
Gradient adhesion modification of polyacrylamide/alginate–calcium tough hydrogels†
Wanglong
Zhang
,
Yiwei
Zhang
,
Yu
Dai
 ,
Fan
Xia
and
Xiaojin
Zhang
*
,
Fan
Xia
and
Xiaojin
Zhang
*
State Key Laboratory of Biogeology and Environmental Geology, Engineering Research Center of Nano-Geomaterials of Ministry of Education, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, China. E-mail: zhangxj@cug.edu.cn
First published on 20th December 2021
Abstract
Strong hydrogel adhesion requires the synergy of adhesion and cohesion. Gradient adhesive–tough hydrogels can balance adhesion and cohesion, however, their construction is still a challenging task. Here, we used ethylenediaminetetraacetic acid (EDTA) on-side coordination-induced diffusion chelating Ca2+ to form an adhesive surface in a polyacrylamide/alginate–calcium (PAAm/Alg–Ca2+) tough hydrogel as a facile method for the construction of gradient adhesive–tough hydrogels. The adhesion energy of a gradient adhesive–tough hydrogel to skin is increased by 128% compared with PAAm/Alg–Ca2+ tough hydrogels and the elongation at break is two times higher than that of PAAm/Alg hydrogels. In addition, gradient adhesive–tough hydrogels also exhibit wide linear sensitivity (the gauge factor (GF) = 0.196 (0% < ε < 400%); GF = 0.260 (400% < ε < 650%)) as a wearable strain sensor to monitor human motions. This work provides a versatile strategy for the design of gradient adhesive–tough hydrogels and also provides a practical model for the development of wearable strain sensors.
1. Introduction
In 2012, Suo et al. reported highly stretchable and tough hydrogels by mixing two types of crosslinked polymer: ionically crosslinked alginate, and covalently crosslinked polyacrylamide.1 The synergy of crack bridging by a covalent crosslinking network and hysteresis by an ionic crosslinking network gave the hydrogels toughness. They proposed the mechanisms of deformation and energy dissipation, which greatly expanded the application scope of hydrogels.2–8 Tough hydrogels have high cohesion, and the movement of polymer chains is limited, leading to a decrease in adhesion. The adhesion strength of hydrogels depends on the interrelation between cohesion and adhesion.9 This leads to the low mechanical properties of many hydrogels with excellent adhesion.10 Although scientists have developed a series of integrated adhesive–tough hydrogels,11–16 it is still difficult to obtain the synergy of adhesion and cohesion.17 Janus hydrogels with a bilayer structure18–20 or gradient structure21–23 have significant advantages in simultaneously obtaining strong adhesion and high toughness of hydrogels.For example, Gao et al. reported bilayer adhesive–tough hydrogels by in situ polymerization of monomers on the surface of tough hydrogels, inspired by the layered structure of skin.24 The hydrogel has good tensile properties, high mechanical strength and strong adhesion to various solid materials and skin. However, the adhesive layer and tough layer of the hydrogel are easily separated.25–27 Moreover, the strains of the adhesive layer and tough layer are different under the same stress, which will affect the sensitivity of the hydrogel strain sensor.28 Compared with a bilayer structure, a gradient structure can avoid these problems.29 Mooney et al. reported gradient adhesive–tough hydrogels by introducing chitosan and other polymers into polyacrylamide/alginate–calcium tough hydrogels to form a positively charged adhesive surface (about 40 μm depth).30 The adhesive surface of the hydrogel adheres strongly to various biological tissues, such as skin, cartilage, heart, artery, and liver, by electrostatic interactions, covalent bonds and physical interpenetration. However, the multiple time-consuming chemical reactions make it impossible to apply. The strong chemical bridging adhesion is prone to irreversibility when the interface is broken.
Here, we report gradient adhesive–tough hydrogels by gradient adhesion modification of polyacrylamide/alginate–calcium tough hydrogels, whose adhesive surface is constructed by EDTA chelating Ca2+. The gradient adhesive–tough hydrogel can form strong adhesion and realize energy dissipation simultaneously. The gradient adhesive–tough hydrogel can avoid the problem of uneven stress caused by the difference between the adhesive layer and tough layer of bilayer adhesive–tough hydrogels.31 The gradient adhesion modification is very simple and can be done in a few minutes. The adhesion between the hydrogel and substrate is mainly formed by non-covalent interactions such as hydrogen bonds and electrostatic interactions, and the hydrogel can be repeatedly adhered to substrates. The concept of gradient modification of hydrogels provides a new window for the design of specific functional hydrogels.32
2. Materials and methods
2.1. Materials
Acrylamide (AAm) was purchased from Sangon Biotech Co., Ltd (Shanghai, China). Sodium alginate (SA), ammonium persulfate (APS), calcium sulfate dihydrate (CaSO4·2H2O), sodium carbonate (Na2CO3), sodium hydrogen carbonate (NaHCO3), disodium edetate dihydrate (EDTA–2Na·2H2O), and calconcarboxylic acid were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). N,N′-Methylenebisacrylamide (MBA), N,N,N′,N′-tetramethylethylenediamine (TEMED), sodium hydroxide (NaOH), and lithium chloride (LiCl) were purchased from Aladdin Chemical Reagent Co., Ltd (Shanghai, China). All reagents were of analytical grade and used without further purification. Deionized water (18.2 MΩ·cm) was obtained from a Heal Force water purification system (Shanghai, China).2.2. Preparation of hydrogels
Gradient adhesion modification of PAAm/Alg–Ca2+ hydrogels. PAAm/Alg–Ca2+ hydrogels were placed in a mold (liquid cannot flow down the contact surface). EDTA solution (0.05 M) was added to the mold. After 10 min, the hydrogel was taken out, and the residual solution on its surfaces was removed using N2 gas for 30 s.
