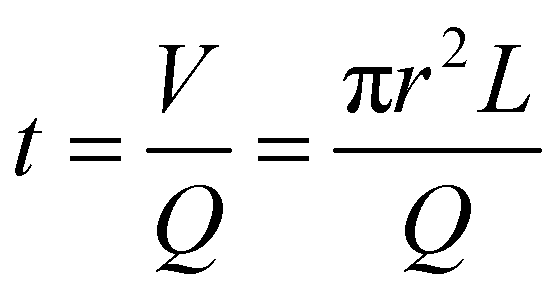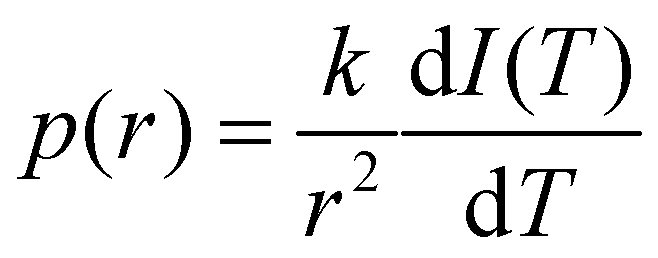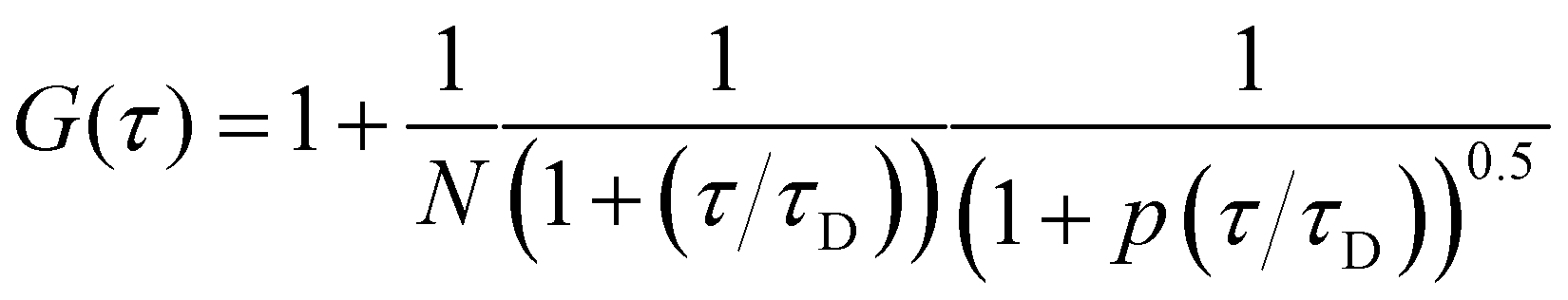DOI:
10.1039/D1TB02751D
(Paper)
J. Mater. Chem. B, 2022,
10, 1980-1990
PEG-based cleavable hydrogel microparticles with controlled porosity for permiselective trafficking of biomolecular complexes in biosensing applications†
Received
14th December 2021
, Accepted 16th February 2022
First published on 16th February 2022
Abstract
In the last decade, PEG-based hydrogels have been extensively used for the production of microparticles for biosensing applications. The biomolecule accessibility and mass transport rate represent key parameters for the realization of sensitive microparticles, therefore porous materials have been developed, mainly resorting to the use of inert porogens and copolymers with different chain lengths. However, very limited information is reported regarding the addition of cleavable crosslinkers to modulate the network porosity. In this scenario, the aim of this work is to design, synthesize and characterize hydrogel microparticles, based on the copolymerization between PEG-diacrylate and N,N′-(1,2-dihydroxyethylene)-bisacrylamide, a cleavable crosslinker that simultaneously produces pores and reactive groups for bioprobe 3D bioconjugation. The results show great accessibility of these microparticles to antibodies and their complexes, without affecting their diffusion rate. Furthermore, the presence of a well-defined number of reactive aldehydes, produced by the cleavage reaction, allows modulating biosensor sensitivity through a fine control of the conjugation degree. The antibody-conjugated microparticles can efficiently capture the analyte down to a few picograms. These novel microparticles could be used as a highly sensitive platform for biomacromolecule detection in complex fluids, exploiting the combined effects of PEG's anti-fouling properties, large network porosity and interconnections, and three-dimensional bioconjugation.
Introduction
Protein-immobilized microparticles have been widely used as a versatile platform for biological assays in biosensing applications.1–4 Compared to plate-based technologies, polymeric microparticles offer a higher surface to volume ratio, which improves scalability, reduces assay time and reagent consumption.5 The realization of hydrogel particles, in contrast with non-porous polymeric particles, allows a three-dimensional bioconjugation of probes which provides their flexible presentation, with consequent reduction of steric limitations and improvement of loading capacity of capture probes.6–10 Furthermore, porous microparticles are able to concentrate the target in a small volume, amplifying the signal and consequently, improving sensitivity and detection limits.11 In addition, PEG-based material properties including biocompatibility, molecular filtering capacity, a solution-like environment and non-fouling nature12–15 render these hydrogels ideal scaffolds to preserve the activity of immobilized proteins and to perform in-gel immunoassays in complex biological fluids.
However, a careful design of the particle layout is required to ensure (i) a fine-tuned nanostructure to facilitate mass transport and analyte diffusion within the polymer network and (ii) mild conditions for chemical ligation of bioprobes.
As network accessibility and mobility of large biomolecules affect key parameters of an in-gel immunoassay, like sensitivity and kinetics, increasing the porosity of the PEG hydrogel represents a valid strategy. A number of different approaches have been reported for porous hydrogels production, such as freeze drying,16 phase separation,17 salt leaching,18,19 3D printing,20 and microgel assembly.21 Whereas these strategies are widely used for the development of porous bulk hydrogels, they are not easily applicable for the preparation of microparticles. One of the simplest strategies to obtain porous particles is to modulate the composition of the pre-polymer solution. Increasing the molecular weight of the PEG chains and/or lowering its concentration are effective ways to increase the gel porosity, but a fine modulation is not achieved.22,23 Another commonly used technique is solvent casting/particulate leaching, where various particle classes are homogeneously distributed into a solution prior to polymerization. Once polymerization is completed, inactive porogens, typically inert PEG, are subsequently leached or dissolved away with a selective solvent, resulting in a porous network.24–27 Although porous particles can be effectively generated with this method, the use of a porogen requires many washing steps and a long time (up to 48 hours) to eliminate excess.28
Concerning bioprobe immobilization within the support material, currently the most used approaches involve the introduction of reactive groups (like acrylate moieties) on the biomolecule through the use of heterobifunctional linkers,29 to obtain covalent immobilization during the chain-growth polymerization step. However, this strategy could lead to biomolecule degradation due to extreme reaction conditions (UV exposure, free radicals, temperature, organic solvents).30,31
Taking into account both requirements, the use of a cleavable crosslinker, which once cleaved produces pores and functional groups, could represent a suitable route to simultaneously overcome issues related to the optimization of polymer nanostructures and conjugation of capture probes. Among cleavable crosslinkers, N,N′-(1,2-dihydroxyethylene)-bisacrylamide (DHEBA) has been used in the literature to produce hollow particles,32 thermoresponsive membranes33 or shell responsive particles,34 but it has never been used to partially degrade the network for generating new pores and reactive groups, while preserving the mechanical integrity of the hydrogel.
