
Mitochondrion-targeting PEGylated BODIPY dyes for near-infrared cell imaging and photodynamic therapy†
Isabel Wen
Badon‡
 a,
Chanwoo
Kim‡
b,
Jong Min
Lim‡
c,
Duy Khuong
Mai‡
ad,
Temmy Pegarro
Vales
e,
Dongho
Kang
b,
Sung
Cho
d,
Joomin
Lee
*f,
Ho-Joong
Kim
*a and
Jaesung
Yang
*b
a,
Chanwoo
Kim‡
b,
Jong Min
Lim‡
c,
Duy Khuong
Mai‡
ad,
Temmy Pegarro
Vales
e,
Dongho
Kang
b,
Sung
Cho
d,
Joomin
Lee
*f,
Ho-Joong
Kim
*a and
Jaesung
Yang
*b
aDepartment of Chemistry, Chosun University, Gwangju 61452, Korea. E-mail: hjkim@chosun.ac.kr
bDepartment of Chemistry, Yonsei University, Wonju, Gangwon 26493, Korea. E-mail: jaesung.yang@yonsei.ac.kr
cCenter for Molecular Spectroscopy and Dynamics, Institute for Basic Science (IBS), Seoul 02841, Korea
dDepartment of Chemistry, Chonnam National University, 99 Daehak-ro, Yuseong-gu, Gwangju 61186, Korea
eDepartment of Natural Sciences, Caraga State University, Butuan City 8600, Philippines
fDepartment of Food and Nutrition, Chosun University, Gwangju 61452, Korea
First published on 17th January 2022
Abstract
A series of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-based photosensitizers (AmBXI, X = H, M, Br) featuring a cationic mitochondrion-targeting group and near-infrared (NIR) absorption was synthesized. After extending the photosensitizers’ π conjugation via Knoevenagel reaction, both the absorbance and emission maxima of AmBXI shifted to the phototherapeutic wavelength range (650–900 nm). Theoretical computations indicate that the introduction of bromine atoms promotes spin–orbit coupling, so that for each additional bromine atom in AmBXI an increase in singlet oxygen quantum yield would be expected (0.3%, 2.2%, and 4.1%, for AmBHI, AmBMI, and AmBBrI, respectively). Moreover, AmBXI photosensitizers exhibited low cytotoxicity in the dark and high phototoxicity, with the half maximal inhibitory concentrations of AmBBrI found to be 46.93 nM and 22.84 nM, while those of AmBMI were 129.7 nM and 58.34 nM in HeLa and MCF-7 cancer cells, respectively. Notably, introduction of a single bromine atom was enough to produce a cytotoxic effect. Furthermore, the presence of a quaternary ammonium group in AmBXI enabled the dyes to localize and stain the negatively charged mitochondria. The results presented herein indicate the straightforward and facile synthesis of NIR-light triggered mitochondrion-targeting photosensitizers.
Introduction
Photodynamic therapy (PDT) is a photochemistry-based health treatment that is minimally invasive, causes low toxicity in healthy tissues, and does not produce drug resistance, even after repeated implementations.1,2 PDT has been proven effective against malignant and benign tumors, whether implemented as the primary treatment or in combination with other therapies.3–5 In PDT, cytotoxic singlet oxygen (1O2) is generated as a result of the interaction of the three key components of the therapeutic approach: a photosensitizer (PS), light, and molecular oxygen (3O2). Initially, PSs are introduced in the target tissue; the said tissue is then subjected to light irradiation, triggering the excitation of the PS from the singlet ground state (S0) to the singlet excited state (Sn). Sn can then relax back to the ground state or undergo intersystem crossing (ISC) to the triplet excited state (T1), which can either activate the photochemical processes leading to the production and release of reactive free radicals or transfer its energy to ground-state 3O2 species to produce 1O2.6 The 1O2 thus generated can cause cancer cell death either by directly attacking these cells or by causing tumor vascular occlusions.7 However, the 1O2 quantum yield (ϕΔ) largely depends on the efficiency of the S1 → T1 electronic transition, which is a spin-forbidden process. Attaching heavy atoms like iodine, bromine, or transition metals to the PS molecule promotes spin–orbit coupling (SOC) and increases the likelihood of the S1 → T1 ISC. However, transition metals like iridium or ruthenium are expensive, and the ϕΔ value achieved using them is no higher than that obtained using halogens.8 Moreover, the