2.3. Characterization of hydrogels
The morphology was characterized using a field-emission scanning electron microscope (SU8010, Hitachi, Japan) with an accelerating voltage of 10 kV. Fourier transform infrared spectroscopy (Nicolet6700, Thermo Nicolet, USA), X-ray photoelectron spectroscopy (K-ALPHA, Thermo Nicolet, USA) with monochromatized AlKα radiation, drop shape analyzer (DSA100, KRÜSS, Germany), differential scanning calorimeter (DSC 3, Mettler Toledo, Germany), and thermogravimetry (STA449F3, Netzsch, Germany) were used to characterize the hydrogel.2.4. Visualized observation of EDTA diffusion in hydrogels
To quantify the degree of EDTA diffusion in hydrogels, calconcarboxylic acid was used as an indicator. The colorimetric solution was prepared by adding 1.0 mg of calconcarboxylic acid into 2.5 mL of buffer solution (PBS, pH 10.0). The PAAm/Alg hydrogel and PAAm/Alg–Ca2+ hydrogel were prepared in a centrifuge tube (Φ = 14.0 mm). The colorimetric solution was added on top of the hydrogel to characterize the EDTA diffusion. The photos of EDTA diffusion at different time points were collected, and the diffusion distance was measured by ImageJ. The linear correlation between the diffusion time and mean square displacement (MSD) behavior was obtained and described by mathematical equation.To eliminate the influence of gravity on the EDTA diffusion, the colorimetric solution was added on top of the PAAm/Alg–Ca2+ hydrogel. After 5 min, the colorimetric solution was removed and one of the hydrogels was inverted. The photos of EDTA diffusion at different time points were collected and the diffusion distance was compared.
To explore the reason for the red line broadening in the experiment, two control groups were set up. 0.2 mL of the colorimetric solution was added to a centrifuge tube and 10-fold diluted with water, AAm solution, or SA solution. The color change of the solution was observed to verify the influence of dilution and gel composition. 0.2 mL of the colorimetric solution was added in a centrifuge tube, and flowed with CO2 gas for 0.5 min, O2 gas for 5 min, or stayed as an open system for 12 h. The color change of the solution was observed to verify the influence of external environmental conditions.
2.5. Evaluation of the structural change of alginate
To evaluate the changes in the structure of alginate after EDTA diffusion, three groups of comparative experiments were designed. (1) CaSO4·2H2O was added to SA solution, and the sample was incubated at 50 °C for 2 h. (2) CaSO4·2H2O and EDTA–2Na were added to SA solution, and the sample was incubated at 50 °C for 6 h. (3) CaSO4·2H2O was added to SA solution, and the sample was incubated at 50 °C for 2 h. EDTA–2Na was then added, and the sample was incubated at 50 °C for 6 h. The final concentrations of SA, EDTA–2Na and Ca2+ were 20 mg mL−1, 86.4 mg mL−1, and 2.5 mg mL−1.2.6. Peeling test of hydrogels
The adhesion of the hydrogel to glass and skin was studied by peeling test on an electronic universal testing machine (E43.104, MTS) at room temperature. The peeling process adopted the 90° peeling method. The clean pigskin needed to be fixed on a solid substrate to ensure the completion of the peeling process. The back of the hydrogel was lined with PET film to prevent its stretching. The tensile speed was set at 20 mm min−1, and the fracture energy was calculated.2.7. Mechanical measurement of hydrogels
A uniaxial tensile test was carried out at room temperature using an electronic universal testing machine (E43.104, MTS). The tensile speed was set at 100 mm min−1. The hydrogel sample was made into a rectangular shape (gauge length of 20 mm, width of 20 mm, thickness of 5 mm). The fracture toughness was estimated by the stress–strain curve.For the tensile loading–unloading cycle test, the tensile speed was set at 100 mm min−1. Because of the good water retention of the hydrogel, each loading and unloading test was carried out at room temperature. The dissipated energy was calculated from the area between the loading-unloading curves.
2.8. Sensing measurements of hydrogels
The conductive hydrogel was obtained by adding LiCl into the hydrogel. Under the condition of 0.25 V, the current of the hydrogel was determined using an electrochemical workstation. The relative resistance change was determined by the following equation:where R0 is the strain free resistance and R is the real-time resistance.