In this work, we describe a one-step synthesis of porous and reactive PEG-based hydrogel microparticles, through the copolymerization between PEGDA (Mn = 700) and DHEBA at different ratios. After photo-polymerization, the vicinal diols of the cross-linker can be turned into aldehydes through oxidative cleavage,35 using stoichiometric amounts of sodium periodate.36 Upon chemical cleavage, the crosslinking density reduces, the pore size distribution widens, and, simultaneously, two aldehyde groups are formed that are useful for further functionalization with wild-type bioprobes via reductive amination (Fig. 1a). Schiff base formation between aldehydes and amine-containing biomolecule (i.e. antibody) occurs readily in aqueous solutions, and this bond is stabilized by adding a reductant compound (i.e. sodium cyanoborohydride). Microparticles’ swelling behaviour and aldehydes amount have been analysed to optimize cleavage conditions and the bioconjugation degree. Moreover, to gain information about the biomolecules’ accessibility within the polymeric network, porosity and diffusion of fluorescently labelled biomolecules have been studied by NMR cryoporometry, equilibrium partitioning and fluorescence correlation spectroscopy.
 |
| | Fig. 1 (a) Schematic illustration of hydrogel microparticles’ chemical composition and cleavage reaction: microparticles’ network is based on a copolymer between PEG diacrylate and the cleavable crosslinker N,N′(1,2-dihydroxyethylene)bisacrylamide (DHEBA). The vicinal diols in the crosslinker can be cleaved by stoichiometric amounts of sodium periodate, simultaneously producing pores and reactive aldehydes, useful for further functionalization with biomolecules such as antibodies (R = antibody). (b) Cleavable hydrogel microparticle production and morphological characterization: microparticles synthesized via droplet generation in a microfluidic T-junction chip, optical image of polymerized and swollen hydrogel microparticles, SEM image of dried microparticles. | |
We found that the addition of DHEBA induced highly porous morphology in hydrogel microparticles, assuring full accessibility of large proteins to immobilized probes. Furthermore, as a proof of concept for the development of in-gel immunoassays, the optimized microparticles were functionalized with antibodies able to bind human IgG, and have been shown to efficiently capture the target in the picomolar range.
Results and discussion
Hydrogel microparticle synthesis and morphological characterization
The 3D PEG-based hydrogels represent a perfect platform for bead-based assays because of their hydrophilicity, chemical flexibility and anti-fouling properties. However, the crosslinking density creates a diffusion barrier for large biomolecules, limiting their penetration within the hydrogel. To overcome this issue, a cleavable crosslinker was introduced to PEGDA solution to increase the porosity of the polymeric network, thus improving the reaction/capture kinetics. In this study, the microparticles were formed into a microfluidic chip with a T-junction geometry (Fig. S1, ESI†). Four different pre-polymer solutions were used to create microparticles with four different compositions (Table S1, ESI†). In particular, we prepared two pre-polymer solutions using 10 wt% PEGDA700 with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and 1:40 DHEBA:PEGDA ratios (respectively named as 10R4 and 10R40), and other two pre-polymer solution mixing PEGDA700 at 15 wt% with 1:4 and 1:40 DHEBA:PEGDA ratios (named respectively 15R4 and 15R40). Light mineral oil with 5% SPAN80 is used as a continuous phase, while the interfacial tension with the dispersed phases ranges from 1.8 to 3 mN m−1. Droplet size and generation rate were regulated by different parameters. We applied different outer to inner flow rate ratios to test sizes and generation rates during the droplet generation process (Table S2, ESI†). In detail, the monodisperse droplets have been generated using a flow rate of 0.25 μL min−1 for the dispersed phase and 2.5 μL min−1 for the oil, resulting in a production of ∼20 droplets per second. The histogram in Fig. S1 (ESI†) shows the frequency distribution of the droplet diameter. About 90% of the droplets falls into the range between 72.6 and 77.4 μm (Fig. S1, ESI†). Optical images (Fig. 1b) confirm that the microparticles, after the photopolymerization, are monodisperse without any contraction. SEM images (Fig. 1b) demonstrates that the microparticles retain their spherical shape even in the dried state, with a diameter of about 30 μm. The SEM micrograph confirms the uniformity and fine globular shape of the PEG polymer beads, which exhibit a robust solid structure, an important feature for biosensing applications.
:4 and 1:40 DHEBA:PEGDA ratios (respectively named as 10R4 and 10R40), and other two pre-polymer solution mixing PEGDA700 at 15 wt% with 1:4 and 1:40 DHEBA:PEGDA ratios (named respectively 15R4 and 15R40). Light mineral oil with 5% SPAN80 is used as a continuous phase, while the interfacial tension with the dispersed phases ranges from 1.8 to 3 mN m−1. Droplet size and generation rate were regulated by different parameters. We applied different outer to inner flow rate ratios to test sizes and generation rates during the droplet generation process (Table S2, ESI†). In detail, the monodisperse droplets have been generated using a flow rate of 0.25 μL min−1 for the dispersed phase and 2.5 μL min−1 for the oil, resulting in a production of ∼20 droplets per second. The histogram in Fig. S1 (ESI†) shows the frequency distribution of the droplet diameter. About 90% of the droplets falls into the range between 72.6 and 77.4 μm (Fig. S1, ESI†). Optical images (Fig. 1b) confirm that the microparticles, after the photopolymerization, are monodisperse without any contraction. SEM images (Fig. 1b) demonstrates that the microparticles retain their spherical shape even in the dried state, with a diameter of about 30 μm. The SEM micrograph confirms the uniformity and fine globular shape of the PEG polymer beads, which exhibit a robust solid structure, an important feature for biosensing applications.
