
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiolated cationic poly(aspartamides) with side group dependent gelation properties for the delivery of anionic polyelectrolytes†
Aysel
Mammadova
a,
Benjámin
Gyarmati
 *a,
Kitti
Sárdi
a,
Adrien
Paudics
a,
Zoltán
Varga
b and
András
Szilágyi
a
*a,
Kitti
Sárdi
a,
Adrien
Paudics
a,
Zoltán
Varga
b and
András
Szilágyi
a
aDepartment of Physical Chemistry and Materials Science, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3, H-1111 Budapest, Hungary. E-mail: gyarmati.benjamin@vbk.bme.hu
bResearch Centre for Natural Sciences, Institute of Materials and Environmental Chemistry, Magyar Tudósok Körútja 2, 1117 Budapest, Hungary
First published on 1st June 2022
Abstract
In situ gellable polymers have potential applications as injectable formulations in drug delivery and regenerative medicine. Herein, thiolated cationic polyaspartamides were synthesised via two different approaches to correlate the side group structure with gelation properties, gel strength and drug release kinetics. Cysteamine (CEA) was used as a thiolating agent to prepare thiolated cationic polyaspartamide groups with short thiolated side groups. As a new pathway, thiolactone chemistry was integrated with cationic modification of polyaspartamides to prepare thiolated derivatives with longer, flexible side groups using N-acetyl-DL-homocysteine (NAH) thiolactone. Both types of thiolated polyaspartamides could be converted into stiff hydrogels under mild reaction conditions through oxidation-induced intermolecular disulfide formation. We confirmed that the longer side groups largely accelerated gelation and the stiffness of the resultant hydrogels was higher than that of the CEA-modified counterparts. Both the gelation time and stiffness could be adjusted by the degree of thiolation. Poly(aspartic acid) (PASP) derivatives with a controlled concentration of anionic groups were entrapped in the hydrogels during the in situ gelation. Based on the possible electrostatic interaction between the linear anionic polyelectrolytes and the cationic polymer network, we hypothesized that the release of the encapsulated material is controlled by the charge density. In accordance, fully anionic PASP was entrapped completely in the hydrogels, whereas a reduction in the number of anionic groups caused the partial release of PASP derivatives. NAH- and CEA- modified cationic polyaspartamide hydrogels showed distinct release rates, indicating the interplay between cationic and thiol functionalities in release kinetics.
1 Introduction
Stimuli-responsive hydrogels for in situ gelling formulations have gained much attention in several biomedical applications, particularly in controlled and targeted drug delivery.1–7 Their tailor-made structure makes them suitable as delivery vehicles for drug molecules, nucleic acids, biologics, growth factors, or cells according to the scope of application. Particular efforts are dedicated to develop in situ gelling formulations for loading of high molecular weight drugs, such as peptide and protein drugs, or for improved bioadhesion.8 These formulations often utilize thiol–disulfide conversion to form a reversibly cross-linked network. It was shown that thiolated hyaluronic acids can form hydrogels within a short time (as fast as 5 min),9 similarly to thiolated collagen,10 which has great importance to avoid the loss of drugs from the ocular surface where liquid drug formulations are generally used. Bernkop-Schnürch and co-workers11 emphasized the importance of reduced gelation time and showed that the chitosan–thioglycolic acid conjugate gelled without any chemical oxidant within 40 min, while the gelation time was reduced to a few minutes by using oxidants. The thiolation of hyaluronic acid is also used to produce disulfide cross-linked formulations, e.g., for wound healing applications.10 Disulfide groups can be cleaved in a reducing environment, so a great effort has been devoted to utilize it for intracellular delivery in cancer treatment.12 In such applications, non-viral gene delivery is of particular interest.13,14 Priya et al.15 reported a pullulan-based cationic polymer which established electrostatic interactions with DNA, but the interactions were readily reversible to release the encapsulated material in cells. Neda et al.16 reported the synthesis of cationic dextran nanogels showing triggered release of covalently conjugated peptides under reducing conditions. The effect of network charge on the immobilization and release of proteins from chemically cross-linked dextran hydrogels was also investigated and it was found that both negatively and positively charged proteins were fully immobilized in the networks due to electrostatic interactions with opposite charges at low ionic strength. In contrast, at physiological ionic strength, the percentage of immobilized proteins depended on the charge density of the hydrogel.17 The cationic, thiol, and other functionalities affect the properties of drug delivery vehicles, including stability, encapsulation and release efficiency as well as toxicity and biodegradability. We earlier reported that poly(aspartic acid) and polyaspartamide derivatives and their hydrogels are promising candidates as biocompatible, chemically versatile, and responsive excipients.18–23 We also showed that the non-enzymatic degradation rate of polyaspartamide hydrogels could be controlled by the side group structure,24 making disulfide cross-linked PASP hydrogels good candidates for human biological applications where in situ gelation and/or bioadhesion are beneficial.Due to the advantages of thiolated polymers for in situ gelling applications, extensive research has been dedicated to developing new strategies for the synthesis of thiolated polymers.8,25–27 Free thiol-containing compounds including the most frequently used thiolating agents, cysteine and cysteamine, are sensitive to oxidation in aqueous solution and thus have a poor shelf life, and several reagents provide limited conversion.28 To overcome these hindrances, Du Prez and co-workers developed synthetic approaches based on “thiolactone” derivatives, which are thiolating agents without free thiol groups prior to the reaction. By the nature thiolactones are highly reactive compounds29 and undergo nucleophilic ring-opening under slightly alkaline conditions, resulting in the release of a free sulfhydryl (thiol) group. Additional functionalities can be incorporated by thiol click-reactions.30 In particular, N-acetyl-homocysteine thiolactone, also known as citiolone, is a derivative of homocysteine-γ-thiolactone, a commercial compound that was introduced as a thiolating agent for proteins, antioxidants, and mucolytic drugs.31 Despite many synthetic protocols reported about the synthesis of thiolactone-based polymers for various applications, their use in drug delivery applications is limited.32–35 For instance, bifunctionalized redox-responsive layers were developed, which can be used to produce bioactive surfaces with drug loading and release properties.32 Polymer prodrugs with tumour acidity-promoted cellular internalization and intracellular drug release34 can be achieved through the modification of drug molecules with thiolactone moieties. However, there is still a huge demand to develop thiolated and cationic polymer derivatives for the efficient delivery of anionic macromolecular compounds, e.g., nucleic acid derivatives. In this sense, careful consideration of the formation of the disulfide cross-linked network and its interaction with encapsulation materials is required. To the best of our knowledge, in situ gellable formulations synthesised by thiolactone chemistry have not been reported yet, and thus our goal was to integrate this approach into the synthesis of thiolated cationic polyaspartamides. Both a conventional (cysteamine-based) and a thiolactone-based synthetic strategy were used to reveal the correlation between the gelation properties and side group composition, including the thiol content and the length of thiolated side groups. As a model for anionic macromolecular drugs, poly(aspartic acid) derivatives with a controlled negative charge density were used as the entrapped material to study the release kinetics from disulfide cross-linked hydrogels.
