated at 24 h after seeding cells, under an inverted microscope equipped with a camera using a 4x objective and bright contrast.
000) suspended in DMEM medium, in the presence or absence of 30 μg mL−1 MIP-NGs or NIP-NGs, were seeded on Matrigel-coated inserts, whereas the lower compartment contained DMEM medium supplemented with 10% FBS as chemoattractant. After 24 h incubation at 37 °C, 5% CO2 and 100% humidity, the upper surface of the insert was cleared from non-migratory cells with a cotton swab and washed with PBS. The remaining (invasive) HeLa cells that attached to the lower surface of the filter were fixed with cold methanol, stained with Hoechst (at a dilution of 1:2000 in PBS for 10 min) and analyzed with an epifluorescence microscope at 63x of magnification, using the filter A4. Invasive cells were quantified as the number of migrated cells per microscopic field, 10 fields per assay from two independent repetitions. Invasion index was calculated as the ratio between the number of invasive HeLa cells treated with MIP-NGs or NIP-NGs and the number of invasive HeLa (without polymer).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. X. M. R. thanks the Mexican National Council for Science and Technology (CONACYT) and the Instituto para el Desarrollo de la Sociedad del Conocimiento del Estado de Aguascalientes (IDSCEA), for PhD scholarship. A. M. thanks CONACYT and the Instituto de Innovación y Transferencia de Tecnología de Nuevo Leon for PhD scholarship. E. M. thanks the Erasmus+ program for financial support. The authors acknowledge financial support from the Region of Picardy and the European Union (co-funding of equipment under CPER 2007–2020 and project POLYSENSE). B. T. S. B. acknowledges COST Action CA18013 “Innovation with Glycans: new frontiers from synthesis to new biological targets”. K. H. acknowledges financial support from Institut Universitaire de France.References
- C. Yoshida-Noro, N. Suzuki and M. Takeichi, Dev. Biol., 1984, 101, 19–27 CrossRef CAS PubMed.
- T. J. Boggon, J. Murray, S. Chappuis-Flament, E. Wong, B. M. Gumbiner and L. Shapiro, Science, 2002, 296, 1308–1313 CrossRef CAS PubMed.
- K. M. Mrozik, O. W. Blaschuk, C. M. Cheong, A. C.-W. Zannettino and K. Vandyke, BMC Cancer, 2018, 18, 939 CrossRef CAS PubMed.
- M. Takeichi, Curr. Opin. Cell Biol., 1993, 5, 806–811 CrossRef CAS PubMed.
- W. Yu, L. Yang, T. Li and Y. Zhang, Front. Oncol., 2019, 9, 989 CrossRef PubMed.
- V. Padmanaban, I. Krol, Y. Suhail, B. M. Szczerba, N. Aceto, J. S. Bader and A. J. Ewald, Nature, 2019, 573, 439–444 CrossRef CAS PubMed.
- K. Tamura, W. S. Shan, W. A. Hendrickson, D. R. Colman and L. Shapiro, Neuron, 1998, 20, 1153–1163 CrossRef CAS PubMed.
- S. C. Newton, O. W. Blaschuk and C. F. Millette, Dev. Dyn., 1993, 197, 1–13 CrossRef CAS PubMed.
- H. Tanaka, E. Kono, C. P. Tran, H. Miyazaki, J. Yamashiro, T. Shimomura, L. Fazli, R. Wada, J. Huang, R. L. Vessella, J. An, S. Horvath, M. Gleave, M. B. Rettig, Z. A. Wainberg and R. E. Reiter, Nat. Med., 2010, 16, 1414–1421 CrossRef CAS PubMed.
- O. W. Blaschuk, Cell Tissue Res., 2012, 348, 309–313 CrossRef CAS PubMed.
- C. C. Zhang, Z. Yan, Q. Zhang, K. Kuszpit, K. Zasadny, M. Qiu, C. L. Painter, A. Wong, E. Kraynov, M. E. Arango, P. P. Mehta, L. Popoff, G. F. Casperson, G. Los, S. Bender, K. Anderes, J. G. Christensen and T. VanArsdale, Clin. Cancer Res., 2010, 16, 5177–5188 CrossRef CAS PubMed.
- S. M. Brouxhon, S. Kyrkanides, X. Teng, V. Raja, M. K. O’Banion, R. Clarke, S. Byers, A. Silberfeld, C. Tornos and L. Ma, Clin. Cancer Res., 2013, 19, 3234–3246 CrossRef CAS PubMed.
- E. Williams, G. Williams, B. J. Gour, O. W. Blaschuk and P. Doherty, J. Biol. Chem., 2000, 275, 4007–4012 CrossRef CAS PubMed.
- S. M. Burden-Gulley, T. J. Gates, S. E.-L. Craig, S. F. Lou and S. A. Oblander, Peptides, 2009, 30, 2380–2387 CrossRef CAS PubMed.