Characterization of porosity through equilibrium volumetric swelling, NMR cryo-porometry and equilibrium partitioning measurements
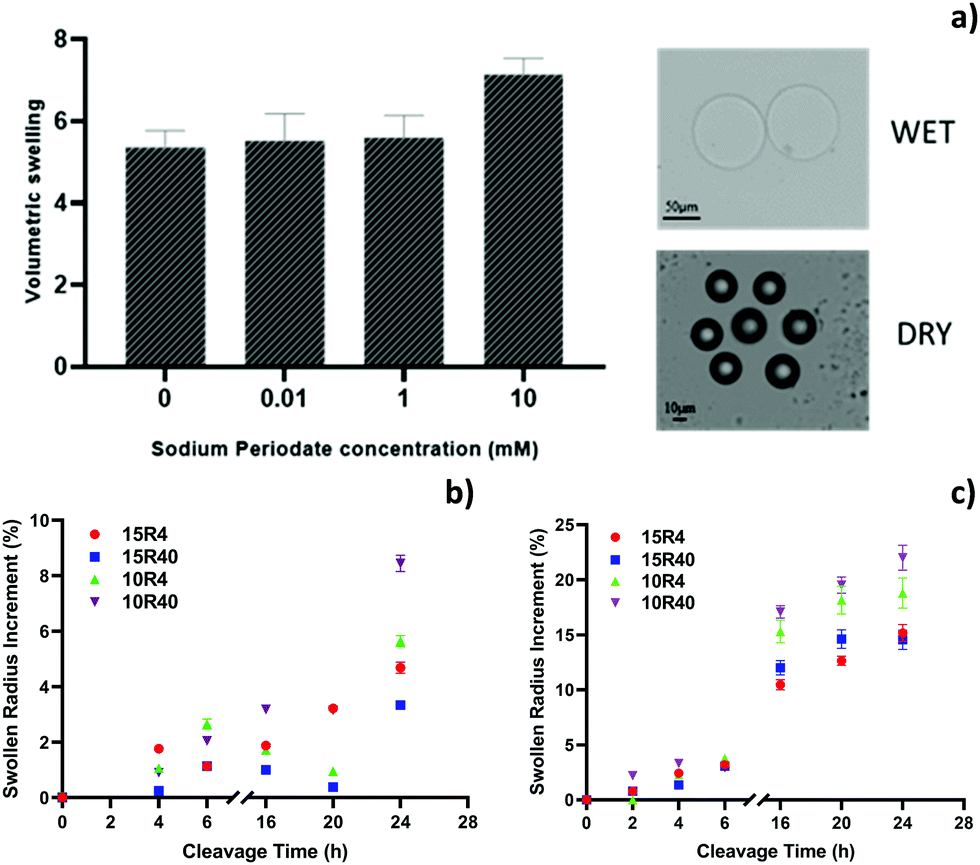
The polymerized cleavable microparticles are predominantly composed of PEGDA with the crosslinker DHEBA as a minor component (at different mol%). As the vicinal diols in the cross-linker can be cleaved by stoichiometric amounts of sodium periodate, the pore size of the gel can be increased by degradation of the cross-linker. An equilibrium volumetric swelling analysis of the hydrogel microparticles is directly related to their porosity. The higher the swelling the greater the amount of water absorbed by the hydrogel, suggesting a greater effectiveness of the cleavage reaction. To optimize sodium periodate concentrations, changes in the volumetric swelling ratio, Q, of microparticles are analysed, before and after the cleavage reaction. In particular, three different concentrations of sodium periodate (0.1, 1 and 10 mM) have been evaluated for the cleavage of 15R4 microparticles, whose composition presents the higher amount of crosslinker than other particles. Fig. 2a shows the variation in the volumetric swelling of these particles as a function of the periodate concentration. Upon addition of periodate to the solution, the microparticles swell differently, as suggested by the changes in the radius observed with the optical microscope. In particular, 10 mM of sodium periodate leads to a greater volumetric swelling and, consequently, this concentration was used for further characterizations and analysis. In order to optimize cleavage temperature and reaction time, studies on the influence of these two parameters on the swollen particles size were conducted for all the hydrogel microparticle compositions. The percentage of particles radius variations over time at room temperature (Fig. 2b) and at a temperature of 50 °C (Fig. 2c) are reported. At room temperature, the reaction is particularly slow and not very effective, in fact there is no significant increase in the particle’ radius up to 24 hours, showing a maximum variation of only about 8.4%, in the case of 10R40 particles. Whereas, the cleavage reaction conducted at 50 °C is faster, showing a substantial increment in the particle radius in the range of 16–24 hours (up to ≈22%) (Fig. 2c and Table S3, ESI†). However, after 20 hours, most of the microparticles start to lose their spherical shape due to a significant network degradation (data not shown). Thus, the optimal cleavage conditions are found to be 50 °C overnight, a condition that allows for maximum radius increase, without losing the microparticle structural integrity. Also in this case, the 10R40 microparticles presented the highest increment in radius, so suggesting the presence of larger pores (Fig. 2c). 10R4 hydrogel microparticles show lower porosity at 16 hours probably due to its pre-cleavage higher crosslinking density (related to high DHEBA concentration).
 |
| | Fig. 2 (a) Volumetric swelling of hydrogel microparticles 15R4 under different cleavage conditions in terms of sodium periodate concentrations (left) and example of optical microscope images of swollen and dry hydrogel microparticles (right). Swollen radius increment (%) over cleavage time at (b) room temperature and (c) 50 °C for all chemical compositions (15R4, 15R40, 10R4 and 10R40). | |
To provide information about the porosity of microparticles, bulk materials corresponding to microparticle compositions were prepared and analysed through NMR cryoporometry. This technique is based on the suppression of the melting/freezing point in confined liquids as described by equations in the Methods section. Fig. S2a (ESI†) shows the melting of water in all non-cleaved (NC) and cleaved (C) samples, with varying PEGDA concentrations and PEGDA/cross-linker ratios (IT curve). In all samples, the NMR signal intensity increases with temperature, as the water confined within the cross-linked networks melts.
No further increase is observed above the bulk melting temperature suggesting that no excess water was present on the exterior of the gels. An increase in signal intensity is visible in the melting curve data of all cleaved samples compared to their non-cleaved bulks, suggesting a higher water content confined within the cleaved networks. Moreover, as the PEGDA concentration is decreased (10%), the density of larger pores also increases. This behaviour is revealed in Fig. S2a (ESI†), where the portion of water present inside the larger pores melts at a higher temperature. These data confirm 10R40 as the most porous sample, with a higher water content than other compositions. This result is more evident differentiating the curves (Fig. S2b, ESI†). Using eqn (2) and (4), the I(t) curves have been analysed in order to obtain a pore size distribution for all the hydrogel compositions. Water as probe fluid for NMR cryoporometry has been largely studied in the literature and its constant kc is known (30 nm K).23 The Fig. S2b (ESI†) shows the derivative of the NMR signal as function of the inverse of temperature (1000 T−1 = X) and the data are fitted with a Gaussian. The intensity of this derivative is strictly connected with the quantity of pores and the parameter X is inversely proportional to the pore radius. Thus, lower values of X reflect a higher pore size,37 while a value of 3.66 K−1 (origin of the x axes) corresponds to free water. Through eqn (5), the best fitting values for each curve in the graph can be used to evaluate the mean pore size. For all the compositions, the peak of Gaussian curves lies in the range between 3.85 and 3.87 K−1, which corresponds to a mean pore size of about 2.2 nm. This result is in accordance with the mean mesh size of the PEGDA700 at a polymer concentration of 10–15%.23 Furthermore, for X values lower than 3.7 K−1 (pore size larger than 10 nm), the derivative dI/dX results to be more intense for the 10R40 and 10R4 samples, reflecting the presence of larger pores, in accordance with the equilibrium volumetric swelling analysis.