2 Experimental
2.1 Materials
L-Aspartic acid (99.5%) and phosphoric acid (99%) were bought from Merck. Deuterium oxide (D2O, 99.9 atom% D, containing 0.05 wt% 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt) and ethanolamine (HE; 99%) were bought from Sigma-Aldrich. Cysteamine (CEA) (95%), dithiothreitol (DTT; 99%), dibasic sodium phosphate monohydrate (99.5%), sodium chloride, hydrochloric acid (37%), sulfuric acid (96%), N,N-dimethylformamide (DMF), sodium bromate (99%), and sodium hydroxide (99%) were bought from Reanal Hungary. Butane-1,4-diamine (DAB), N-acetyl-DL-homocysteine thiolactone (NAHT), L-tryptophan methyl ester hydrochloride (Trp) and dibutylamine (DBA, 99%) (T) were bought from TCI. Imidazole (99%, crystalline) was bought from Thermo Scientific Chemicals. Acetate acid (96%) was bought from Molar Chemicals. Sodium acetate trihydrate (pure) was bought from Lach-Ner. A dialysis tubing cellulose membrane (avg. flat width: 43 mm, molecular weight cut-off: 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was bought from Merck. All reagents were used without any purification and their quality was “for analysis” unless otherwise noted. Syntheses and measurements were done at 25 °C unless otherwise indicated. Ultrapure water (ρ > 18.2 MΩ cm, Millipore) was used for the preparation of aqueous solutions.
000) was bought from Merck. All reagents were used without any purification and their quality was “for analysis” unless otherwise noted. Syntheses and measurements were done at 25 °C unless otherwise indicated. Ultrapure water (ρ > 18.2 MΩ cm, Millipore) was used for the preparation of aqueous solutions.
Phosphate-buffered saline (PBS) solution (pH = 7.4) was prepared by dissolving 8.00 g of NaCl, 0.20 g of KCl, 1.44 g of Na2HPO4·2H2O and 0.12 g of KH2PO4 in 1 L of water, the pH being adjusted with 0.1 mol L−1 HCl.
Imidazole buffer (pH = 8) was prepared by dissolving 6.80 g of imidazole, 10.38 g of KCl and 10.73 ml of 1 mol L−1 HCl in 1 L of water.
Acetic acid/sodium acetate buffer (pH = 4.5, I = 0.5 M) was prepared by dissolving 33.34 g sodium acetate trihydrate and 22.97 g acetic acid in 1 L of water. The ionic strength of buffer solution was adjusted to 0.5 M with 14.89 g of NaCl.
2.2 Synthesis
 | ||
| Scheme 1 Synthesis of thiolated cationic polyaspartamides from polysuccinimide (PSI) with an amine substituent, butane-1,4-diamine (DAB), and a thiolating agent (A), cysteamine (CEA) or (B) N-acetyl-DL-homocysteine (NAH) thiolactone. | ||
PASP-DAB-NAH polymers with different degrees of thiolation (NAH 2, 4, 6, 8, and 10) were prepared by the reaction of PASP-DAB with NAH thiolactone. For this purpose, 0.5 g (2.1 mmol repeating units) PASP-DAB was dissolved in 5.34 ml imidazole buffer (pH = 8) and stirred under an argon atmosphere for 15 min before adding NAH thiolactone, then the solution was stirred for 48 h to complete the reaction. PASP-DAB-NAH solution was dialyzed against water until the conductance dropped below 10 μS cm−1. Then, the pH of the sample was adjusted to 3 with 1 M sulfuric acid and dialyzed again for several days. The sample was dialyzed again after adding DTT (half of NAH repeating units) for one day, then concentrated to 10 ml using a rotary evaporator (40 °C, 30 mbar) and freeze-dried to yield a solid, off-white product.
:1 molar ratios, were prepared (Scheme 2). At first, PSI was modified with the fluorescent L-tryptophan methyl ester (Trp). 0.5 g (5.15 mmol succinimide repeating units) PSI was dissolved in 4.00 g DMSO, and after dissolution 0.013 g (0.051 mmol) tryptophan methyl ester hydrochloride was added to the mixture. Then, 0.25 g of 4 wt% H3PO4/DMSO solution and 0.026 g of DBA (0.23 mmol) were added dropwise, and the mixture was stirred at room temperature for 48 h. After the reaction was completed, the polymer (PSI-Trp) was modified as described below.