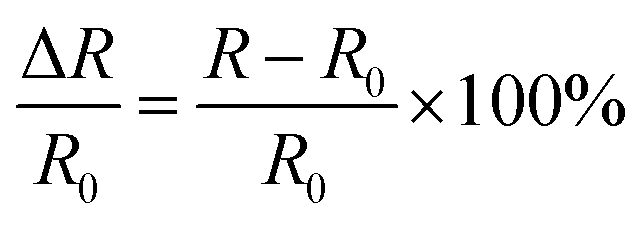
The conductivity (σ, mS cm−1) was calculated according the equation:
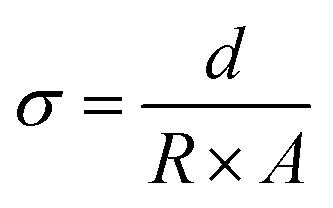
3. Results and discussion
3.1. Hydrogel fabrication
Here, we fabricated a gradient adhesive–tough hydrogel by strong chelating force between EDTA and Ca2+ (Fig. 1). Typically, in the presence of sodium alginate (SA) and calcium sulphate slurry (CaSO4·2H2O), PAAm/Alg–Ca2+ hydrogel was prepared by free radical polymerization of acrylamide (AAm) with N,N′-methylenebisacrylamide (MBA) as a crosslinker, ammonium persulfate (APS) as an initiator, and N,N,N′,N′-testramethylethylenediamine (TEMED) as a crosslinking accelerator at 50 °C heated in a water bath. After 24 h curing at room temperature, the PAAm/Alg–Ca2+ hydrogel formed an ionically crosslinked first network and covalently crosslinked second network.1 After EDTA diffusion, the quasi-gradient distribution of Ca2+ in the hydrogel was constructed through the strong chelating action of EDTA on Ca2+.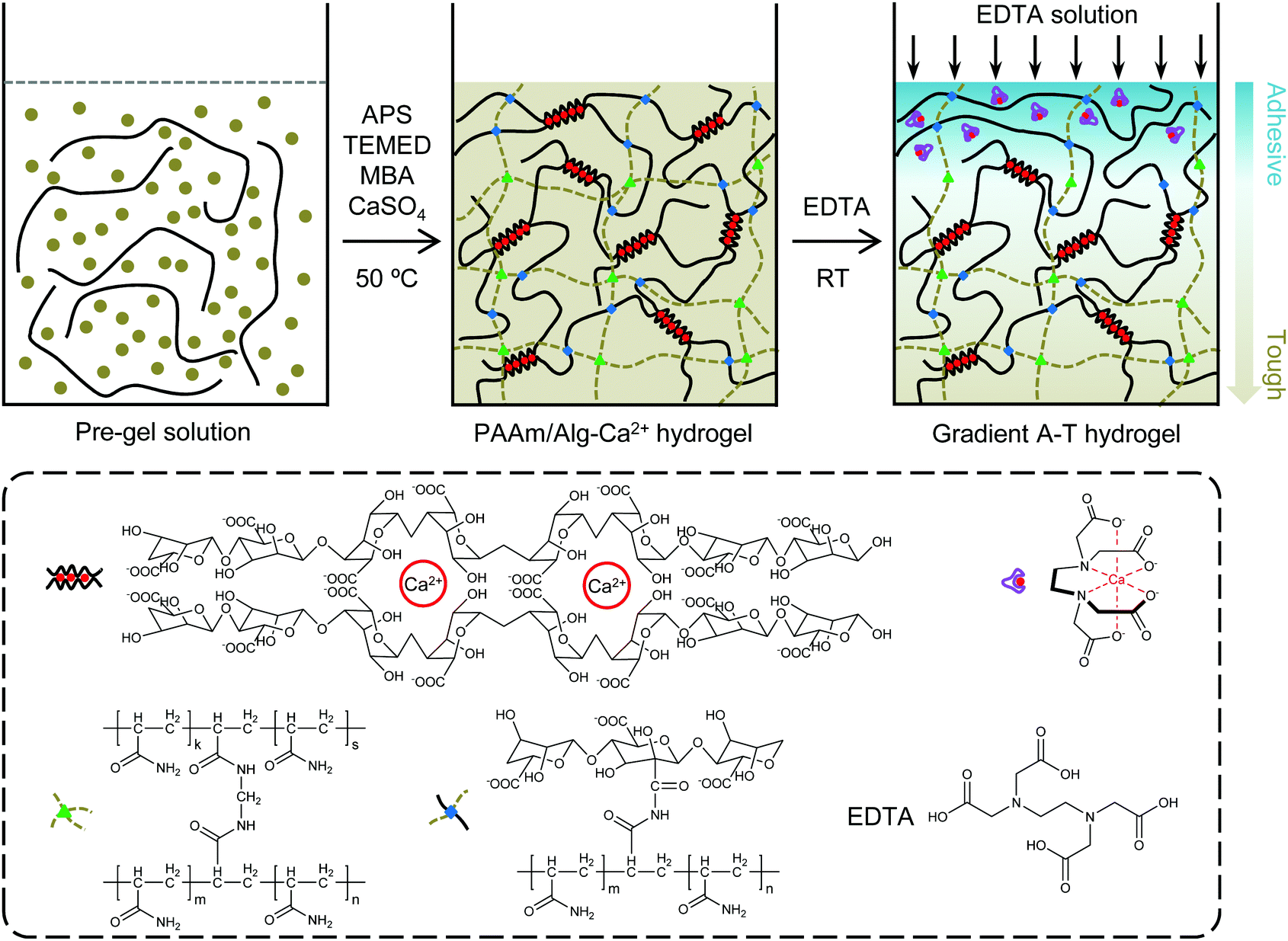 | ||
| Fig. 1 Schematic illustration of the preparation of a gradient adhesive–tough hydrogel. | ||
The influence of diffusion time on the diffusion distance of EDTA was first studied by columnar hydrogel. To further quantify the permeation and diffusion of EDTA in hydrogels, calconcarboxylic acid was used as an indicator. With the passage of time, the diffusion distance of the blue solution containing EDTA increases gradually in the PAAm/Alg hydrogel (Fig. S1a, ESI†) and PAAm/Alg–Ca2+ hydrogel (Fig. S1b, ESI†). The blue height was measured using ImageJ software. The diffusion distance of EDTA in the PAAm/Alg–Ca2+ hydrogel with diffusion time was obtained (Fig. S2a, ESI†). According to the relationship between mean square displacement (MSD) behavior and the diffusion time, EDTA diffusion in the PAAm/Alg–Ca2+ hydrogel conforms to Fick's second law (Fig. S2b, ESI†).