The protein diffusion through the polymer network of microparticles may be hindered if the network nanostructure is comparable to the hydrodynamic diameter of biomolecules.38 To investigate the effect of different PEGDA/DHEBA ratios on the network permeability to large biomacromolecules, the equilibrium partitioning of fluorescently labelled IgG antibodies (Rh, hydrodynamic radius of about 5.6–5.9 nm39) within the microparticles has been evaluated. First, the 10R40 microparticles were incubated with fluo-IgG, and the particle fluorescence signals were recorded over time using confocal microscopy to determine time needed to reach partition equilibrium. From Fig. S3 (ESI†), we see that a plateau is reached in 90–120 min. So once the time was set, the same experiment was carried out on the other particles, and the equilibrium partitioning coefficient has been calculated as IN/OUT ratio of fluorescence signal recorded after 120 min of incubation (Fig. 3a). As expected, the network permeability to IgG molecules is higher for cleaved particles with respect to the corresponding non-cleaved samples, as an antibody is significantly larger than the mean mesh size of the polymer network at 10 and 15% PEGDA. In particular, in accordance with the previous experiments, the 10R40 hydrogel microparticles present the higher partitioning coefficient, suggesting that the PEGDA:DHEBA ratio at 40:1 leads to a more porous material. Moreover, the uniform fluorescence distribution within the 10R40 particles indicates the ability of the protein to access the core of the microparticle, also providing a proof of the overall pore interconnectivity (Fig. S4, ESI†). The large network porosity is further verified by evaluating the network accessibility to fluorescent silica nanoparticles with a diameter of 50 nm, comparable to the space occupied by a ternary complex of antibodies (about ∼45 nm in its longest direction), required for the development of in-gel sandwich immunoassays. The diffusion of the labelled nanoparticles within the network of 10R40 cleaved microparticles was performed and fluorescence IN/OUT percentage ratio was calculated (Fig. 3b). The results confirm the microparticle permeability to nanoparticles, as the fluorescence recorded within the hydrogel particles is higher after the cleavage (accessible about 6-fold more compared with non-cleaved microparticles). Definitely, these 10R40 microparticles showed significantly greater permeability to large macromolecules, up to 50 nm, and for this reason used for further studies. Furthermore, to understand the possibility of modulating the porosity and consequently the accessibility of these 10R40 microparticles to three labelled biomolecules with different sizes (permiselectivity) by simply controlling the cleavage time, we examined the extent of their permeation into non-cleaved and cleaved microparticles at 2 h, 4 h, 8 h and 16 h. In addition to the immunoglobulin (Rh = 5.6 nm), a small molecule of oligonucleotide (17nt) (Rh = 1.5 nm40) and a molecule of intermediate size such as ovalbumin (OVA) (Rh = 2.7 nm41) have been chosen. Fig. 3c–e shows normalized fluorescence values (as I/Imax, where Imax is the fluo signal at equilibrium, 90 min) within the microparticles as a function of incubation time. As the cleavage time increased, we observed a faster diffusion for all biomolecules. In detail, non-cleaved and 2 h-cleaved microparticles contained about 30% of fluo-oligonucleotide at 30 minutes of incubation (Fig. 3c). However, at the same incubation time the oligonucleotide penetration within the hydrogels is higher than 75% for 4 h, 8 h and 16 h-cleaved microparticles, indicating a larger pore size. A similar behaviour was observed for ovalbumin, since at 30 minutes the percentage of diffused protein within the microparticles is comparable to the oligonucleotide (Fig. 3d). Whereas at 60 minutes, the percentage of penetration increase from 65% to up to almost 100% as the cleavage time increases. In this case the OVA diffusion depends upon all the different degrees of porosity generated at different times. As for IgG (Fig. 3e), which is the largest biomolecule explored, it can be noticed how the percentage of penetration at 60 minutes goes from about 65% to 100% between 8 and 16 hours of cleavage. These data demonstrate how the protein diffusion through the 16 h-cleaved network is not hindered, even at short incubation time. In conclusion, these analyses demonstrated the ability of these porous particles to function as molecular filters, based on biomolecule size, to allow the selective passage of the molecule of interest.
 |
| | Fig. 3 (a) Fluorescence partition coefficient at equilibrium (120 min) for the diffusion of a labelled IgG inside the hydrogel microparticles, before and after the cleavage; (b) fluorescence partition coefficient at equilibrium (120 min) for the diffusion of labelled silica nanoparticles (diameter 50 nm) inside the 10R40 hydrogel microparticles, before and after the cleavage; Diffusion kinetics of fluorescent probes: (c) 17nt-oligonucleotide, (d) ovalbumin and (e) IgG performed on 10R40 microparticles with different cleavage times (non cleaved = NC, 2 h, 4 h, 8 h and 16 h cleaved MPs). | |
Fluorescence correlation spectroscopy measurements
Equilibrium partitioning measurements have shown that the porous nanostructure created in 10R40 microparticles leads to large macromolecule permeability, but the rate of molecule penetration and consequently the assay kinetics, depend on its diffusion coefficient through the polymer mesh. The mass transfer rate of molecules through hydrogels is an important design parameter for in-gel immunoassays, since rapid transport to the core of the microparticle would allow greater biomacromolecular interactions with immobilized antibodies in a shorter time. In order to optimize the incubation times within these microparticles, fluorescence correlation spectroscopy analysis was conducted to evaluate the diffusion coefficient of a biomolecule at high molecular weight (human IgG) within the hydrogel microparticles. The diffusivity of labelled human IgG (atto647N–hIgG) in 10R40 microparticles was measured after 2 h of incubation. The small molecule atto647N was used as the calibrating agent for the confocal volume, with the know diffusion coefficient.42 The average autocorrelation functions of hIgG and atto647N, as free molecules in the solution and within the hydrogel microparticles, are presented in Fig. 4. The autocorrelation functions reveal the time required for a molecule to diffuse through a defined confocal volume. The diffusion coefficients of the molecules, both in solution and within the hydrogel matrix, were determined through the fitting (eqn (7)) of the autocorrelation functions. The D0 values, summarized in Table of Fig. 4, are in line with that reported in the literature.43 According to data for the hydrogel, the results demonstrate that the Dgel value of IgG molecules is comparable to the corresponding D0 value, with a decrement in diffusion coefficient of about 8%. Considering the large size of IgG molecules, the decrement in diffusion is very low. This proves that the hydrogel microparticles are porous enough to perform an immunoassay rather quickly. In fact, assuming a unidirectional diffusion of the antibody molecules inside the microparticles with a 45 μm average radius, the time needed to reach the core of the particle is less than 1 minute. Furthermore, the diffusivity value Dgel/D0 for the human IgG can be used to get an idea of the mesh size range of microparticle networks. Using eqn (10), the mesh size ξ of the cleaved hydrogel microparticles is about 64 μm. Although this value cannot be considered as robust evidence, it confirms a porosity equal or greater than 50 nm, as revealed by all previous experiments.