low tissue penetration depth achieved by visible light presents another practical limitation to the use of PDT in biological applications. In fact, tissue penetration is maximal for light characterized by wavelengths in the near-infrared (NIR) spectrum.9 In this regard, PSs that can be activated by electromagnetic waves falling within the “phototherapeutic window (650–900 nm)” are preferable.Touted as “porphyrin's little sister,” derivatives of 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) boast excellent photophysical properties, such as sharp absorption bands, high thermal and photochemical stability, and high fluorescence quantum yields, which make them ideal platforms for optical imaging and lasing, as well as for the manufacture of light-emitting diodes and dye-sensitized solar cells.10–14 Their structures can also be tuned to accommodate synthetic demands. Needless to say, numerous BODIPY-based PSs have been developed.15–22 Specifically, NIR-absorbing BODIPY derivatives are readily accessible via extension of the π-conjugation of the BODIPY core structure.23 However, as a result of this strategy, the hydrophobicity of the overall structure is inadvertently increased, which is an undesirable outcome from the standpoint of biological applications, as rigid and lipophilic BODIPY-based compounds tend to localize in the intracellular membrane.24,25 This problem can be overcome by introducing hydrophilic groups in the structure of the PS, such as polyethylene glycol, phosphonates, and sulfonates, whose presence offsets the increase in hydrophobicity associated with π-conjugation extension and prevents dye molecule aggregation within the cell.26,27
Nonspecific binding and off-target toxicity of compounds are serious bottlenecks in the development and approval of novel cancer drugs. The risk of causing deleterious effects on healthy tissues highlights the importance of identifying biomarkers or developing targeting strategies.28–30 However, the variability in the degree of expression of genes and the presence of mutations even within the same tumor renders the targeting of a single gene or a single pathway a complex task to realize.31,32 Notably, mitochondria are membrane-bound organelles that in healthy cells are mainly in charge of energy production, reactive oxygen species (ROS) production, macromolecular synthesis, and cell signaling activation.33,34 In cancer cells, mitochondrial dysfunction is common, and it is associated with compromised oxidative phosphorylation, excess ROS production, calcium accumulation and inflammation. Mitochondrial dysfunction also promotes tumorigenesis and cancer cell progression and recurrence. Cancer cells are also characterized by a higher mitochondrial membrane potential than normal cells.35 These distinctive features of dysfunctional mitochondria render them potential targeting sites for cancer treatment. In fact, several mitochondrion-targeting PSs feature cations in their structure, such as triphenylphosphonium and quaternary ammonium groups, which can exploit the mentioned difference in mitochondrial membrane potential to drive the preferential accumulation of the PSs in the mitochondria of cancer cells.36,37 For example, molybdenum-based complexes functionalized with either TPP or methylpyridinium ligands have been shown to accumulate in HeLa cell mitochondria and exhibit phototoxicity toward the cells. However, these PSs have limitations, such as insufficient water solubility and poor cellular uptake.36–44
In view of the described concepts, we herein report the synthesis of NIR-absorbing BODIPY-based dyes featuring good water solubility and mitochondrion selectivity. The spectroscopic properties and 1O2 generation efficiency of the mentioned fluorophores were also investigated. Computational studies were also conducted to understand the effects of the structural modifications of the dyes. The synthesized compounds were tested in vitro to investigate the practicality of using the dyes in living systems. Evidence suggested that they may enable users to achieve light-controlled cell death and a clear visualization of mitochondria within cells.