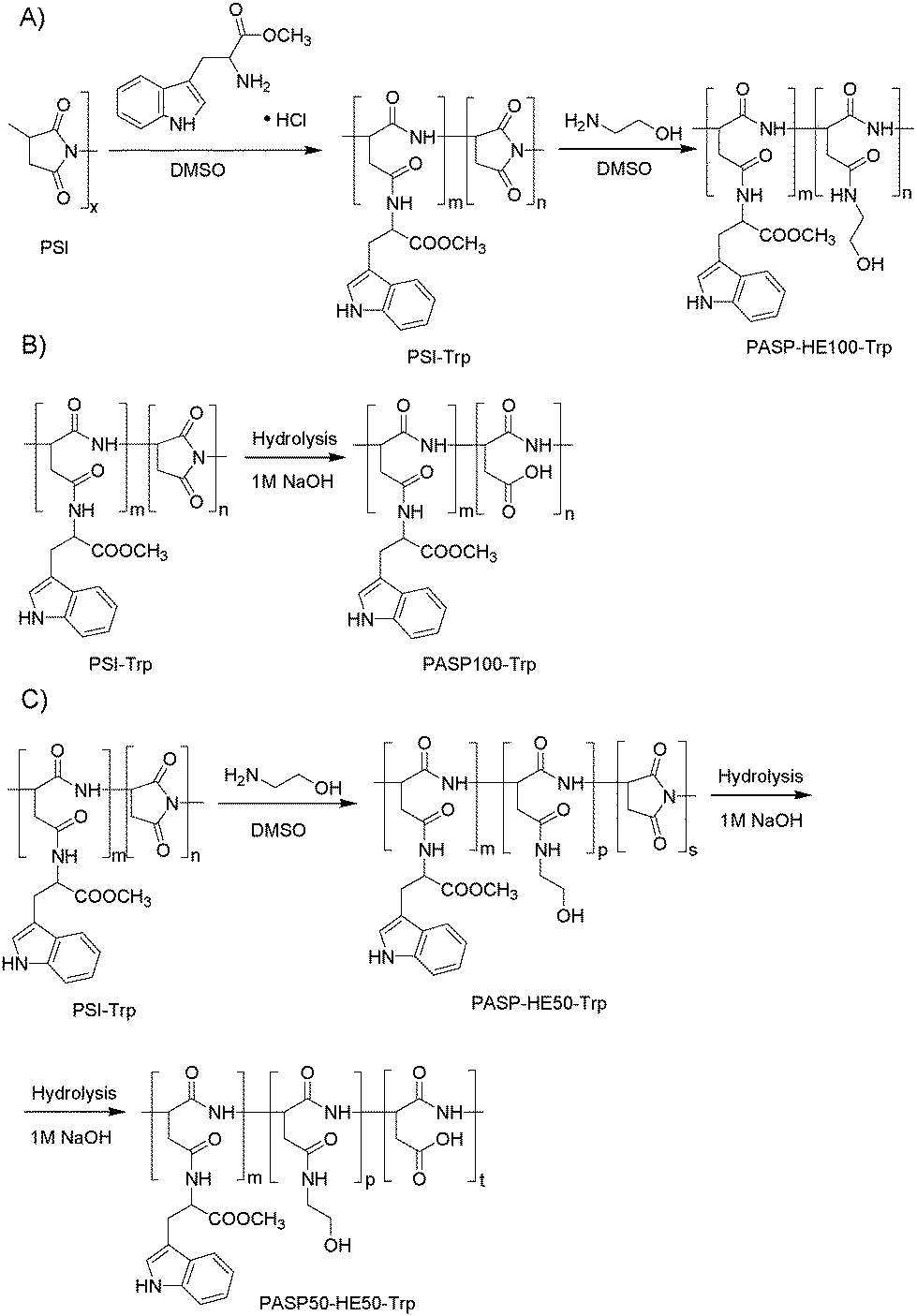 | ||
| Scheme 2 Synthesis of polyaspartamides labeled with the fluorescent tryptophan methyl ester: (A) neutral, (B) anionic, and (C) partially anionic derivatives. | ||
2.2.4.1 Preparation of fluorescence marked polymers with neutral side groups (PASP-HE100-Trp). A PASP derivative with neutral side-groups (PASP-HE100-Trp) was prepared (Scheme 2A) by adding 0.47 g (7.7 mmol) ethanolamine (HE) to the solution of Trp modified PSI and stirred for further 48 h. HE was added in 1.5-fold excess to modify all succinimide units. After the reaction was completed, the polymer was precipitated in ethyl acetate (the volume ratio of ethyl acetate to the polymer solution was 10
:1) and washed five times with ethyl acetate (50 ml) and three times with acetone (50 ml). The polymer was dissolved in water and dialyzed against water for one week with daily change of water until the conductance dropped below 10 μS cm−1. The solution was concentrated to 20 ml using a rotary evaporator (40 °C, 30 mbar) and freeze-dried to yield a solid, off-white product.
2.2.4.2 Preparation of fluorescence marked polymers with anionic charges (PASP100-Trp). A polymer with anionic side groups (PASP100-Trp) was prepared (Scheme 2B) by adding the solution of PSI-Trp to 100 ml of 0.1 M NaOH and stirring for 48 h. NaOH was used in two-fold excess for complete hydrolysis. After hydrolysis, the polymer was dialyzed against water for one week with daily change of water until the conductance dropped below 10 μS cm−1. The solution was concentrated to 20 ml using a rotary evaporator (40 °C, 30 mbar) and freeze-dried to yield a solid, off-white product.
2.2.4.3 Preparation of fluorescence marked polymers with anionic and neutral groups (PASP50-HE50-Trp). In the preparation of the polymer with both anionic and neutral side groups (PASP50-HE50-Trp) (Scheme 2C), 0.16 g of HE (2.6 mmol) was added to the solution of PSI-Trp to modify half of the succinimide units with HE and stirred for two days. Then the polymer (PSI50-HE50-Trp) was hydrolyzed with 52 ml of 0.1 M NaOH for 48 h. PASP50-HE50-Trp was dialyzed against water for one week with daily change of water until the conductance dropped below 10 μS cm−1. The solution was concentrated to 20 ml using a rotary evaporator (40 °C, 30 mbar) and freeze-dried to yield a solid, off-white product.
The estimated degree of modification for Trp groups in each fluorescent PASP derivative is 0.5–1.0% based on 1H NMR spectra (not shown).