 |
| | Fig. 4 Top: FCS average correlation functions G(t) for atto647N and hIgG–atto647N in buffer and inside the hydrogel microparticles. Bottom: mean diffusion coefficients in buffer (D0) and in gel (Dgel) and their ratios Dgel/D0 for atto647N and for hIgG–atto647N. * Calibration value from the literature;42 ** diffusion coefficient in line with the literature reports.44 | |
Aldehydes’ titration
After demonstrating the generation of a porous network accessible to large biomolecules, the number of aldehydes formed after the treatment with periodate has been calculated. This step is fundamental for optimizing the immobilization of primary antibodies (via reductive amination) and thus developing an effective in-gel immunoassay. A titration of the aldehydes formed over time was performed using a colorimetric aldehyde quantitation kit. After the cleavage of 1000 microparticles (10R40), the kit solutions were added to the particles and the manufacturer's instructions were followed. As shown in Fig. 5a, the samples absorbance at 550 nm, which is proportional to the amount of aldehydes formed, increases overtime until a plateau is reached between 16 and 24 hours, confirming the cleavage time set by other previous experiments (overnight). From the fitting of the standard curve using linear regression analysis (Fig. 5b inset), a quantification of aldehydes can be found. In particular, the quantity of aldehydes formed in 16 hours is about 4 nmol in 1000 particles and, scaling for a single particle it is about 4 pmol, in accordance with the amount of the cross-linker DHEBA present during the microparticle synthesis (Fig. 5b). This quantification allows fine control of the antibody conjugation degree in in-gel immunoassays, tuning in this way biosensor performance in terms of sensitivity, as well as minimizing biosensor costs and avoiding waste.
 |
| | Fig. 5 (a) Aldehyde absorbance at 550 nm over time evaluated by using 1000 particles 10R40 per sample. (b) Quantity of aldehydes per particle over cleavage time, evaluated through the standard curve. Inset: Aldehydes colorimetric kit standard curve. | |
Activity and accessibility of antibody-conjugated microparticles
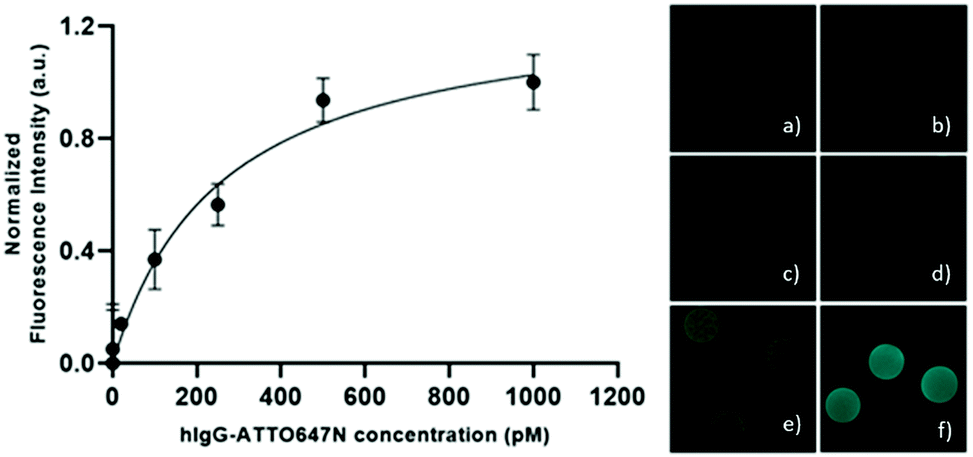
10R40 microparticles have proven to have the most suitable composition for the development of effective in-gel immunoassays thanks to (i) the ability to allow the antibody diffusion in short time, (ii) the presence of pores large enough to accommodate a ternary complex and, (iii) the possibility to fine control the degree of conjugated bioprobes. As proof of concept, we tested the detection capabilities of these hydrogel microparticles against human IgGs. For the purpose, the capture antibodies (anti-hIgG) have been firstly conjugated within the polymeric networks. In detail, the aldehydes formed within the polymer network react with the amino group of antibodies to form Schiff's base, which is stabilized by reduction with sodium cyanoborohydride. In order to make all the immobilized antibodies accessible, and to prevent a steric hindrance to the diffusion of targets and reporter biomolecules, we conjugate 0.1 pmol of anti-hIgG per particle (even if 4 pmol of aldehydes per particle are available). After washing the microparticles, 10 anti-hIgG-conjugated microparticles were incubated with different concentrations of atto647N–hIgG exploring a range from 0.1 to 1000 pM. After washing the microparticles to remove the unbound fluorescent target, the microparticles were analysed with a confocal microscope. The fluorescence signal is generally uniform throughout the microparticles, as shown in the right panel of Fig. 6, suggesting a homogeneous distribution of immobilized antibodies and targets. After the subtraction of the background signal, the normalized fluorescence signal versus the target atto647N–hIgG concentration is plotted (Fig. 6), showing a specific dose–response binding curve as the fluorescent signal increases with increasing hIgG concentration. These results confirm antibodies immobilized within these porous PEG microparticles retain their structure and are accessible for interaction with targets, showing the ability to capture the target down to few picograms with this degree of immobilized antibody. Thus, the developed microparticles represent promising tools to perform fast and sensitive in-gel sandwich immunoassays.
 |
| | Fig. 6 Left: Normalized fluorescence signal intensity of hIgG–ATTO647N captured by functionalized hydrogel microparticles with 0.1 pmolpart−1 of anti-hIgG antibody; right: confocal fluorescence images of functionalized microparticles detecting atto647N–hIgG at different concentrations: (a) 1, (b) 10, (c) 100, (d) 250, (e) 500, and (f) 1000 pM, as shown in the side graph. | |
Conclusions
Here we report the microfluidic synthesis and physicochemical characterization of porous hydrogel microparticles. We successfully synthesized monodisperse PEG-based microparticles through the addition of a cleavable cross-linker DHEBA to the pre-polymer solution in a very versatile manner. At the initial cross-link density, the pore size of the microparticles is too small to allow the diffusion of large bioprobes and targets within the entire microparticle. Upon chemical cleavage of the cross-links, several features for the development of sensitive microparticles are satisfied such as the simultaneous creation of large pores and reactive functional groups and the possibility to functionalize with any biomolecule, without changing the pre-polymer solution and/or modifying the biomolecule. As a result, these porous microparticles show the ability to function as molecular filters, based on the biomolecule size, to allow the selective passage of the molecule of interest. This can be addressed to the tuneable network porosity and interconnection and to PEG's anti-fouling properties. Moreover, through the quantification of aldehydes groups, it is possible to finely control the conjugation degree of particles with bioprobes, like antibodies, in order to opportunely tune the biosensor performance according to the target concentration range. Finally, we have applied this tool to develop an assay for human IgG recognition, resulting in a detected range comparable to available commercial kits for the total IgG detection.45 The increase in transport to the core of the microparticle would allow greater biomacromolecular interactions with immobilized antibodies and promises in enhancing the sensitivity of immunoassays based on porous microparticles. This strategy could be applied to many types of biomacromolecules of interest to medicine, environment or food fields, thanks to a simple and highly versatile synthesis strategy.