The EDTA diffusion solution is blue, which is the color of the indicator (Fig. S3a, ESI†). The red line appears at the diffusion front (Fig. S3b, ESI†), where the EDTA concentration is very low, calconcarboxylic acid and Ca2+ combine to form the wine-red complex. The effect of the gravity field on EDTA diffusion is excluded (Fig. S4, ESI†). To explain the phenomenon of red line broadening with time, the effects of experimental components and external environmental conditions were explored (Fig. S5, ESI†). The results show that the dilution and gel composition are independent of red line broadening. The red line broadening is due to the influence of CO2 in the external environment. The amount of CO2 entering the hydrogel increases gradually with time, and the pH value of the hydrogel exposed to the environment gradually decreases, so the indicator changes from blue to wine red.33
To explore the change in alginate structure during the diffusion process of EDTA in the PAAm/Alg–Ca2+ hydrogel, control experiments were designed. When CaSO4 was added, the alginate solution easily formed the gel after 2 h at 50 °C (Fig. S6a, ESI†). When CaSO4 and EDTA were added to the alginate solution at the same time, the solution was still in sol state after 6 h at 50 °C (Fig. S6b, ESI†). CaSO4 was added in the alginate solution, and the solution changed to a gel state after 2 h at 50 °C. At the same temperature, EDTA slurry was added to the gel, and the gel changed to sol state after 6 h (Fig. S6c, ESI†). Through the state transition of alginate sol–gel–sol, we speculate that EDTA in the PAAm/Alg–Ca2+ hydrogel can effectively change the structure of alginate.
To explore EDTA–Ca2+ chelation in the hydrogel, the adhesive side and tough side of the gradient adhesive–tough hydrogel were analyzed by X-ray photoelectron spectroscopy (XPS). From the wide-scan spectra (Fig. S7, ESI†), the molar ratio of C/O decreases from 1.70 (tough side) to 1.42 (adhesive side) because of the chelation of EDTA with Ca2+. Moreover, the molar ratio of O/Ca decreases from 27.27 (tough side) to 21.87 (adhesive side), indicating that Ca2+ is partially chelated by EDTA. The O1s core-level XPS spectra could be curve-fitted with three peaks at the binding energies of 531.18 eV, 532.58 eV, and 533.32 eV, corresponding to the carbon of N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, and O–CO, respectively (Fig. S8a, ESI†).34 The relative peak area of O–CO on the adhesive side increases obviously with the introduction of Ca2+ (Fig. S8b, ESI†). The relative peak area of Ca2p on the adhesive side is similar to that on the tough side (Fig. S9, ESI†). The relative peak area of C–N/C–OH decreases by 5.63% (Fig. S10, ESI†). These results indicate that EDTA has successfully chelated Ca2+. At the same time, the relative content of N atom remains unchanged (Fig. S11, ESI†), indicating that there is no other reaction except chelation in the process of EDTA diffusion.
O, C–O, and O–CO, respectively (Fig. S8a, ESI†).34 The relative peak area of O–CO on the adhesive side increases obviously with the introduction of Ca2+ (Fig. S8b, ESI†). The relative peak area of Ca2p on the adhesive side is similar to that on the tough side (Fig. S9, ESI†). The relative peak area of C–N/C–OH decreases by 5.63% (Fig. S10, ESI†). These results indicate that EDTA has successfully chelated Ca2+. At the same time, the relative content of N atom remains unchanged (Fig. S11, ESI†), indicating that there is no other reaction except chelation in the process of EDTA diffusion.