Results and discussion
In Scheme 1 is outlined the synthetic approach adopted to obtain BODIPY-based, NIR-absorbing dyes endowed with mitochondrion-targeting ability to be used in PDT. The applied design strategy was intended to circumvent the general limitations associated with the use of BODIPY-based PSs by making full use of the sites available for modification. Initially, 2,4-dimethylpyrrole and (4-dimethyl)benzaldehyde were made to react at room temperature in the presence of trifluoroacetic acid acting as a catalyst. The reaction reached completion following the addition of 1,3-dichloro-5,6-dicyano-benzoquinone (DDQ), TEA, and BF3·OEt2 under controlled conditions. The lateral 2,6 positions of the BODIPY core were then subjected to monobromination or dibromination. Benzaldehydes conjugated with two triethylene glycol (TEG) chains were made to react with the two acidic bottom methyl groups of the BODIPY core via a Knoevenagel condensation. Finally, a quantitative methylation of the dimethylanilino moiety produced an ammonium cation, which enhances the hydrophilicity of the dye and acts as mitochondrion-targeting active group. The three compounds thus obtained, listed in order of increasing number of conjugated bromine atoms, are referred to as AmBHI (0 bromine atoms), AmBMI (1) and AmBBrI (2).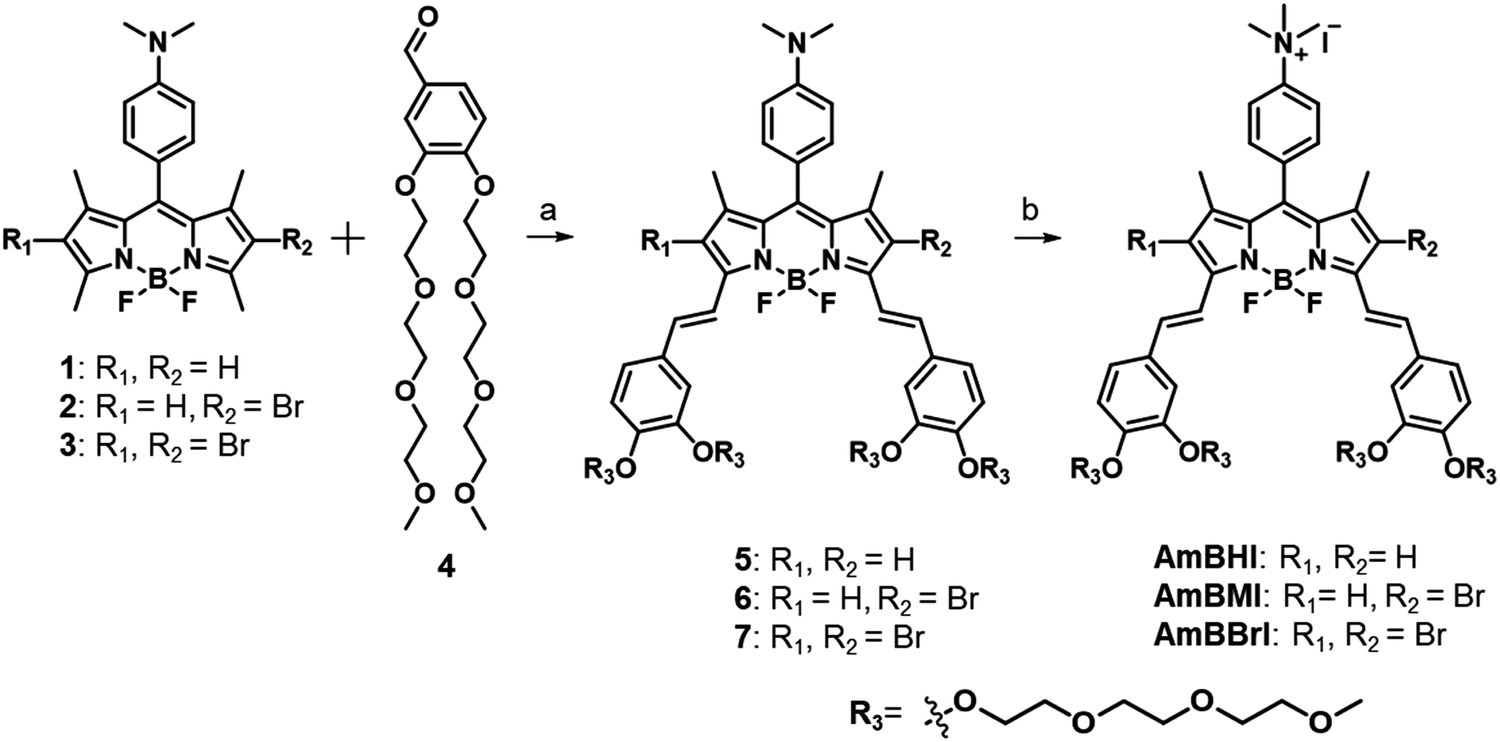 | ||
| Scheme 1 Synthesis of mitochondrion-targeting 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-based photosensitizers AmBXI (X = H, M, Br). (a) Piperidine, acetic acid, dimethylformamide, reflux. (b) Iodomethane, acetonitrile, room temperature. | ||
Because of the presence of multiple TEG chains and ammonium cation, AmBXI dyes were easily soluble in water such that sample solutions with concentration as high as sub-millimolar could be prepared with little precipitation as assessed by eyes. Furthermore, dynamic light scattering measurements of AmBXI dyes in phosphate-buffered saline at concentration of 250 μM revealed no appreciable signal, indicating the absence of nanometer-sized particles resulting from dye aggregation and assuring the high solubility of the dyes in aqueous media. While discernible aggregates are not formed, π–π interactions between numbers of AmBXI dye molecules exist, particularly, at high concentrations, as reflected by concentration-dependent spectral broadening and saturation in intensity of fluorescence spectra (see Fig. S1, ESI†).
The steady-state absorption and emission spectra of the synthesized BODIPY-based dyes are reported in Fig. 1a and b. Compared with compounds 1–3 (see Scheme 1), AmBHI, AmBMI, and AmBBrI exhibited a very dramatic redshift in absorption, so that the maximum absorbance was observed within the phototherapeutic window (650–850 nm). The observed λmax value is in agreement with that previously reported for distyryl-BODIPY derivatives modified in the same fashion.45 Notably, this success in the preparation of NIR-activatable PSs displaying excellent water solubility was achieved at the expense of the intensity of fluorescence emission. The attachment of two styryl units to the BODIPY core plays an essential role in the extension of π-conjugation, which led to the observed spectral redshift. However, free rotation is possible around the resultant vinyl bonds, which provides undesirable nonradiative decay pathways and induces fluorescence quenching. This rotation is likely to be boosted by a bundle of long, flexible TEG chains dangling from the styryl moieties. Hence, the fluorescence quantum yield (ϕF) of AmBHI was determined to be 0.18 (Table 1), which is half the typical value observed for unsubstituted BODIPY derivatives.46,47 The further gradual decrease in ϕF value in going from AmBHI to AmBMI (0.11), and AmBBrI (0.06) is attributable to ISC, which becomes more efficient as the number of bromine atoms increases. We stress that despite the low value for ϕF, the halogenated BODIPY derivatives can still be used for cell imaging, as the evidence discussed below will indicate.