Experimental section/methods
Hydrogel microparticle synthesis
Polyethylene glycol diacrylate (PEGDA700 by Sigma-Aldrich) solutions were prepared, as shown in Table S1 (ESI†), with 4 different amounts of DHEBA. The prepolymer solution was vortexed for 1 min at room temperature, adding 0.5% v/v of Darocur1173 (Sigma-Aldrich) as the photo initiator. All preliminary steps were performed minimizing sample exposure to light, to avoid any undesired polymerization. The droplet generation was performed starting from the microfluidic emulsification of pre-polymer solution in a continuous phase (oil). A T-junction glass chip (Dolomite Microfluidics) was used to obtain 75 μm diameter droplets, using light mineral oil (LMO) with 5% (v/v) SPAN 80 as continuous phase. The continuous and disperse phases were pushed using high precision syringe pumps (Nemesys-low pressure) to ensure a reproducible, stable flow. Drops were polymerized in flow by means of a UV lamp. On the basis of the flow rates, it is possible to estimate the permanence time of a particle in the portion of the outlet tube illuminated by the UV lamp. The internal radius of the outlet tube r measure 0.38 mm, the average length of the illuminated portion L is 3.8 cm and the total flow rate Q (PEGDA + oil) is 3 μL min−1, so according to this equation:| | 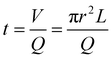 | (1) |
the permanence time is about 6 min, with a lamp power density of 1.8 W cm−2 and a resulting exergy flux of about 250 J cm−2, which is enough to fully crosslink the polymer. After photo-polymerization, hydrogel microparticles were collected and washed. Videos of the production of about 1000 droplets were acquired and analysed using droplet monitor software to evaluate the droplet diameter distribution.
Morphological characterization
Morphological characterization was carried out collecting images of the hydrogel microparticles in the dried state using a scanning electron microscope (SEM) and swollen state using an optical microscope (IX 71 Olympus, DRY 10× objective). SEM analysis were performed on a FE-SEM Ultra Plus (Zeiss) microscope at 5 kV, 4.5k× magnification, 20 μm aperture size and a 6.3 mm working distance. For sample preparation, 50 μL of the microparticle suspension was spotted on a thin glass slide, air-dried for 16 hours at room temperature and then sputtered with a 10 nm thin gold layer. After these steps, images of the dried microparticles were acquired.
Equilibrium volumetric swelling measurements
Equilibrium swelling experiments were performed to investigate the influence of sodium periodate concentration on the cleavage reaction. Equilibrium swelling ratio Q is defined as eqn (2):| |  | (2) |
where Vs is the volume of the water-swollen microparticles at room temperature and Vd is the volume of the dried microparticles, assuming that the particles remain spherical during the drying process. The effect of different concentrations of sodium periodate (0.1 mM, 1 mM, 10 mM) on microparticles’ swelling was investigated. The microparticles were cleaved overnight at room temperature and then washed to stop the reaction. The experiments were performed recording hydrogel microparticle images in the swollen state and in the dry state with an inverted microscope (IX 71 Olympus) equipped with a 10× objective. Cleaved hydrogel microparticles were soaked at room temperature in water with 0.05% tween (v/v) for 24 h and then the images were recorded. The average radius of the swollen particles was estimated using ImageJ software. The same samples were then dried in an oven at 40 °C under vacuum for 24 h, and their average radius, in the dry state, was measured.
Oxidative cleavage kinetics
In order to optimize cleavage conditions with sodium periodate, several studies were carried out concerning the time and temperature setting, following the variation of particle size over time. Regarding the particle radius, images of swollen particles were acquired and analysed using an inverted microscope (IX 71 Olympus) equipped with a 10× objective and ImageJ software for the analysis. The reaction was carried out at two different temperatures (25 °C and 50 °C) and followed over time up to 24 hours for the higher temperature reaction and up to 48 hours for reactions at room temperature. For every time step, a small amount of particles were collected, washed with a solution of sodium bicarbonate 0.1 M and then visualized at the microscope.
Aldehydes’ titration
In order to follow the generation of aldehydes inside the particles during the reaction, a commercial colorimetric aldehyde assay kit (Sigma Aldrich) was used. Hydrogel microparticles were cleaved with a 10 mM sodium periodate solution at 50 °C and kept under agitation at 500 rpm, avoiding any exposition to light that could slow the reaction. For every time step, about 1000 particles were collected, washed with a solution of sodium bicarbonate 0.1 M to stop the reaction and then analysed with a colorimetric kit, also including a sample containing non-cleaved particles (time zero). A standard calibration curve was realized with known concentrations of aldehydes (0, 1, 3.3, 10, 33, 100, 333, and 1000 mM). The assay was performed in a 96-well plate, following the manufacturer's instructions. The 550 nm absorbance of all samples was recorded, using a plate reader (2300 EnSpire multilabel reader, PerkinElmer) and the number of aldehydes was calculated through the calibration curve. All experiments were carried out in triplicate and the error was represented by the standard deviation.
Equilibrium partitioning measurements and diffusion measurements
Equilibrium partitioning of both FITC-labelled IgG antibodies (Sigma Aldrich) and FITC-labelled Silica nanoparticles (size 50 nm) (Sigma Aldrich) was measured for both cleaved and not-cleaved microparticles. Fluorescence signals inside and outside the particles were recorded and the average IN/OUT fluorescence ratio (ratio of average fluorescent intensity within each particle to the average intensity in the surrounding area) was calculated. The experiments were performed by dropping the particle suspension (corresponding to about 100 particles) in a “IBIDI μ-Slide 18 Well – Flat” and then adding the solution of 20 μM IgG-FITC or silica nanoparticle suspension (obtaining a final concentration of 2 μM for each well). After 120 minutes all images were acquired using a confocal microscope (CLSM Leica TCS SP5, using an argon laser line 488 nm, objective 25× water, scan speed 400 Hz, λem range 500–550 nm). To evaluate the diffusion kinetics for fluorescent oligonucleotide, ovalbumin and IgG in microparticles with different cleavage times, aliquots of microparticles were cleaved and the reaction was stopped at 2, 4, 8 and 16 hours by washing with 0.1 M TRIS buffer. The experiments were performed by dropping all the particle suspensions together with an aliquot of non-cleaved particles (corresponding to about 50 particles) in a “IBIDI μ-Slide 18 Well – Flat” and then oligonucleotide, ovalbumin and IgG labelled with atto647N (obtaining a final concentration of 1 μM for each well). Images of the microparticles were acquired over time (0, 30, 60 and 90 minutes) using a confocal microscope (CLSM Leica TCS SP5, using a 633 nm laser, objective 25× water, scan speed 400 Hz, λem range 650–700 nm). For all the experiments with cleaved particles, aldehydes have been previously blocked to avoid any interaction between biomolecules and hydrogel microparticles during the diffusion. In particular, the cleaved particles were capped with TRIS buffer 0.1 M (under stirring for 30 min at room temperature) and then washed. The IBIDI slides were pre-treated with 1% bovine serum albumin (BSA, Sigma Aldrich) to avoid non-specific adsorption of biomolecules on the glass slide.