3.2. Structure and properties of gradient adhesive–tough hydrogel
The hydrogel has good adhesion on different substrates (plastic, wood, rubber, steel, copper, stone, ceramics, and glass) (Fig. 2a). Compared with the PAAm/Alg hydrogel (Movie 1, ESI†) and PAAm/Alg–Ca2+ hydrogel (Movie 2, ESI†), the adhesive property of the adhesive side and tough side of the gradient adhesive–tough hydrogel is significantly different (Movies 3 and 4, ESI†). There is no residue after peeling from skin (Fig. 2b). The adhesion and peeling process does not cause significant pain or discomfort in the volunteers. The overall feeling can be digitalized at 2 if 0 is no feeling and 10 is unbearable pain (Fig. S12, ESI†). The hydrogel also shows good toughness (Fig. 2c). The hydrogel exhibits a pore structure at both the adhesive side and tough side (Fig. 2d). However, the pore structure of the adhesive side is obviously different from that of the tough side. The transition from the dual-network structure of the tough side to the interpenetrating network structure of the adhesive side can be observed by the local enlarged SEM image. We also investigated the hydrophilic–hydrophobic property of the adhesive surface and tough surface by the hanging drop method (Fig. S13, ESI†). The EDTA–Ca2+ chelation can effectively increase the hydrophilicity of the hydrogel. | ||
| Fig. 2 Structure and properties of gradient adhesive–tough hydrogels. (a) Images of gradient adhesive–tough hydrogels adhered to various materials (plastic, wood, rubber, steel, copper, stone, ceramics, and glass). Scale bar: 1 cm. (b) Peeling process of the gradient adhesive–tough hydrogel onto skin. (c) Image of the gradient adhesive–tough hydrogel displaying its stretchable ability. (d) SEM image of the gradient adhesive–tough hydrogel and partial magnification of the adhesive side and tough side. | ||
To determine the structural difference between the adhesive side and tough side, the thermal degradation process in a nitrogen atmosphere was studied. Each peak in the DSC curves represents the temperature at which the degradation rate reaches the maximum at each degradation stage in the whole process. There are three pyrolysis stages (Fig. S14, ESI†). The weight loss in the first stage is attributed to the presence of moisture in the sample. The peak temperature of the first stage of the tough side is slightly higher than that of the adhesive side. The possible reason is that the dual-network structure of the tough side is more conducive to water retention than the interpenetrating network structure of the adhesive side. The weight loss in the second stage is mainly attributed to the degradation of the carboxyl group and the release of one ammonia molecule per two amide groups to form imide. The peak temperature of this stage increases from 253.2 °C for the tough side to 256.8 °C for the adhesive side. The possible reason is that the binding force of the carboxyl group in EDTA to Ca2+ is greater than that of alginate to Ca2+. The third stage is polymer depolymerization and carbonaceous residue formation.
3.3. Adhesion and mechanical properties of hydrogels
The adhesive strength of hydrogels on glass and skin was quantitatively determined using a peeling test. Compared with the PAAm/Alg–Ca2+ hydrogel, the adhesion force of the gradient adhesive–tough hydrogel on glass (Fig. 3a) and skin (Fig. 3b) increases significantly. The hydrogel adhesion is primarily attributed to non-covalent interactions, such as the hydrogen bond between the functional groups (e.g. carboxyl and hydroxyl) of the flexible alginate chain (after EDTA–Ca2+ chelation) and the substrate surface,35 electrostatic interaction between Alg–Ca2+ from the adhesive side of the gradient adhesive–tough hydrogel and negative charge from substances.36 The mechanical properties of the hydrogels were studied using a tensile test (Fig. 3c). The PAAm/Alg hydrogel has a poor mechanical property with the fracture stress of 17.5 kPa at a strain of 1044%, while the fracture stress of the PAAm/Alg–Ca2+ hydrogel reaches 126.5 kPa. Compared with the PAAm/Alg hydrogel, the gradient adhesive–tough hydrogel exhibits high mechanical strength with the fracture stress of 93.0 kPa. Besides, the elongation, stress, and toughness of the gradient adhesive–tough hydrogel also have obvious advantages. The elongation at break is two times higher than that of the PAAm/Alg hydrogel, and the fracture energy is much higher than that of the PAAm/Alg hydrogel under the condition that the stress is 26.5% lower than that of the PAAm/Alg–Ca2+ hydrogel (Fig. 3d and Fig. S15, ESI†).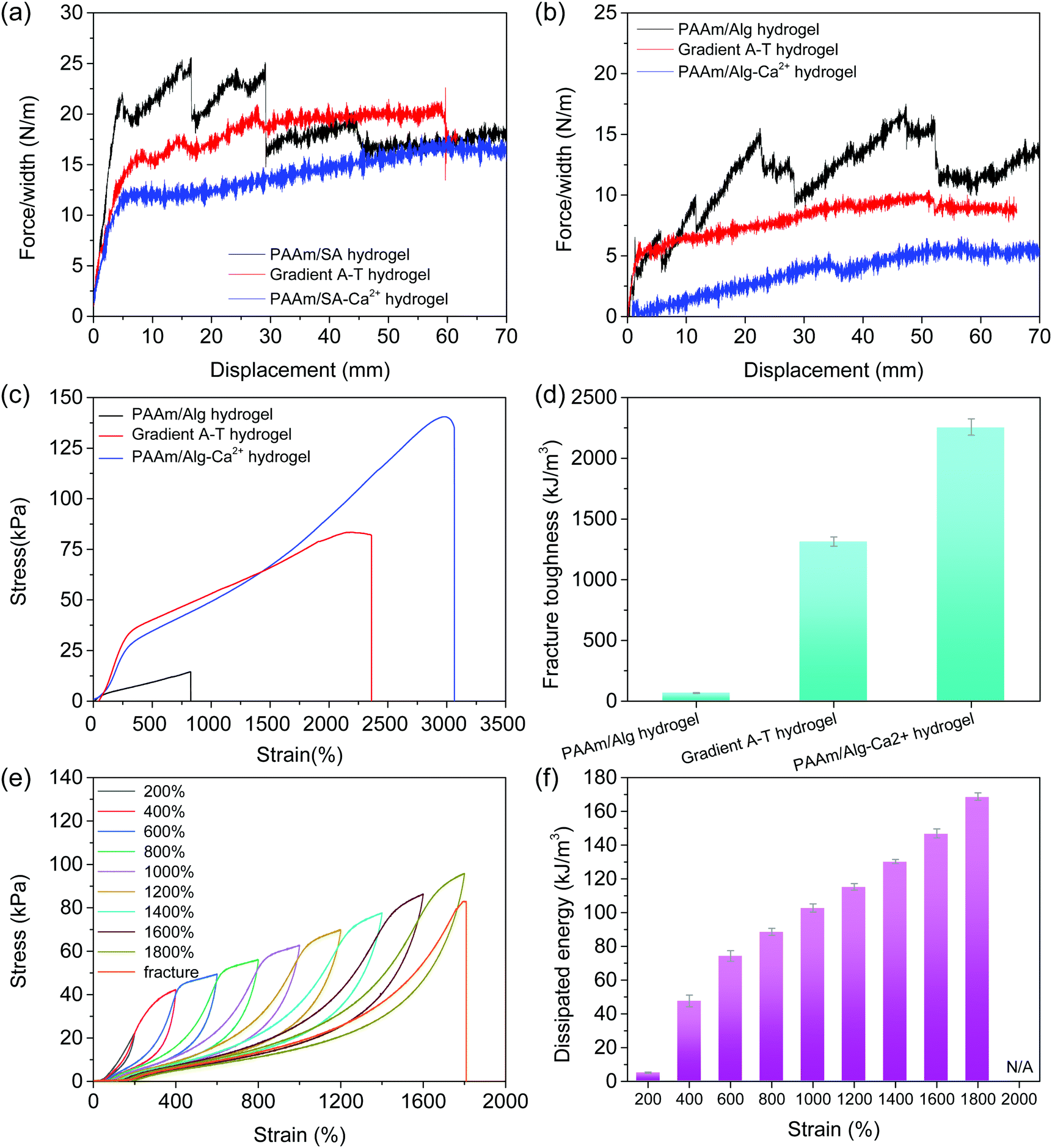 | ||