2.3 Chemical characterization
The structures of both CEA-modified and NAH-modified polyaspartamides were analyzed by using 1HNMR spectroscopy (500 MHz, 16 scans). Solutions for the measurements were prepared by dissolving 20 mg of freeze-dried polymers in 1 ml of D2O.2.4 Gel permeation chromatography measurements
The number averaged molecular weight (Mn), weight averaged molecular weight (Mw) and polydispersity index (PDI) of freeze-dried thiolated polyaspartamides at a concentration of 2 mg ml−1 in acetate buffer (pH = 4.5, I = 0.5 M containing 10 mM DTT) were determined by size exclusion chromatography (SEC). A Nucleogel GFC-300-8 column was used with a Jasco HPLC system (Jasco, Tokyo, Japan) consisting of a PU-4180 pump (the flow rate was 1.0 ml min−1) with a UV-4075 UV/Vis detector (λ = 254 nm). β-Galactosidase, bovine serum albumin and wheat germ agglutinin (all were bought from Sigma-Aldrich) were used as protein standards to estimate the molar mass distribution of polyaspartamides.2.5 Rheological measurements
Rheological measurements were done on an Anton Paar Physica MCR301 rheometer equipped with a cone-plate geometry (diameter: 25 mm; cone angle: 1°; gap: 54 μm). 15 wt% solutions were prepared by dissolving 20 mg of each polymer in 93 mg of PBS buffer (pH = 7.4) containing DTT (half of the thiolated repeated groups), and 20 μl of the oxidant (0.5 M NaBrO3) was added to the polymer solutions on the plate of the rheometer. The change of dynamic moduli (storage and loss moduli, G′ and G′′, respectively) was monitored at constant strain and angular frequency (γ = 1%, ω = 1 rad s−1) during gelation. After gelation, the frequency dependence of the dynamic moduli (γ = 1%, ω = 0.5–500 rad s−1) was measured.2.6 Dynamic light scattering and zeta potential measurements
The size and zeta potential of PASP100-Trp, PASP50-HE50-Trp, and PASP-HE100-Trp were measured using a Malvern Nano-ZS Zetasizer (Malvern Instruments). For dynamic light scattering (DLS) measurements, polyaspartamides were dissolved in PBS buffer with pH = 7.4 at a concentration of 10 g L−1. For zeta potential measurements, the polyaspartamides were dissolved in PBS buffer with pH = 7.4 at a concentration of 1 g L−1. The electrophoretic mobility was measured for each sample. The Smoluchowski model was used to calculate the zeta potential from mobility values.2.7 Potentiometric titration
0.05 M polymer solutions of PASP-DAB, PASP100-Trp, and PASP50-HE50-Trp were prepared by dissolving the polymers in water. The inert salt NaCl was added to the polymer solutions to a final concentration of 50 mM. The pH of the polymer solutions was set to 2 with 1 M HCl. A single-junction glass pH electrode (Metrohm pH electrode, pH range: 1–13, filled with 3 M KCl) was calibrated using commercial pH buffers (Merck, Certipur® buffer solutions with pH = 4, 6, and 9). The polymer solutions were added to the flask equipped with a Teflon stir bar, a gas bubbler, and a titrant dispenser. The flask was then sealed with Parafilm® and purged with N2 for 15 minutes prior to the titration and also during the titration. The titrations were performed using an auto-titrator (Metrohm 808 Titrando, Tiamo, version 1.2) set to deliver 0.1 M NaOH with a dissolution amount of 50 mM NaCl (prepared freshly before the measurement) in automatic additions from pH 2 to 12, with the stability criterion of signal change slower than 25 mV min−1 between additions. A sufficient time (about 30 s) was allowed to reach a reasonably stable pH reading before the next volume of the base was added. Each titration lasted approximately 1 h. The sample temperature was kept constant (23 ± 1 °C).2.8 Turbidimetric measurements
Turbidimetric measurements were performed to confirm the interactions between cationic polyaspartamide (PASP-DAB) and polyaspartamides with different surface charges. A 1 g L−1 polymer solution of amine-modified cationic PASP-DAB and 1 g L−1 solutions of negatively charged poly(aspartic acid) (PASP100-Trp), neutral polyaspartamide (PASP-HE100-Trp), and partially negatively charged PASP (PASP50-HE50-Trp) were prepared in PBS buffer solution (pH = 7.4). PASP100-Trp, PASP-HE100-Trp, and PASP50-HE50-Trp solutions were added to a 2 ml solution of PASP-DAB stepwise, and after each step the absorbance spectra of the solutions were recorded between 300 and 800 nm using a UV-Vis spectrometer (Agilent Cary 60). Absorbance values at 400 nm were recorded to evaluate the turbidimetric data. Results are the means of triplicate measurements.2.9 Scanning electron microscopy (SEM) imaging
The morphology of polyaspartamide hydrogels with or without fluorescent polymers (PASP100-Trp, PASP-HE100-Trp, and PASP50-HE50-Trp) was investigated by SEM (FEI Inspect S50, accelerating voltage: 20kV). The hydrogels were prepared in the same composition as for release experiments, and after gelation the samples were kept in water to remove salts. The samples were kept in liquid nitrogen for 15 min before freeze-drying. Before taking the images, solid specimens were coated with gold (Emitec K550x, 1.5min, 35mA).
2.10 Release experiments
Drug release experiments were performed to study the release profiles of PASP derivatives from disulfide cross-linked polyaspartamide hydrogels and the correlation between release kinetics and polymer interactions. 18 mg of the thiolated polymer, PASP-DAB-CEA10 or PASP-DAB-NAH10, was dissolved in 82 μl PBS buffer (pH = 7.4) containing 0.56 mg DTT (44 mM). Separately, each fluorescently labelled polymer (PASP100-Trp, PASP-HE100-Trp, and PASP50-HE50-Trp) was dissolved in 15 μl of 0.5 M NaBrO3. The solutions of PASP-DAB-CEA10 and PASP-DAB-NAH10 were transferred onto a glass plate bordered with 2 mm thick silicone frames (gels formed in holes with a diameter of 7 mm), and under stirring each Trp modified polymer was added to the solution of PASP-DAB-CEA10 or PASP-DAB-NAH10 (six compositions in total). Stirrer bars were then removed from the precursor solutions, and the frames were covered with silicon sheets. After the gelation was complete, the gels were placed into dissolution jars containing 20 ml PBS buffer solution (pH = 7.4). 2 ml of the solution was withdrawn at 0, 15, 30, 60, 120, 180, and 240 min from every dissolution jar and an equal amount of fresh buffer was replaced after each sampling. The emission spectra of the samples were recorded with an Edinburgh Instruments FS5 fluorescence spectrometer in the emission wavelength range of 290–460 nm (dwell time = 0.5 s, xenon lamp, excitation at 270 nm, scan slit = 1.499, temperature = 20 °C). Release of the model drug from the chemically cross-linked hydrogels was tested in triplicate for all compositions. The emission values at 350 nm were used to calculate the amount of drug released from the hydrogels at predetermined times after calibration was done with aqueous solutions of marked polymers. To show the efficiency of the drug release from the hydrogels, the cumulative release (CR) for each time point was calculated.3 Results and discussion
3.1 Synthesis of cationic thiolated polyaspartamides
Cationic thiolated polyaspartamides were designed to encapsulate anionic poly(aspartic acid) derivatives during gelation as a result of oxidant-induced disulfide formation. Two different thiolation strategies were applied. In the first pathway (Fig. 1(A)), PSI was thiolated by CEA18,24 and primary amine groups were immobilized by using a large excess of DAB. We expected the formation of a cross-linked network upon the addition of a chemical oxidant (for the reaction scheme see Scheme S1(A), ESI†). In the second and completely new pathway (Fig. 1(B) and Scheme S1(B), ESI†) for preparing in situ gellable derivatives, primary amine groups were first immobilized, and a thiolactone-type agent (NAH thiolactone) was used to attach thiol groups as longer, flexible side groups to enhance gelation and avoid problems with free thiol-containing reagents. | ||
| Fig. 1 The effect of the side group structure on oxidation-induced gelation and release of anionic and neutral PASP. (A) Thiolation by CEA; (B) thiolation using NAH thiolactone. | ||
The structure of the synthesised polymers was confirmed by 1H NMR spectroscopy (the 1H NMR spectra, details on peak assignment are shown in Fig. S1 and S2, ESI†). Results showed that the actual composition of polyaspartamides correlated well with the feed composition in each case. Thus all side groups can be attached to polysuccinimide with high conversion (Table S1, ESI†).