NMR cryoporometry measurements
NMR cryoporometry is a technique for the determination of the pore size distribution via the observation of the solid–liquid phase transition temperature of a medium (water) confined in the pores. Water trapped in small pores melts at lower temperature, while the bigger the pore the most water trapped behaves as free water, liquefying near 0 °C. Jackson and McKenna46 expressed the shift of the pore melting point Tm as:| |  | (3) |
where r is the pore radius, kc is a material constant that depends on the molar volume v, the solid–liquid surface energy γsl, the enthalpy ΔH and the bulk melting point T0, as eqn (4) indicates.| |  | (4) |
The resulting NMR signal I(T) varies smoothly with temperature T and its shape reflects the pore size distribution. By assuming I(T) ∼ V(T), where V(T) is the pore volume that contains molten liquid at a given temperature T, it can be simply obtained as pore-size distribution p(r) as follows:| |  | (5) |
Basing on this equation, the higher the derivative of the NMR signal the more porous the material. NMR spectra were recorded using an Agilent 600 MHz (14 Tesla) spectrometer equipped with a DD2 console and an OneNMR HX probe. The NMR cryoporometry measurements were performed to study the influence of the PEGDA/DHEBA ratio on the pore size distribution before and after the cleavage. The following hydrogel bulk samples with 10% (v/v) D2O were prepared: (i) 15R4, (ii) 15R40, (iii) 10R4, and (iv) 10R40 (Table S1, ESI†). The obtained solutions were degassed with nitrogen, and then transferred into NMR glass tubes (5 mm of diameter) reaching a height of 5 cm. The NMR tubes were fixed to a support and the pre-polymer solutions were then polymerized using a UV lamp for 30 minutes with a 365 nm light. Regarding the post-cleavage samples, the hydrogels were dried in an oven (overnight, 60 °C) and then a 10 mM solution of sodium periodate was added in order to completely cover the samples. The cleavage reaction took place overnight at 50 °C. After that, NMR cryoporometry measurements were performed on fast frozen samples, using a temperature ramp going from −45 °C to 5 °C with a velocity of 0.1 °C per minute. Signals were acquired every 50 minutes (every 5 °C).
Fluorescence correlation spectroscopy measurements
In order to estimate the diffusion coefficient of antibodies inside the hydrogel microparticles, fluorescence correlation spectroscopy (FCS) experiments were carried out. Microparticles were cleaved (overnight, 50 °C, 500 rpm) and washed with a 0.1 M sodium bicarbonate solution to stop the reaction. The cleaved microparticles were incubated (24 h, RT) with 10 nM atto647N and 10 nM atto647N-labelled human IgG and, then, analysed with a confocal microscope (Zeiss Confocor LSM 700, using a 633 laser 2% in the total reflection mode, objective 40× water). The IgG was labelled with the fluorophore, ATTO647N-N-hydroxysuccinimide (3-fold excess), for 30 min at room temperature, and then dialysed and characterized through spectrophotometric measurement to calculate the concentration and degree of functionalization. The FCS experiments were performed in an “IBIDI μ-Slide 8 Wells”, pre-treated with 1% BSA solution to avoid any interaction between the glass slide and the fluorescent probes during the acquisitions. For fluorescence excitation a low-noise laser was used (633 nm wavelength). Determination of the confocal volume was established through calibration with a 10 nM phosphate buffered saline (PBS) solution of atto647N, assuming D0 = 160 μm2 s−1, as reported in the literature.42 All FCS measurements were performed at room temperature and, for each sample, 10 FCS correlation functions were acquired for 10 s in three different particles. From the mathematical point of view, the time sequence of the detected fluorescence intensity, I(t), emitted by the molecules in the confocal volume (V) at time t, is time-correlated to generate the autocorrelation function defined as:| |  | (6) |
where δI(t) = I(t) − 〈I(t)〉 denotes the deviation of the measured intensity from the average intensity 〈I(t)〉. Analysis of G(τ) provides information about the underlying mechanisms for the intensity fluctuations. For an ideal case of freely diffusing monodisperse and uniformly bright fluorescent particles, an expression for the correlation function can be derived as:| |  | (7) |
where N denotes the average number of particles in the excitation volume, τ the delay time, τD the characteristic diffusion time, and p an instrumental constant. Assuming that the fluorescent diffusing molecules are excited by a 3D Gaussian beam, the characteristic time for fluorescent particles diffusing along the lateral width (r0) of the focused incident laser beam is defined as:| |  | (8) |
where D denotes the translational diffusion coefficient of the particles in the host medium. To interpret the data obtained from FCS experiments, several assumptions were made in order to obtain these correlation functions. First, since the porosity of the hydrogel was larger than the hydrodynamic radius of the diffusing solutes, the case of freely diffusing monodisperse fluorescent solutes was applied. Second, because of low laser intensity, the excitation of molecular triplet states was not taken into consideration. For the labelled IgG, we have also assumed that only one fluorophore was conjugated to each protein molecule. In fact, after protein labelling, we have calculated a labelling ratio (fluorophore molecule per protein molecule) of 1.1 (data not shown). Furthermore, as the fraction of the free Atto647 molecules remaining in solution after the IgG labelling process is very low (<5%), we assume that it is negligible for the fitting. Furthermore, antibody diffusion coefficients in water (D0) and in gel (Dgel) were used to predict the mesh size ξ of the 10R40 polymeric network, through the use of the hydrodynamic theory introduced by Cukier.47 According substitutions of the scaling relationship,44 the original equation can be simplified as follows:| |  | (9) |
where Rh is the hydrodynamic radius of the diffusing molecule. From eqn (9), the following formula to estimate the mesh size can be derived:| |  | (10) |
Immobilization of anti-hIgG and accessibility/activity analysis
To evaluate activity and accessibility of antibody-conjugated microparticles to large proteins, cleaved hydrogel microparticles were firstly conjugated adding anti-human immunoglobulin type G (anti-hIgG) at a ratio of 0.1 pmol per particle in the presence of a stabilizing agent (sodium cyanoborohydride) overnight at room temperature. Then the conjugated microparticles were washed and incubated for 90 minutes, at 37 °C and 500 rpm with different concentrations of fluorescent human immunoglobulin G (atto647N–hIgG) by exploring a range between 0 and 1 nM. After the incubation time with fluorescent h-IgG and subsequent washing, images of the particles were acquired using a confocal microscope (CLSM Leica SP5, Objective 20× DRY, scan speed of 400 Hz, excitation wavelength 633, emission wavelength 648–710 nm) to evaluate the relative fluorescence intensities. The fluorescence signal indicates the binding between the anti-hIgG-conjugated microparticles and fluorescent h-IgG. Fluorescence intensities were normalized by subtracting the signals of control samples for all the atto647N–hIgG concentrations. Control samples were prepared by incubating cleaved hydrogel microparticles, whose aldehydes were blocked with TRIS buffer, with fluorescent h-IgG at the same concentrations and conditions.
Author contributions
A. D. M. and P. L. S. designed and performed all the experiments. E. B. and F. C. conceived the idea, P. N. contributed to the discussion and F. C. coordinated the activities. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors acknowledge Dr. Valentina Mollo for the preparation and the analysis of SEM samples.
References
- Y. Ma, Y. Mao, D. Huang, Z. He, J. Yan, T. Tian, Y. Shi, Y. Song, X. Li, Z. Zhu, L. Zhou and C. J. Yang, Lab Chip, 2016, 16, 3097–3104 RSC.
- S. C. Chapin and P. S. Doyle, Anal. Chem., 2011, 83, 7179–7185 CrossRef CAS PubMed.
- R. L. Srinivas, S. C. Chapin and P. S. Doyle, Anal. Chem., 2011, 83, 9138–9145 CrossRef CAS PubMed.