- G. M. Beasley, J. C. Riboh, C. K. Augustine, J. S. Zager, S. N. Hochwald, S. R. Grobmyer, B. Peterson, R. Royal, M. I. Ross and D. S. Tyler, J. Clin. Oncol., 2011, 29, 1210–1215 CrossRef CAS PubMed.
- R. M. Mege, D. Goudou, C. Diaz, M. Nicolet, L. Garcia, G. Geraud and F. Rieger, J. Cell Sci., 1992, 103, 897–906 CrossRef CAS PubMed.
- B. D. Cosgrove, K. L. Mui, T. P. Driscoll, S. R. Caliari, K. D. Mehta, R. K. Assoian, J. A. Burdick and R. L. Mauck, Nat. Mater., 2016, 15, 1297–1306 CrossRef CAS PubMed.
- R. Arshady and K. Mosbach, Makromol. Chem., 1981, 182, 687–692 CrossRef CAS.
- G. Wulff, A. Sarhan and K. Zabrocki, Tetrahedron Lett., 1973, 14, 4329–4332 CrossRef.
- K. Haupt, A. V. Linares, M. Bompart and B. Tse Sum Bui, Top. Curr. Chem., 2012, 325, 1–28 CrossRef CAS PubMed.
- P. X. Medina Rangel, E. Moroni, F. Merlier, L. A. Gheber, R. Vago, B. Tse Sum Bui and K. Haupt, Angew. Chem., Int. Ed., 2020, 59, 2816–2822 CrossRef CAS PubMed.
- K. Haupt, P. X. Medina Rangel and B. Tse Sum Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef CAS PubMed.
- L. O’Driscoll, N. Engl. J. Med., 2020, 383(9), 871–873 CrossRef PubMed.
- J. Xu, F. Merlier, B. Avalle, V. Vieillard, P. Debré, K. Haupt and B. Tse Sum Bui, ACS Appl. Mater. Interfaces, 2019, 11, 9824–9831 CrossRef CAS PubMed.
- A. Mier, I. Maffucci, F. Merlier, E. Prost, V. Montagna, G. U. Ruiz-Esparza, J. V. Bonventre, P. K. Dhal, B. Tse Sum Bui, P. Sakhaii and K. Haupt, Angew. Chem., Int. Ed., 2021, 60, 20849–20857 CrossRef CAS PubMed.
- S. Ambrosini, S. Beyazit, K. Haupt and B. Tse Sum Bui, Chem. Commun., 2013, 49, 6746–6748 RSC.
- J. Xu, S. Ambrosini, E. Tamahkar, C. Rossi, K. Haupt and B. Tse Sum Bui, Biomacromolecules, 2016, 17, 345–353 CrossRef CAS PubMed.
- A. Poma, A. Guerreiro, M. J. Whitcombe, E. V. Piletska, A. P.-F. Turner and S. A. Piletsky, Adv. Funct. Mater., 2013, 23, 2821–2827 CrossRef CAS PubMed.
- P. X. Medina Rangel, S. Laclef, J. Xu, M. Panagiotopoulou, J. Kovensky, B. Tse Sum Bui and K. Haupt, Sci. Rep., 2019, 9, 3923 Search PubMed.
- R. A. Reid and J. J. Hemperly, Nucleic Acids Res., 1990, 18, 5896 CrossRef CAS PubMed.
- L. Cenci, G. Guella, E. Andreetto, E. Ambrosi, A. Anesi and A. M. Bossi, Nanoscale, 2016, 8, 15665–15670 RSC.
- S. V. Litvinov, M. Balzar, M. J. Winter, H. A.-M. Bakker, I. H. Briaire-de Bruijin, F. Prins, G. J. Fleuren and S. O. Warnaar, J. Cell Biol., 1997, 139, 1337–1348 CrossRef CAS PubMed.
- A. S. Ribeiro, A. Albergaria, B. Sousa, A. L. Correia, M. Bracke, R. Seruca, F. C. Schmitt and J. Paredes, Oncogene, 2010, 29, 392–402 CrossRef CAS PubMed.
- Y. Dong, W. Li, Z. Gu, R. Xing, Y. Ma, Q. Zhang and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 10621–10625 CrossRef CAS PubMed.
- T. Takeuchi, K. Mori, H. Sunayama, E. Takano, Y. Kitayama, T. Shimizu, Y. Hirose, S. Inubushi, R. Sasaki and H. Tanino, J. Am. Chem. Soc., 2020, 142, 6617–6624 CrossRef CAS PubMed.
- Z. El-Schich, M. Abdullah, S. Shinde, N. Dizeyi, A. Rosén, B. Sellergren and A. Gjörloff Wingren, Tumor Biol., 2016, 37, 13763–13768 CrossRef CAS PubMed.
- Y. Ma, Y. Yin, L. Ni, H. Miao, Y. Wang, C. Pan, X. Tian, J. Pan, T. You, B. Li and G. Pan, Bioact. Mater., 2021, 6, 1308–1317 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00680d |
| ‡ Present address: Department of Internal Medicine, Yale University School of Medicine, New Haven, Connecticut, USA. |
| This journal is © The Royal Society of Chemistry 2022 |