The molecular weight of thiolated polyaspartamides was determined from SEC (for SEC traces see Fig. S3, ESI†) measurements by converting the elution time to molar mass. Mn, Mw and PDI values are listed in Table 1 (molar mass values must be only considered as a rough estimation as polyaspartamide standards are not available for SEC).
| Composition | M n (kDa) | M w (kDa) | PDI (−) |
|---|---|---|---|
| PASP-DAB | 108 | 116 | 1.07 |
| PASP-DAB-CEA2 | 89 | 94 | 1.06 |
| PASP-DAB-CEA4 | 116 | 125 | 1.07 |
| PASP-DAB-CEA6 | 114 | 124 | 1.08 |
| PASP-DAB-CEA8 | 110 | 120 | 1.08 |
| PASP-DAB-CEA10 | 113 | 122 | 1.07 |
| PASP-DAB-NAH2 | 113 | 123 | 1.08 |
| PASP-DAB-NAH4 | 115 | 125 | 1.08 |
| PASP-DAB-NAH6 | 114 | 123 | 1.07 |
| PASP-DAB-NAH8 | 116 | 125 | 1.07 |
| PASP-DAB-NAH10 | 115 | 125 | 1.08 |
The molecular weight of thiolated polyaspartamides showed similar values for all compositions, and a slightly lower mass was obtained for PASP DAB-CEA2. The analogous numbers in the molecular weight of thiolated polyaspartamides suggest that the properties of polymers will not affected by the difference in molecular weight.
3.2 Gelation properties of disulfide cross-linked hydrogels
The effect of two different thiolating agents (CEA and NAH) on the hydrogel formation and mechanical properties was studied via rheological measurements by following the changes in dynamic moduli for one hour during gelation and directly after gelation by the frequency dependence of moduli. We showed earlier that the oxidation-induced conversion of thiolated poly(aspartic acid) derivatives to disulphide bridges is accompanied by the gel formation in aqueous medium.18 Accordingly, the gels formed on the plate of the rheometer from thiolated polyaspartamides upon oxidation (Scheme S1, ESI†), and aqueous solutions of all the investigated polymers formed a gel within 20 min, except PASP-DAB-CEA2 and PASP-DAB-CEA4, which did not form hydrogels within one hour. It should be noted that the rheological measurement could be started only 50 s after the addition of the oxidizing agent, which explains the comparable storage (G′) and loss (G′′) moduli at the beginning.38 Nevertheless, the storage and loss moduli are very small in the beginning and the moduli are comparable, suggesting a viscoelastic liquid state of the mixture in its sol state. As the cross-linking reaction proceeds, the gel point is reached when the mixture suddenly loses its fluidity under the effect of the interconnection between sol components through the formation of disulfide linkages. The storage modulus becomes much higher than the loss modulus (G′ > G′′), and the system progressively loses its viscosity, while its elasticity increases significantly. The gelation is close to complete where G′ reaches a plateau. After that point, G′ still increases slightly, which can be due to post-cross-linking reactions, including dangling chain formation.24 The gelation was accompanied by a significant increase of the storage modulus and the gelation time was calculated from the inflection point of the storage modulus (Fig. 2). | ||
| Fig. 2 Time-dependent moduli (solid symbols represent the storage modulus G′ and open symbols represent the loss modulus G′′) for thiolated cationic polyaspartamides: (A) PASP-DAB-CEA and (B) PASP-DAB-NAH. | ||
In general, NAH-modified polyaspartamides formed gels faster than CEA-modified counterparts for each degree of thiolation (Fig. 3). The range of gelation time was below 20 min for all gellable compositions and was as short as 12 min for NAH derivatives which can be considered as fast-gelling formulation.39 Furthermore, NAH-modified derivatives formed gels even at the two lowest degrees of thiolation, contrary to the lack of gelation for CEA counterparts, clearly indicating the enhanced disulfide formation of NAH derivatives. For PASP-DAB-CEA4, only an increase in viscosity was observed which is shown by increased dynamic moduli and will be further analyzed by the data of frequency sweep measurements. PASP-DAB-CEA2 did not show any cross-linking process indicated by the moduli remaining constant.
 | ||
| Fig. 3 Gelation times of thiolated cationic polyaspartamides in the case of both thiolating agents as a function of the degree of thiolation. | ||
The frequency-dependence of moduli was determined after one hour of gelation to investigate the mechanical properties of the hydrogels (Fig. 4). Among CEA-modified polyaspartamides, only PASP-DAB-CEA10 and PASP-DAB-CEA8 showed constant storage moduli over the whole frequency range which is the requirement of a stable gel structure. The loss modulus of PASP-DAB-CEA10 was almost frequency independent, only showing frequency dependence at high frequencies, while for the rest of the polymers, the loss modulus was frequency-dependent. For PASP-DAB-CEA4 and PASP-DAB-CEA2 polymers, both storage and loss moduli were frequency-dependent, indicating the absence of gel formation. At lower frequencies, the storage modulus of PASP-DAB-CEA4 was constant (below 5 rad s−1) and showed frequency dependence at higher frequencies, proving that even though complete gel formation did not happen, the cross-linking reaction proceeded to some extent. NAH-modified polyaspartamides showed constant storage modulus over the whole frequency range. Only a slight increase was observed at higher frequencies for PASP-DAB-NAH4 and PASP-DAB-NAH2 due to chain entanglements of hydrogels (Fig. 4).24 The loss modulus of all compositions was frequency-dependent, indicating the presence of a relaxation process in the frequency window of the measurement.
 | ||
| Fig. 4 Frequency dependence of dynamic moduli after 1 h of gelation (solid symbols: storage modulus, G′, and open symbols: loss modulus, G′′) for thiolated cationic polyaspartamides: (A) PASP-DAB-CEA and (B) PASP-DAB-NAH. | ||
We used a power-law equation (eqn (1)) in the frequency range of 0.5 to 500 rad s−1 to quantify the frequency dependence of the storage modulus.