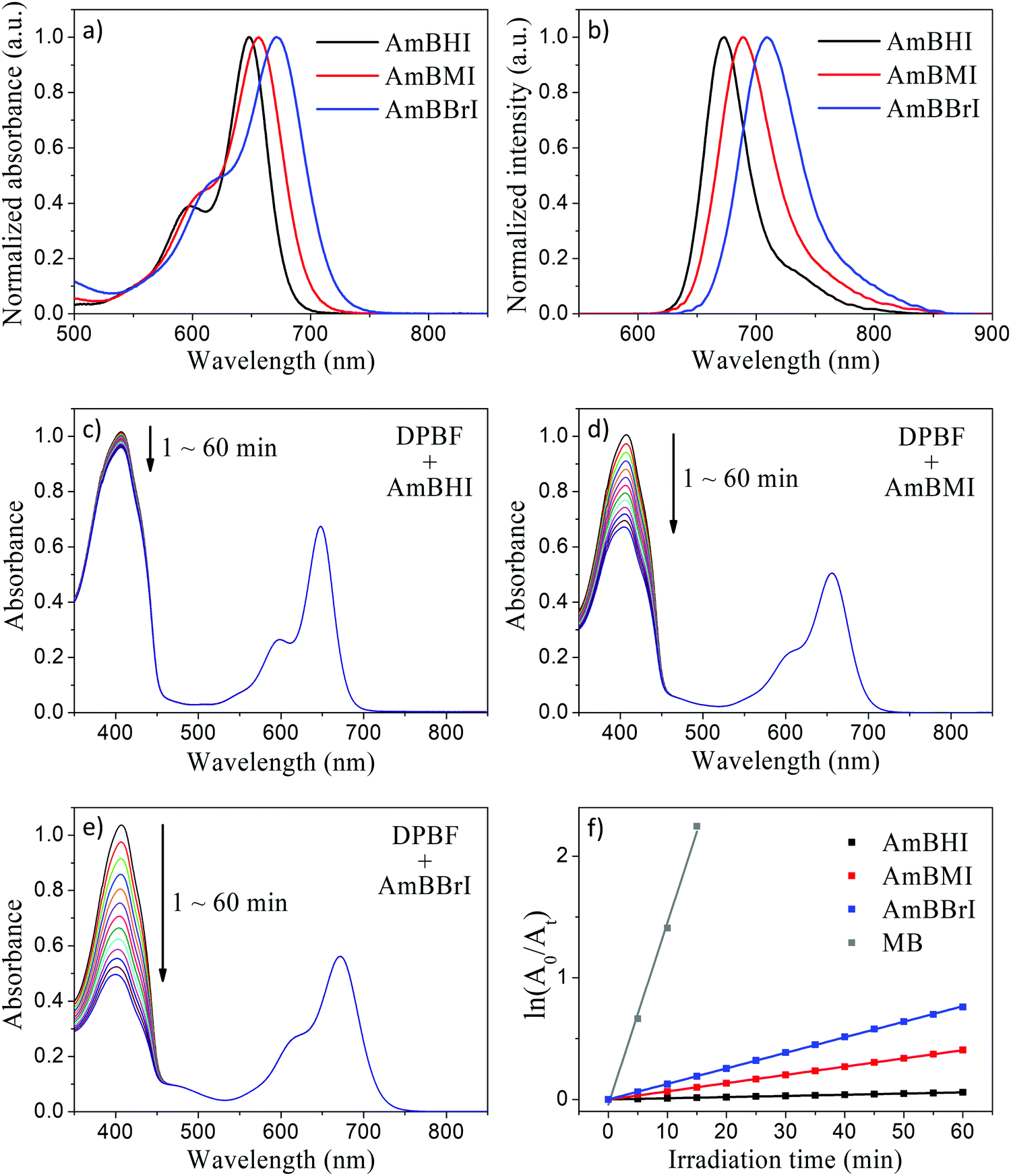 | ||
| Fig. 1 Steady-state absorption (a) and emission (b) spectra of AmBXI dyes in methanol. Time-dependent absorption spectra of air-saturated DPBF methanol solutions containing AmBHI (c), AmBMI (d), and AmBBrI (e) obtained under the irradiation of 660 nm-LED light. (f) Temporal decrease in the absorbance of DPBF at 410 nm drawn according to the first-order kinetics (square dots) with linear fits (solid line) in semilogarithmic scale. MB: methylene blue. | ||
| Experimental data | Calculated data | ||||||
|---|---|---|---|---|---|---|---|
| λ abs (nm) | λ em (nm) | ϕ F | ϕ Δ | S1e (eV) | f | Major contribution to the transition | |
| a Wavelength of the maximum absorption. b Wavelength of the maximum fluorescence emission. c Fluorescence quantum yield determined with respect to rhodamine 6G in ethanol (ϕF = 0.95). d Singlet oxygen quantum yield determined using the DPBF bleaching method with methylene blue in methanol as standard (ϕΔ = 0.50). e Vertical excitation energy computed in water using B3LYP/6-31g(d). f Oscillator strength. | |||||||
| AmBHI | 648 | 673 | 0.18 | 0.004 | 2.09 | 1.0576 | HOMO → LUMO (100%) |
| AmBMI | 659 | 689 | 0.11 | 0.025 | 2.06 | 0.9907 | HOMO → LUMO (100%) |
| AmBBrI | 671 | 709 | 0.06 | 0.045 | 2.03 | 0.9410 | HOMO → LUMO (100%) |
In order to evaluate the 1O2-generation efficiency of AmBXI,1,3-diphenylisobenzofuran (DPBF) bleaching experiments were performed. Indeed, DPBF reacts with 1O2 to yield 1,2-dibenzoylbenzene via a cycloaddition reaction. In turn, the oxidation of DPBF is made evident by a decrease in DPBF absorbance, which can be recorded as a simple strategy for quantifying the amount of 1O2 species generated by PSs. In the relevant experiments, air-saturated methanol solutions of DPBF containing AmBXI were prepared, whereby the concentrations of both compounds were carefully controlled. The solutions were then irradiated with an LED light, while the absorption spectrum of DPBF was measured periodically. As can be evinced from Fig. 1c–e, a dramatic temporal decrease in the absorbance of DPBF was observed in the presence of AmBMI and AmBBrI. By contrast, only a trivial decrease was observed in the presence of AmBHI. The 1O2ϕΔ value was calculated assuming first-order kinetics using methylene blue (ϕΔ = 0.50 in methanol) as standard (Fig. 1f and Table 1). Compared to AmBHI, for which ϕΔ was as low as 0.4%, AmBMI and AmBBrI exhibited an order of magnitude enhancement in ϕΔ, with AmBBrI displaying the greatest efficiency (∼4.5%). Consistent trend was observed for 1O2 generation efficiency in water (Fig. S2, ESI†). Notably, the efficiency was observed to be