- S. C. Chapin, D. C. Appleyard, D. C. Pregibon and P. S. Doyle, Angew. Chem., Int. Ed., 2011, 50, 2289–2293 CrossRef CAS PubMed.
- A. M. Cusano, F. Causa, R. Della Moglie, N. Falco, P. L. Scognamiglio, A. Aliberti, R. Vecchione, E. Battista, D. Marasco, M. Savarese, U. Raucci, N. Rega and P. A. Netti, J. R. Soc., Interface, 2014, 11, 20140718 CrossRef PubMed.
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- D. C. Appleyard, S. C. Chapin, R. L. Srinivas and P. S. Doyle, Nat. Protoc., 2011, 6, 1761–1774 CrossRef CAS PubMed.
- M. N. Hsu, S.-C. Wei, D.-T. Phan, Y. Zhang and C.-H. Chen, Adv. Healthcare Mater., 2019, 1801277 CrossRef PubMed.
- E. Battista, P. L. Scognamiglio, N. Di Luise, U. Raucci, G. Donati, N. Rega, P. A. Netti and F. Causa, J. Mater. Chem. B, 2018, 6, 1207–1215 RSC.
- C. Di Natale, G. Celetti, P. L. Scognamiglio, C. Cosenza, E. Battista, F. Causa and P. A. Netti, Chem. Commun., 2018, 54, 10088–10091 RSC.
- J. Kim, J. Heo and R. M. Crooks, Langmuir, 2006, 22, 10130–10134 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- N. A. Peppas, K. B. Keys, M. Torres-Lugo and A. M. Lowman, J. Controlled Release, 1999, 62, 81–87 CrossRef CAS PubMed.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- Y. Kim, S. Son, C. Chun, J. T. Kim, D. Y. Lee, H. J. Choi, T. H. Kim and E. J. Cha, Biomed. Eng. Lett., 2016, 6, 287–295 CrossRef.
- N. Broguiere, A. Husch, G. Palazzolo, F. Bradke, S. Madduri and M. Zenobi-Wong, Biomaterials, 2019, 200, 56–65 CrossRef CAS PubMed.
- J. S. Park, D. G. Woo, B. K. Sun, H. M. Chung, S. J. Im, Y. M. Choi, K. Park, K. M. Huh and K. H. Park, J. Controlled Release, 2007, 124, 51–59 CrossRef CAS PubMed.
- Y.-C. Chiu, J. C. Larson, A. Isom and E. M. Brey, Tissue Eng., Part C, 2010, 16, 905–912 CrossRef CAS PubMed.
- S. Naficy, T. Y. L. Le, F. Oveissi, A. Lee, J. C. Hung, S. G. Wise, D. S. Winlaw and F. Dehghani, Adv. Mater. Interfaces, 2020, 7, 1901770 CrossRef CAS.
- S. Xin, O. M. Wyman and D. L. Alge, Adv. Healthcare Mater., 2018, 7, 1800160 CrossRef PubMed.
- R. Randriantsilefisoa, J. Luis Cuellar-Camacho, M. S. Chowdhury, P. Dey, U. Schedler and R. Haag, J. Mater. Chem. B, 2019, 7, 3220 RSC.
- S. Lee, X. Tong and F. Yang, Acta Biomater., 2014, 10, 4167–4174 CrossRef CAS PubMed.
- Y. H. Wu, H. B. Park, T. Kai, B. D. Freeman and D. S. Kalika, J. Membr. Sci., 2010, 347, 197–208 CrossRef CAS.
- D. Choi, E. Jang, J. Park and W. G. Koh, Microfluid. Nanofluid., 2008, 5, 703–710 CrossRef CAS.
- N. W. Choi, J. Kim, S. C. Chapin, T. Duong, E. Donohue, P. Pandey, W. Broom, W. A. Hill and P. S. Doyle, Anal. Chem., 2012, 84, 9370–9378 CrossRef CAS PubMed.
- S. G. Lee, H. Lee, A. Gupta, S. Chang and P. S. Doyle, Adv. Funct. Mater., 2016, 26, 4896–4905 CrossRef CAS.
- M. A. Al-Ameen, J. Li, D. G. Beer and G. Ghosh, Analyst, 2015, 140, 4530–4539 RSC.
- M. A. Al-Ameen and G. Ghosh, Biosens. Bioelectron., 2013, 49, 105–110 CrossRef CAS PubMed.
- G. C. Le Goff, R. L. Srinivas, W. A. Hill and P. S. Doyle, Eur. Polym. J., 2015, 72, 386–412 CrossRef CAS PubMed.
- A. Salvi, P. A. Carrupt, J. P. Tillement and B. Testa, Biochem. Pharmacol., 2001, 61, 1237–1242 CrossRef CAS PubMed.
- S. Nayak, D. Gan, M. Serpe and L. Andrew Lyon, Small, 2005, 1, 416–421 CrossRef CAS PubMed.
- B. P. Tripathi, N. C. Dubey and M. Stamm, ACS Appl. Mater. Interfaces, 2014, 6, 17702–17712 CrossRef CAS PubMed.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2004, 43, 6706–6709 CrossRef CAS PubMed.
-
R. J. Ouellette and J. D. Rawn, Organic Chemistry, Elsevier, 2018, pp. 463–505 Search PubMed.
- Y.-L. Zhong and T. K. M. Shing, J. Org. Chem., 1997, 62, 2622–2624 CrossRef CAS PubMed.
- G. H. Findenegg, S. Jähnert, D. Akcakayiran and A. Schreiber, ChemPhysChem, 2008, 9, 2651–2659 CrossRef CAS PubMed.
- L. M. Weber, C. G. Lopez and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2009, 90A, 720–729 CrossRef CAS PubMed.
- Y. Zhou, J. Li, Y. Zhang, D. Dong, E. Zhang, F. Ji, Z. Qin, J. Yang and F. Yao, J. Phys. Chem. B, 2017, 121, 800–814 CrossRef CAS PubMed.
- A. Werner, Nucleic Acids Res., 2011, 39(3), 17 CrossRef PubMed.
- M. A. Qasim and A. Salahuddin, J. Biochem., 1979, 85, 1029–1035 CrossRef CAS PubMed.
- M. Leutenegger, C. Ringemann, T. Lasser, S. W. Hell and C. Eggeling, Opt. Express, 2012, 20, 5243 CrossRef CAS PubMed.
- K. Engberg and C. W. Frank, Biomed. Mater., 2011, 6, 055006 CrossRef PubMed.
- T. Fujiyabu, X. Li, U. Il Chung and T. Sakai, Macromolecules, 2019, 52, 1923–1929 CrossRef CAS.
- Human IgG, Total, ELISA kit, https://www.cygnustechnologies.com/human-igg-total-elisa-kit.html, (accessed 7 June 2021).
- O. V. Petrov and I. Furó, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 54, 97–122 CrossRef CAS.
- R. I. Cukier, Macromolecules, 1984, 17, 252–255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional methods and data regarding the hydrogel microparticle synthesis and cryoporometry characterization. See DOI: 10.1039/d1tb02751d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc
abc