| Fig. 3 Adhesion and mechanical properties. (a) Peeling curves of the PAAm/Alg hydrogel, gradient adhesive–tough hydrogel and PAAm/Alg–Ca2+ hydrogel onto glass. The inserted picture shows that the gradient adhesive–tough hydrogel is clamped by two pieces of glass, and then the glass is pulled out. The adhesive side can still adhere to the glass, indicating the adhesion difference between the adhesive side and tough side. During the peeling process, the adhesive surface of the PAAm/Alg–Ca2+ hydrogel adhered to the glass. (b) Peeling curves of the PAAm/Alg hydrogel, gradient adhesive–tough hydrogel and PAAm/Alg–Ca2+ hydrogel onto skin. During the peeling process, the adhesive surface of the PAAm/Alg–Ca2+ hydrogel adhered to the skin. (c) Stress–strain curves of the hydrogels and (d) the corresponding fracture toughness. (e) Successive loading–unloading curves at different strains with no resting time and (f) the corresponding dissipated energy. The PAAm/Alg–Ca2+ hydrogel was cyclically loaded at different strains, increasing by 200% sequentially. | ||
Then, the energy dissipation and anti-fatigue behavior of the hydrogels were analyzed using a tensile cycling test. After the first cycle, the hysteresis loop basically overlaps and the dissipated energy remains almost unchanged under the continuous cyclic tensile curve with a strain of 1500% (Fig. S16, ESI†). The results show that the gradient adhesive–tough hydrogel has good fatigue resistance. An appropriate interval time is beneficial to the recovery of a dynamic sacrificial bond, but the overall trend remains unchanged (Fig. S17, ESI†). By comparing the cyclic tensile curves of the PAAm/Alg–Ca2+ hydrogel, the fatigue properties of the gradient adhesive–tough hydrogel originate from the dual-network structure and the dynamic sacrificial bond of ionic coordination (Fig. S18 and S19, ESI†). Compared with the cyclic tensile curves of the PAAm/Alg hydrogel, the ion binding sacrificial bond effect is obvious (Fig. S20, ESI†).
3.4. Application as a wearable strain sensor
Tissue adhesive hydrogels have great potential applications in wearable strain sensors to monitor human motions.37–39 Because of the existence of free ions in porous networks, hydrogels have good electrical conductivity. The addition of LiCl can improve the conductivity of hydrogels. With the increase of LiCl concentration, the ionic conductivity of the hydrogel gradually increases, and the maximum conductivity reaches 0.232 mS cm−1 (Fig. S21, ESI†). The signal of the hydrogel strain sensor comes from the change of ion migration distance and ion concentration per unit volume during the deformation process, so that the resistance of the hydrogel in the constant voltage circuit changes (Movie 5, ESI†). The change rate of relative resistance of the hydrogel increases with the increase of tensile strain, showing a step-by-step linear proportional relationship. The gauge factor (GF) is one of the important indexes of the sensitivity of strain sensors.40 From the slope of the fitting curve at stage A (0% < ε < 400%), it can be determined that the GF of the hydrogel is 0.196, and then increases to 0.260 (stage B, 400% < ε < 650%) (Fig. S22, ESI†). This good wide strain linearity may be related to the ion water channel in the hydrogel. The gradual increase of GF is also closely related to the narrowing of the channel in the hydrogel.41The periodic change of relative resistance of the hydrogel provides the possibility of strain sensing. During the stretching and recovery of the hydrogel, the internal channel deformation causes the relative resistance of the hydrogel to change with strain (Fig. 4a). In a closed circuit, the brightness of the light-emitting diode (LED) alternately changes (Movie 6, ESI†). The rotational distortion of the hydrogel will also change the internal channel, and the relative resistance of the hydrogel will change accordingly (Fig. 4b). Such a sensor is adhered to the finger to monitor the motion. Body movement affects the deformation of the hydrogel, causing the relative resistance of the hydrogel to change orderly, and monitoring the movement of finger, wrist, elbow joint, and knee joint (Fig. 4c–f).