directly proportional to the degree of bromination. The observed increase in ϕΔ of the halogenated BODIPY derivatives can be attributed to the heavy-atom effect.48 Heavy atoms, like bromine employed here, boost the ISC process to bring more electrons into the triplet excited states through vibronic SOC.45 The excited electrons, if necessary, relax to the lowest-energy triplet state and then collide with 3O2 molecules to generate the highly reactive 1O2 species, which ultimately destroy tumor cells. Essentially, the generation of 1O2 depends on the efficiency of the ISC process.
In order to correlate the spectral properties of AmBXI dyes with their electronic structures, time-dependent density functional theory (TD-DFT) calculations were carried out at the B3LYP/6-31G(d) level of theory (see the Methods for computational details). As can be evinced from Fig. 2, the optimized geometries of the three dyes reveal that the BODIPY core and the two styryl moieties are almost coplanar in all compounds, resulting in HOMOs and LUMOs that are completely delocalized across both styryl moieties. The said coplanarity is barely affected by the bulky peripheral chains, as indicated by the results of the geometry optimization of the complete structures of the AmBXI dyes (Fig. S3, ESI†). These data confirm the effective π-conjugation over the whole BODIPY backbone, which is the essential factor that shifts the spectral window of AmBXI dyes toward the NIR region. By taking a closer look at the HOMO of the halogenated BODIPY derivatives, one is able to confirm that the electron density spreads over the bromine atoms (Fig. 2), explaining the gradual bathochromic spectral shift observed upon going from AmBHI to AmBMI and AmBBrI (see Fig. 1a and b). Notably, with its orthogonal geometry, the trimethylanilino group in the meso position thwarts orbital overlap and π–π intramolecular interactions.49,50
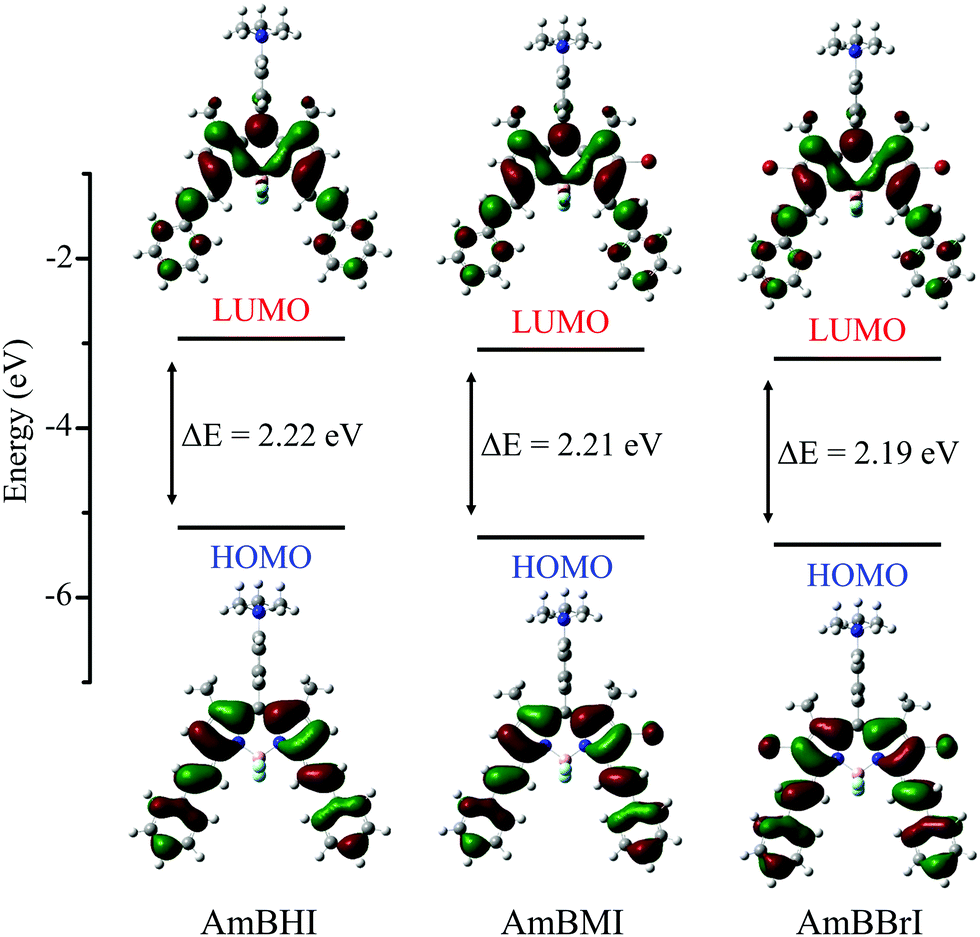 | ||
| Fig. 2 Energy level diagrams and frontier molecular orbitals for the lowest-energy transitions of AmBXI dyes computed in water solvent using the B3LYP/6-31G(d) level of theory. | ||
In triplet PSs, 1O2 generation must be preceded by the generation of triplet states, by which ϕΔ is directly related to the rate of ISC (kISC). A simplified description of kISC can be given as:51