| G′ = kωn | (1) |
 | ||
| Fig. 5 The strength of frequency dependence (n) as a function of the degree of thiolation. | ||
To compare the stiffness of the hydrogels formed from CEA- and NAH-modified polyaspartamides, the storage moduli of polymers at a frequency of 0.5 rad s−1 were plotted as a function of the degree of thiolation (Fig. 6). Although the storage moduli of PASP-DAP-CEA10 and PASP-DAB-NAH10 gels were similar, by reducing the degree of thiolation, the stiffness of PASP-DAP-CEA hydrogels decreased largely in comparison with NAH-modified counterparts. We can assume from the storage modulus values and the frequency dependence of dynamic moduli that the longer side group length of NAH thiolactone can lead to faster gelation and a higher cross-linking density compared to CEA modification.
 | ||
| Fig. 6 Storage modulus (G′) as a function of the degree of thiolation at a frequency of 0.5 rad s−1 in the case of both thiolating agents. | ||
3.3 Mesh size of hydrogels and particle size of model drugs
The mesh size, the average distance between consecutive cross-links, is an important hydrogel feature, it is correlated with the mechanical strength and degree of swelling of the hydrogels, and it controls many important properties of drug release, such as the diffusion coefficient and the rate of release of entrapped drugs/proteins.40,41 To this end, the mesh size of hydrogels was calculated from the storage modulus values (G′) of disulfide cross-linked cationic polyaspartamides at the lowest angular frequency measured (0.5 rad s−1), which is considered to be of an equilibrium value (eqn (2)):17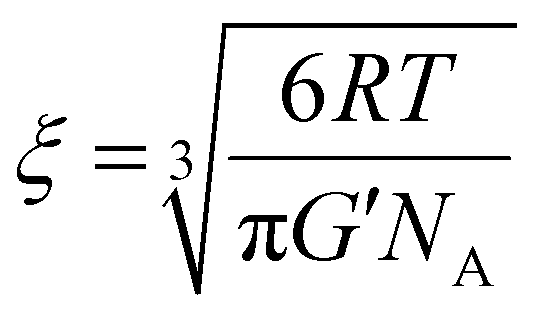 | (2) |
The calculated mesh size of PASP-DAB-NAH10 were 11.3 nm and that of PASP-DAB-CEA10 was 10.8 nm, which are in the range for typical synthetic hydrogels and can be ideal for sustained release of macromolecular drugs, e.g., proteins or nucleic acids, which are usually in the range of a few nm. We emphasize that the mesh size can be a determining factor in the release of encapsulated compounds; however, a general porous structure on various length scales was observed for all gels, as proven by the SEM images (Fig. S4, ESI†).
We synthesised fluorescently labelled model polyelectrolytes to study drug release. Fully and partially anionic as well as neutral PASP derivatives with tryptophan groups were synthesised (Scheme 2) to test drug encapsulation and release. The particle size of model drugs determined by DLS was generally smaller than the estimated mesh size of the hydrogel but was in the same magnitude (Fig. S5, ESI†). The particle sizes of PASP100-Trp, PASP50-HE50-Trp, and PASP-HE100-Trp were 3.1 ± 0.9 nm, 2.5 ± 0.5 nm, and 4.7 ± 1.2 nm, respectively (Table 2), which suggested the lack of steric hindrance for the release of the chosen model polymer drugs, but we also expected that the charge significantly affects the release rates.
| Composition | pKa | Zeta potential (mV) | Particle size (nm) |
|---|---|---|---|
| PASP-DAB | 9.6 | 15 ± 0.1 | — |
| PASP100-Trp | 4.8 | −10 ± 1.8 | 3.1 ± 0.9 |
| PASP50-HE50-Trp | 4.8 | −12 ± 1.8 | 2.5 ± 0.5 |
| PASP-HE100-Trp | — | −5 ± 1 | 4.7 ± 1.2 |
3.4 Protonation state of the polymers used and their electrostatic interactions
To reveal the protonation and charge state of model drugs and the hydrogels, acid–base titration and zeta potential measurements were performed. Titration was carried out for PASP-DAB, PASP100-Trp and PASP50-HE50-Trp and the number of protons bound to the basic group (COO- or NH2) was determined by converting titration curves to Bjerrum type plots (Fig. 7) using the following formula (eqn (3)):42| nH = (V0cHCl − VtcNaOH) − ([H+] − Kw[H+]−1)(V0 + Vt) + NH | (3) |
 | ||
| Fig. 7 Bjerrum curves of PASP-DAB, PASP100-Trp and PASP50-HE50-Trp. | ||
The pKa values of PASP100-Trp, PASP-DAB and PASP50-HE50-Trp were calculated from the inflection point by plotting the second derivative plot of nH over pH and the zero point was the pKa. The pKa value was 9.6 for PASP-DAB, 4.8 for aspartic acid (PASP100-Trp), and 4.8 for PASP50-HE50-Trp, which were in good agreement with the literature.42 Results suggest that all amine groups of cationic polyaspartamide (PASP-DAB) at pH = 7.4 are protonated, while the protonation state of PASP is in the acidic range. Thus, PASP-DAB is positively charged at pH = 7.4, while PASP100-Trp is negatively charged. The zeta potential measurements confirmed the positive surface charge of PASP-DAB and the negative charge of PASP100-Trp at pH = 7.4 (Table 2).
The possible electrostatic interactions between the cationic polyaspartamide, PASP-DAB and the PASP derivatives with different surface charges were tested by turbidimetric measurements (Fig. 8).43 The interaction can be detected by the appearance of turbidity as a result of polyplex formation between oppositely charged polyelectrolytes. No signal was observed between the cationic PASP-DAB and the neutral PASP-HE100-Trp, while a strong interaction was detected between oppositely charged PASP-DAB and PASP100-Trp. As expected, by replacing half of the negative charges with neutral charges, the curve shifted to a higher mass ratio of anionic to cationic derivatives, suggesting weaker interactions. The weaker interactions were also indicated by lower maximum absorbance values for the mixed composition compared to fully anionic PASP.