 | ||
| Fig. 4 Wearable strain sensor. (a) The relative resistance changes of the gradient adhesive–tough hydrogel under the stretching. (b) The relative resistance changes of the gradient adhesive–tough hydrogel under torsion (0°, 90°, and 180°). (c) The relative resistance changes of the gradient adhesive–tough hydrogel with the bending angle (0°, 30°, 60°, and 90°) of the finger. The real-time monitoring of various human motions with the hydrogel directly adhered to (d) wrist, (e) elbow joint, and (f) knee joint. During human motion monitoring, the adhesive surface of the PAAm/Alg–Ca2+ hydrogel adhered to the skin. | ||
4. Conclusions
In summary, a gradient adhesive–tough hydrogel was successfully prepared by unidirectional coordination-induced diffusion of the tough hydrogel. EDTA is used to chelate Ca2+, forming an adhesive side in tough hydrogels (e.g., PAAm/Alg–Ca2+ hydrogels), thereby increasing the adhesion to various materials such as plastic, wood, rubber, steel, copper, stone, ceramics, glass, and skin. The gradient adhesive–tough hydrogel has wide-ranging linear sensitivity and stability when applied to strain sensors. This function makes it possible to monitor human motions. It is worth noting that the gradient adhesive–tough hydrogel without an obvious heterogeneous interface effectively avoids the stress mismatch during the strain process. Through the concept of biological structure bionics and the facile unidirectional diffusion strategy, the construction of gradient adhesive–tough hydrogels will provide new insights for the new generation of structured materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22090050 and 21874121), the National Key Research and Development Program of China (2018YFE0206900), the Hubei Provincial Natural Science Foundation of China (2020CFA037), the Zhejiang Provincial Natural Science Foundation of China (LD21B050001), and the State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Wuhan University of Technology).Notes and references
- J. Y. Sun, X. H. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- J. C. Fan, Z. X. Shi, M. Lian, H. Li and J. Yin, J. Mater. Chem. A, 2013, 1, 7433–7443 RSC.
- X. P. Morelle, W. R. Illeperuma, K. Tian, R. B. Bai, Z. G. Suo and J. J. Vlassak, Adv. Mater., 2018, 30, 1801541 CrossRef PubMed.
- Y. T. Cai, J. B. Qin, W. M. Li, A. Tyagi, Z. J. Liu, D. Hossain, H. M. Chen, J. K. Kim, H. W. Liu, M. H. Zhuang, J. W. You, F. Xu, X. W. Lu, D. Z. Suna and Z. T. Luo, J. Mater. Chem. A, 2019, 7, 27099–27109 RSC.
- S. O. Blacklow, J. Li, B. R. Freedman, M. Zeidi, C. Chen and D. J. Mooney, Sci. Adv., 2019, 5, eaaw3963 CrossRef CAS PubMed.
- J. Zeng, L. B. Dong, W. X. Sha, L. Wei and X. Guo, Chem. Eng. J., 2020, 383, 123098 CrossRef CAS.
- L. Pan, P. Q. Cai, L. Mei, Y. Cheng, Y. Zeng, M. Wang, T. Wang, Y. Jiang, B. H. Ji, D. C. Li and X. D. Chen, Adv. Mater., 2020, 32, 2003723 CrossRef CAS PubMed.
- Y. Ohm, C. F. Pan, M. J. Ford, X. N. Huang, J. H. Liao and C. Majidi, Nat. Electron., 2021, 4, 185–192 CrossRef CAS.
- J. W. Yang, R. B. Bai, B. H. Chen and Z. G. Suo, Adv. Funct. Mater., 2020, 30, 1901693 CrossRef CAS.
- Y. Hong, F. F. Zhou, Y. J. Hua, X. Z. Zhang, C. Y. Ni, D. H. Pan, Y. Q. Zhang, D. M. Jiang, L. Yang, Q. N. Lin, Y. W. Zou, D. S. Yu, D. E. Arnot, X. H. Zou, L. Y. Zhu, S. F. Zhang and H. W. Ouyang, Nat. Commun., 2019, 10, 2060 CrossRef PubMed.
- L. Han, K. Z. Liu, M. H. Wang, K. F. Wang, L. M. Fang, H. T. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2018, 28, 1704195 CrossRef.
- L. F. Wang, G. R. Gao, Y. Zhou, T. Xu, J. Chen, R. Wang, R. Zhang and J. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 3506–3515 CrossRef CAS PubMed.
- J. J. Xu, G. Y. Wang, Y. F. Wu, X. Y. Ren and G. H. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 25613–25623 CrossRef CAS PubMed.
- X. J. Zhang, K. Wang, J. Y. Hu, Y. C. Zhang, Y. Dai and F. Xia, J. Mater. Chem. A, 2020, 8, 25390–25401 RSC.
- Y. C. Zhang, Q. Chen, Z. W. Dai, Y. Dai, F. Xia and X. J. Zhang, J. Mater. Chem. B, 2021, 9, 585–593 RSC.