In Fig. 3 are reported the SOC data and energy level diagrams with plausible photophysical decay pathways. For all compounds, the SOC values of the S1–T1 and S1–T3 manifolds are greater than that of the S1–T2 manifold. However, the values of ΔES1–T1 and ΔES1–T3 are too large (>0.65 eV) for the ISC process to occur, suggesting that the ISC between S1 and Tm (m = 1, 3) states is improbable.53 Among the triplet states, the T2 state is closest in energy to the S1 state; therefore, the S1–T2 manifold is the most likely candidate for the ISC channel of AmBXI dyes. The magnitudes of ΔES1–T2 are similar across all AmBXI dyes, and they all fall in the 0.22–0.24 eV range, indicating a negligible influence of the energy difference on the kinetics of ISC. Unlike ΔES1–T1, the SOC values for the S1–T2 manifold are markedly different from each other. Although the SOC value of AmBHI is marginal (0.06 cm−1), those of AmBMI and AmBBrI are larger (0.17 cm−1 and 0.28 cm−1, respectively), confirming the heavy-atom effect associated with the introduction of bromine atoms in the BODIPY derivatives. These results indicate that in AmBMI and AmBBrI an ISC occurs efficiently between the S1 and T2 states; results also agree with the experimentally determined ϕΔ values for the AmBXI dyes. Overall, the steady-state spectroscopic and theoretical data indicate that the introduction of bromine atoms in the BODIPY core dramatically increases 1O2 generation efficiency and demonstrate the high potential of the halogenated AmBXI dyes for application as PDT agents.
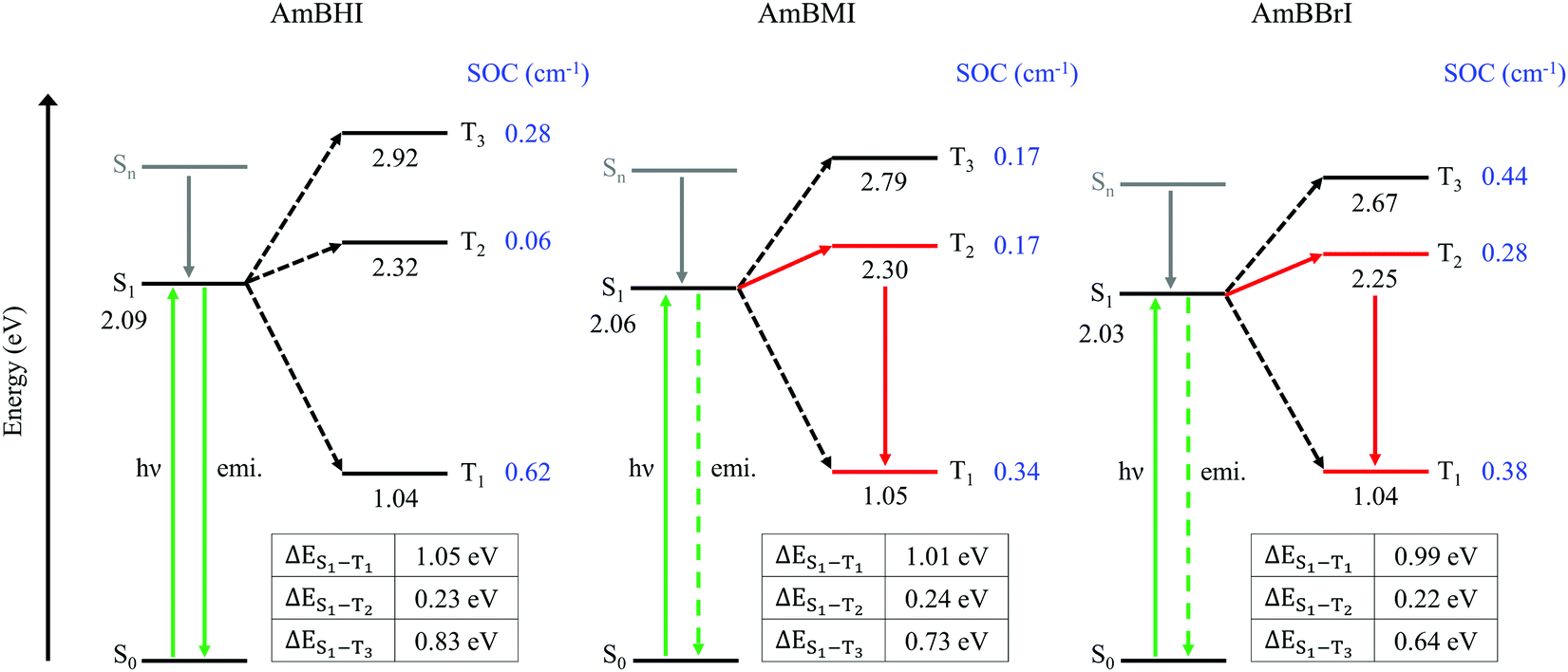 | ||
| Fig. 3 Plausible photophysical decay pathways available for the photoexcited AmBXI dyes assessed on the basis of excited-state energies and spin–orbit coupling (SOC) values. The SOC values between the singlet S1 and the triplet Tm (m = 1–3) states are presented in blue. In the tables are listed the values of the energy gaps between the S1 and Tm (m = 1–3) states for each dye. | ||
A confocal laser scanning microscopy experiment was carried out to determine the localization of AmBXI dyes inside the cell. Human cervical cancer (HeLa) cells were treated with AmBXI 1.0 μM for 24 h. As can be evinced from the images reported in Fig. 4a, the dyes accumulated in the cytoplasm of the HeLa cells (red areas) and were excluded from their nuclei (dark areas). TEG has been shown to significantly improve cellular retention.54 Notably, AmBMI and AmBBrI, which exhibited reduced values for ϕF in aqueous solution relative to AmBHI, exhibited fluorescence inside HeLa cells. Indeed, interactions between the AmBXI dyes and subcellular biomolecules may have hampered free rotation of the TEG chains and increased the molecular rigidity of the dyes, resulting in an enhanced fluorescence.55 In order to verify the mitochondrion selectivity of AmBXI dyes, a multiple staining experiment was conducted utilizing the mitochondrial dye MitoTracker Green (Fig. 4b) and the nuclear dye 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 4c). Merged