 | ||
| Fig. 8 Turbidimetric titration of PASP-DAB solution by PASP100-Trp, PASP50-HE50-Trp, and PASP-HE100-Trp solutions. The concentration of the polymer solution was 1 g L−1. | ||
3.5 Release of model drugs with different surface charges from thiolated polyaspartamides
The role of electrostatic interactions in the drug release profile was studied by the release of neutral (PASP-HE100-Trp), partially anionic (PASP50-HE50-Trp) and completely anionic (PASP100-Trp) PASP derivatives as model drugs from thiolated cationic polyaspartamides. The model drugs were entrapped in PASP-DAB-CEA10 and PASP-DAB-NAH10 hydrogels during in situ gelation, and the effect of network composition, i.e., the type of thiolating agent, was also considered.The drug release profiles from hydrogels are generally governed by the mesh size and the interactions between the components. As previously shown, the hydrodynamic diameters of PASP derivatives used as model drugs are smaller than the estimated mesh size, although it should be noted that the size distribution of mesh size is not known. In this point of view, the release of a significant amount of the entrapped polymers was expected due to the lack of steric hindrance. On the other hand, the anionic polyelectrolytes (PASP100-Trp and PASP50-HE50-Trp) were expected to be retained by the cationic polymer network because of the strong electrostatic forces between the anionic drugs and cationic thiolated polyaspartamides. In accordance with these presumptions, fully anionic PASP100-Trp was completely retained in both cationic hydrogels (Fig. 9). The partial (50%) replacement of anionic groups with neutral ones resulted in the partial release of entrapped PASP50-HE50-Trp with a significant difference between the two hydrogels. The increased release from the PASP-DAB-NAH hydrogel might be explained by the partial screening of primary amine groups from electrostatic interactions by amide groups formed after modification by the thiolactone reagent. The neutral PASP-HE100-Trp showed an increased release from both hydrogels due to the lack of attractive electrostatic interactions with the hydrogel network. Interestingly, the PASP-DAB-NAH10 hydrogel showed somewhat faster release, and thus the screening effect of amide groups as a result of thiolactone modification might play a role here as well through hydrogen bonding.
 | ||
| Fig. 9 Cumulative release of model drugs from thiolated polyaspartamides. | ||
We used the empirical Korsmeyer–Peppas equation (eqn (4)) for describing the release profiles:44
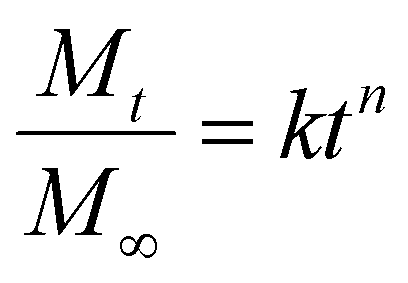 | (4) |
The kinetic constants and exponents are summarized in Table 3, and the percentage release after 4 h is also shown for comparison. For neutral polymers, the exponent is close to 0.5, indicating that the release is diffusion-controlled release. The kinetic constant is affected by both the model drug and network structure. The addition of anionic charges reduced the kinetic constant and, for both drugs, it was higher for NAH-modified gels, suggesting weaker interactions between the model drug and polymer network. In conclusion, the release profile could be controlled by the charge density of the entrapped material, but there is a complex interplay between diffusion limited by mesh size (distribution) and secondary interactions, including electrostatic attraction and hydrogen bonds. The results obtained might be useful in the design of similar cationic polyaspartamides for the encapsulation of nucleic acid derivatives in continuation of the work.
4 Conclusion
Thiolated polyaspartamides with cationic charges were synthesised using two synthetic pathways, such as modification with cysteamine (CEA) and, as a new approach, combining thiolactone chemistry with the cationic modification of polyaspartamides. Primary amine groups were attached to the polymer chain to render cationic properties to all compositions in both cases. Disulfide formation was induced by the addition of an oxidant in the aqueous solution of thiolated polyaspartamides, resulting in an increase of viscosity and, in most cases, the rapid formation of hydrogels. The longer, flexible side group length of NAH-modified polyaspartamides displayed faster gelation than CEA-modified counterparts which also increased the stiffness of the hydrogels gels, proving the enhanced cross-linking reaction of NAH-modified hydrogels over CEA-modified counterparts. We confirmed the electrostatic interaction between the linear anionic PASP derivatives and the cationic thiolated polyaspartamides. The release kinetics of PASP derivatives was followed by the fluorescent labelling of the encapsulated polymers, which showed that fully anionic PASP was entrapped completely in the hydrogels. In contrast, the reduction of the concentration of anionic groups caused a partial release of PASP derivatives. NAH- and CEA-modified cationic polyaspartamide hydrogels showed distinct release rates, indicating the interplay between electrostatic interactions and the chemistry of thiolated side groups. The developed in situ gellable materials have potential applications as injectable, fast gelling formulations to deliver macromolecular drugs of various charges, and possible non-enzymatic degradation of cationic poly(aspartamides) can be combined with these properties for further benefits.Author contributions
Aysel Mammadova: investigation, formal analysis, writing – original draft. Benjámin Gyarmati: conceptualization, methodology, writing – review & editing, supervision. Kitti Sárdi: investigation, formal analysis. Adrien Paudics: formal analysis. Zoltán Varga: investigation, formal analysis. András Szilágyi: conceptualization, methodology, writing – review & editing, resources, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We would like to thank Dr Katalin Kopecskó and Erika Grossmann from the Faculty of Civil Engineering, Budapest University of Technology and Economics (BME), for their help in particle size and zeta potential measurements. The help of Róbert Kovács and Péter Gordon (Faculty of Electrical Engineering and Informatics, BME) in SEM measurements is highly appreciated. The research reported in this paper is part of project no. TKP2021-EGA-02, implemented with the support provided by the Ministry for Innovation and Technology of Hungary from the National Research, Development and Innovation (NRDI) Fund, financed under the TKP2021 funding scheme. Further support was provided by the NRDI Office via grants FK 125074 and FK 138029. B. Gyarmati acknowledges the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The work was also supported by the ÚNKP-21-5 New National Excellence Program of the Ministry for Innovation and Technology from the source of the NRDI Fund. A. Mammadova is grateful for the scholarship of Stipendium Hungaricum.References
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- N. A. Peppas and A. S. Hoffman, Biomaterials Science, An Introduction to Materials in Medicine, 4th edn, 2020, pp. 153–166 Search PubMed.
- A. K. Burkoth and K. S. Anseth, Biomaterials, 2000, 21, 2395–2404 CrossRef CAS.
- M. Li, M. J. Mondrinos, X. Chen, M. R. Gandhi, F. K. Ko and P. I. Lelkes, J. Biomed. Mater. Res., Part A, 2006, 79, 963–973 CrossRef PubMed.
- C. K. Kuo and P. X. Ma, Biomaterials, 2001, 22, 511–521 CrossRef CAS PubMed.
- L. Francis, K. V. Greco, A. R. Boccaccini, J. J. Roether, N. R. English, H. Huang, R. Ploeg and T. Ansari, J. Biomater. Appl., 2018, 33, 447–465 CrossRef CAS PubMed.
- A. Mandal, J. R. Clegg, A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2020, 5, 1–12 Search PubMed.
- S. Bonengel and A. Bernkop-Schnürch, J. Controlled Release, 2014, 195, 120–129 CrossRef CAS.