- W. Cui, R. J. Zhu, Y. Zheng, Q. F. Mu, M. H. Pi, Q. Chen and R. Ran, J. Mater. Chem. A, 2021, 9, 9706–9718 RSC.
- W. L. Zhang, Y. W. Zhang, Y. C. Zhang, Y. Dai, F. Xia and X. J. Zhang, J. Mater. Chem. B, 2021, 9, 5954–5966 RSC.
- L. L. Han, M. F. Liu, B. Yan, Y. S. Li, J. Lan, L. Y. Shi and R. Ran, Mater. Sci. Eng. C: Mater. Biol. Appl., 2020, 109, 110567 CrossRef CAS PubMed.
- X. C. Li, D. Z. Hao, W. J. Hao, X. L. Guo and L. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 51036–51043 CrossRef CAS PubMed.
- Q. L. Wang, Z. Liu, C. Tang, H. Sun, L. Zhu, Z. Z. Liu, K. Li, J. Yang, G. Qin, G. Z. Sun and Q. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 10457–10466 CrossRef CAS PubMed.
- F. B. Li, G. Z. Zhang, Z. S. Wang, H. Y. Jiang, S. Yan, L. Zhang and H. J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 15071–15078 CrossRef CAS PubMed.
- H. Tang, J. X. Sun, X. Shu, Y. Y. Zhao, J. Nie and X. Q. Zhu, ACS Appl. Polym. Mater., 2020, 2, 4140–4148 CrossRef CAS.
- Y. Gao, J. J. Chen, X. Y. Han, Y. D. Pan, P. Y. Wang, T. J. Wang and T. Q. Lu, Adv. Funct. Mater., 2020, 30, 2003207 CrossRef CAS.
- Q. Zhang, X. Liu, L. J. Duan and G. H. Gao, Chem. Eng. J., 2019, 365, 10–19 CrossRef CAS.
- L. Zhao, J. H. Huang, Y. C. Zhang, T. Wang, W. X. Sun and Z. Tong, ACS Appl. Mater. Interfaces, 2017, 9, 11866–11873 CrossRef CAS PubMed.
- X. Z. Wan, Z. Gu, F. L. Zhang, D. Z. Hao, X. Liu, B. Dai, Y. Y. Song and S. T. Wang, NPG Asia Mater., 2019, 11, 49 CrossRef CAS.
- Y. C. Wang, D. Liang, Z. G. Suo and K. Jia, Extreme Mech. Lett., 2020, 39, 100797 CrossRef.
- Y. Ma, Y. Gao, L. Liu, X. Y. Ren and G. H. Gao, Chem. Mater., 2020, 32, 8938–8946 CrossRef CAS.
- Z. Tao, H. L. Fan, J. C. Huang, T. L. Sun, T. Kurokawa and J. P. Gong, ACS Appl. Mater. Interfaces, 2019, 11, 37139–37146 CrossRef CAS PubMed.
- J. Li, A. D. Celiz, J. Yang, Q. Yang, I. Wamala, W. Whyte, B. R. Seo, N. V. Vasilyev, J. J. Vlassak, Z. Suo and D. J. Mooney, Science, 2017, 357, 378–381 CrossRef CAS PubMed.
- X. J. Zhang, Y. C. Zhang, W. L. Zhang, Y. Dai and F. Xia, Chem. Eng. J., 2021, 420, 130447 CrossRef CAS.
- J. J. Liu, S. X. Qu, Z. G. Suo and W. Yang, Natl. Sci. Rev., 2021, 8, nwaa254 CrossRef CAS PubMed.
- J. Q. Wang, China Chlor-Alkali, 2010, 8, 28–29 Search PubMed.
- M. Wang, X. Li, W. K. Hua, L. Deng, P. Y. Li, T. H. Zhang and X. F. Wang, Chem. Eng. J., 2018, 348, 95–108 CrossRef CAS.
- H. A. Afshar and A. Ghaee, Carbohydr. Polym., 2016, 151, 1120–1131 CrossRef PubMed.
- J. W. Seo, H. Kim, K. Kim, S. Q. Choi and H. J. Lee, Adv. Funct. Mater., 2018, 28, 1800802 CrossRef.
- S. N. Li, Y. Cong and J. Fu, J. Mater. Chem. B, 2021, 9, 4423–4443 RSC.
- X. J. Pei, H. Zhang, Y. Zhou, L. J. Zhou and J. Fu, Mater. Horiz., 2020, 7, 1872–1882 RSC.
- X. J. Pei, J. T. Wang, Y. Cong and J. Fu, J. Polym. Sci., 2021, 59, 1312–1337 CrossRef CAS.
- J. Wen, J. Tang, H. M. Ning, N. Hu, Y. Y. Zhu, Y. K. Gong, C. H. Xu, Q. N. Zhao, X. P. Jiang, X. L. Hu, L. Lei, D. Wu and T. Huang, Adv. Funct. Mater., 2021, 31, 2011176 CrossRef CAS.
- H. L. Sun, Y. Zhao, S. L. Jiao, C. F. Wang, Y. P. Jia, K. Dai, G. Q. Zheng, C. T. Liu, P. B. Wan and C. Y. Shen, Adv. Funct. Mater., 2021, 31, 2101696 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02599f |
| This journal is © The Royal Society of Chemistry 2022 |