images of the HeLa cells (Fig. 4d) indicated that the AmBXI colocalized with MitoTracker Green in the same region outside the DAPI-stained nuclei. A closer look at the superimposed fluorescence intensity profiles of AmBXI and MitoTracker Green dyes confirmed the congruence in the region of interest (Fig. 5). This evidence confirms the mitochondrion-specificity of AmBXI dyes. Gunbas et al. synthesized BODIPY-based PSs with cationic methyl pyridinium moieties, which displayed higher affinity for the mitochondria of HeLa cells than those of noncancerous NIH/3T3 cells.44 The fact that the mitochondrial membrane of cancer cells is characterized by a higher potential than that of normal cells accounts for the preferential accumulation of cations in cancer cells.56–58 Fluorescence activated cell sorting (FACS) analysis further evinced the entry of AmBXI dyes the cells (Fig. 6a and b). The corrected median intensity profiles against HeLa and MCF-7 cells shown in Table 2 indicate that the cells treated with AmBXI dyes exhibit more than 200-fold higher fluorescence emission than the untreated cells, with AmBHI producing the highest signal. These data show that the cationic dyes are well taken up by the mitochondria of the cancer cells. Similarly, AmBHI, AmBMI, and AmBBrI are expected to localize preferentially in cancer cells and direct their cytotoxic action against the mitochondria. Having established the cell permeability and biocompatibility of AmBXI dyes, these compounds were subjected to dose-dependent in vitro photosensitizing experiments using HeLa and human breast cancer (MCF-7) cells. The experiments were conducted in triplicate, and the relevant obtained results are summarized in Fig. 7. In the dark, both cell lines incubated with AmBXI dyes consistently exhibited high cell viability, up to a dye concentration of 640 nM; accordingly, AmBXI dyes were concluded to be characterized by low cytotoxicity in the absence of natural light. Note that when the dye concentration was further increased up to 1280 nM, the cell lines incubated with AmBHI exhibited a moderate reduction in cell viability down to 84.6% while incubation with halogenated dyes had little effect. After irradiation at 690 nm for 30 min, the cell viability of HeLa and MCF-7 cells treated with AmBHI remained >80%. By contrast, cells treated with AmBBrI exhibited substantial reductions in viability, with a calculated IC50 values of 46.93 nM and 22.84 nM for HeLa and MCF-7 cells, respectively. In the case of AmBMI-treated cells, the phototoxic effect was moderate in comparison with AmBBrI, with calculated IC50 values of 129.7 nM and 58.34 nM for HeLa and MCF-7 cells, respectively.