- H. J. Lee, G. M. Fernandes-Cunha and D. Myung, React. Funct. Polym., 2018, 131, 29–35 CrossRef CAS PubMed.
- T. I. Zarembinski, N. J. Doty, I. E. Erickson, R. Srinivas, B. M. Wirostko and W. P. Tew, Acta Biomater., 2014, 10, 94–103 CrossRef CAS PubMed.
- D. Sakloetsakun, J. M. R. Hombach and A. Bernkop-Schnürch, Biomaterials, 2009, 30, 6151–6157 CrossRef CAS PubMed.
- P. S. Syamala and R. M. Ramesan, Ther. Delivery, 2018, 9, 751–773 CrossRef CAS PubMed.
- S. Park, S. K. Lee and K. Y. Lee, J. Controlled Release, 2011, 152, e165–e166 CrossRef CAS PubMed.
- E. Wernersson, J. Heyda, A. Kubíčková, T. Křížek, P. Coufal and P. Jungwirth, J. Phys. Chem. B, 2010, 114, 11934–11941 CrossRef CAS PubMed.
- S. S. Priya and M. R. Rekha, Int. J. Pharm., 2017, 530, 401–414 CrossRef CAS PubMed.
- N. Kordalivand, E. Tondini, C. Y. J. Lau, T. Vermonden, E. Mastrobattista, W. E. Hennink, F. Ossendorp and C. F. V. Nostrum, J. Controlled Release, 2019, 315, 114–125 CrossRef CAS PubMed.
- J. P. Schillemans, W. E. Hennink and C. F. van Nostrum, Eur. J. Pharm. Biopharm., 2010, 76, 329–335 CrossRef CAS.
- B. Gyarmati, B. Vajna, Á. Némethy, K. László and A. Szilágyi, Macromol. Biosci., 2013, 13, 633–640 CrossRef CAS PubMed.
- E. Krisch, B. Gyarmati, D. Barczikai, V. Lapeyre, B. Á. Szilágyi, V. Ravaine and A. Szilágyi, Eur. Polym. J., 2018, 105, 459–468 CrossRef CAS.
- B. Á. Szilágyi, B. Gyarmati, G. Horvát, Á. Laki, M. Budai-Szűcs, E. Csányi, G. Sandri, M. C. Bonferoni and A. Szilágyi, Polym. Int., 2017, 66, 1538–1545 CrossRef.
- B. Gyarmati, A. Mammadova, G. Stankovits, D. Barczikai and A. Szilágyi, Period. Polytech., Chem. Eng., 2021, 65, 183–191 CrossRef CAS.
- D. Barczikai, V. Kacsari, J. Domokos, D. Szabó and A. Jedlovszky-Hajdu, J. Mol. Liq., 2021, 322, 114575 CrossRef CAS.
- C. Németh, B. Gyarmati, J. Gacs, D. V. Salakhieva, K. Molnár, T. Abdullin, K. László and A. Szilágyi, Eur. Polym. J., 2020, 130, 109624 CrossRef.
- B. Gyarmati, A. Mammadova, D. Barczikai, G. Stankovits, A. Misra, M. S. Alavijeh, Z. Varga, K. László and A. Szilágyi, Polym. Degrad. Stab., 2021, 188, 109577 CrossRef CAS.
- S. Reinicke, P. Espeel, M. M. Stamenović and F. E. Du Prez, Polym. Chem., 2014, 5, 5461–5470 RSC.
- D. Frank, P. Espeel, S. Claessens, E. Mes and F. E. Du Prez, Tetrahedron, 2016, 72, 6616–6625 CrossRef CAS.
- A. S. Chubarov, Encyclopedia, 2021, 1, 445–459 CrossRef.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1–51 CrossRef PubMed.
- S. Martens, J. O. Holloway and F. E. D. Prez, Macromol. Rapid Commun., 2017, 38, 1–15 CrossRef PubMed.
- S. Martens, F. Driessen, S. Wallyn, O. Türünç, F. E. Du Prez and P. Espeel, ACS Macro Lett., 2016, 5, 942–945 CrossRef CAS PubMed.
- P. Espeel and F. E. Du Prez, Eur. Polym. J., 2015, 62, 247–272 CrossRef CAS.
- C. Chattaway, S. Belbekhouche, F. E. Du Prez, K. Glinel and S. Demoustier-Champagne, Langmuir, 2018, 34, 5234–5244 CrossRef CAS.
- M. R. Molla, A. Böser, A. Rana, K. Schwarz and P. A. Levkin, Bioconjugate Chem., 2018, 29, 992–999 CrossRef CAS PubMed.
- W. Ke, W. Yin, Z. Zha, J. F. Mukerabigwi, W. Chen, Y. Wang, C. He and Z. Ge, Biomaterials, 2018, 154, 261–274 CrossRef CAS PubMed.
- S. Reinicke, H. C. Rees, P. Espeel, N. Vanparijs, C. Bisterfeld, M. Dick, R. R. Rosencrantz, G. Brezesinski, B. G. De Geest, F. E. Du Prez, J. Pietruszka and A. Böker, ACS Appl. Mater. Interfaces, 2017, 9, 8317–8326 CrossRef CAS PubMed.
- E. Krisch, D. Balogh-Weiser, J. Klimkó, B. Gyarmati, K. László, L. Poppe and A. Szilágyi, Express Polym. Lett., 2019, 13, 512–523 CrossRef CAS.
- J. Vlasák, F. Rypáček, J. Drobník and V. Saudek, J. Polym. Sci., Polym. Symp., 2007, 66, 59–64 CrossRef.
- B. Gyarmati, E. Krisch and A. Szilágyi, React. Funct. Polym., 2014, 84, 29–36 CrossRef CAS.
- E. Jain, L. Hill, E. Canning, S. A. Sell and S. P. Zustiak, J. Mater. Chem. B, 2017, 5, 2679–2691 RSC.
- S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285–10291 CrossRef CAS PubMed.
- L. Pescosolido, L. Feruglio, R. Farra, S. Fiorentino, I. Colombo, T. Coviello, P. Matricardi, W. E. Hennink, T. Vermonden and M. Grassi, Soft Matter, 2012, 8, 7708–7715 RSC.
- V. Torma, T. Gyenes, Z. Szakács and M. Zrínyi, Acta Biomater., 2010, 6, 1186–1190 CrossRef CAS PubMed.
- B. Á. Szilágyi, A. Mammadova, B. Gyarmati and A. Szilágyi, Colloids Surf., B, 2020, 194, 111219 CrossRef PubMed.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00674j |
| This journal is © The Royal Society of Chemistry 2022 |