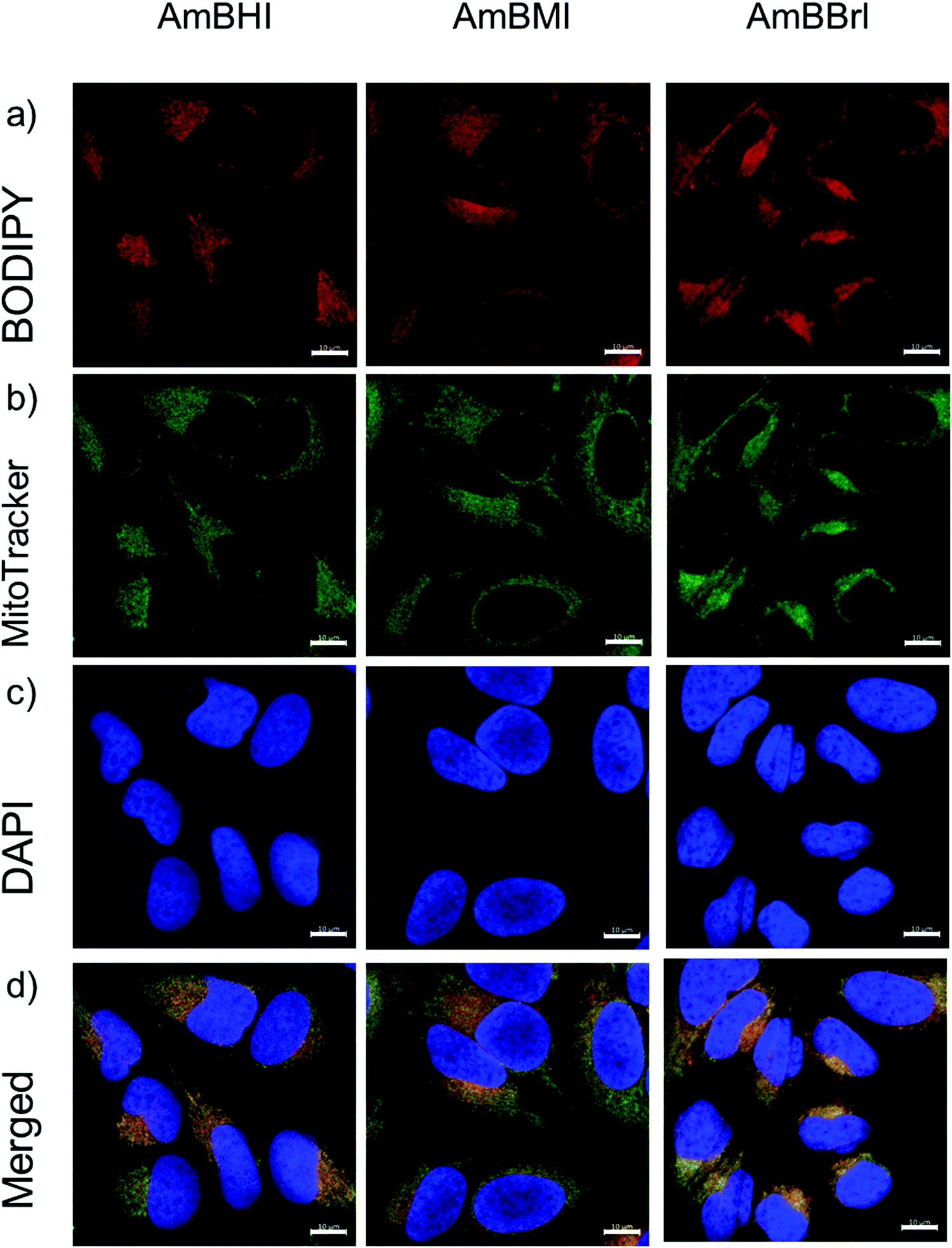 | ||
| Fig. 4 Confocal laser scanning microscopy images of HeLa cells colabeled with AmBHI, AmBMI, and AmBBrI (a), MitoTracker Green (b), 4′,6-diamidino-2-phenylindole (DAPI) (c), and merged images (d). In all cases, the dye concentration was 1 μM. For cellular experiments, unless otherwise noted, the inactivated FBS and antibiotics were included when incubating cells with AmBXI dyes (see the Experimental section for more details). Scale: 10 μm. | ||
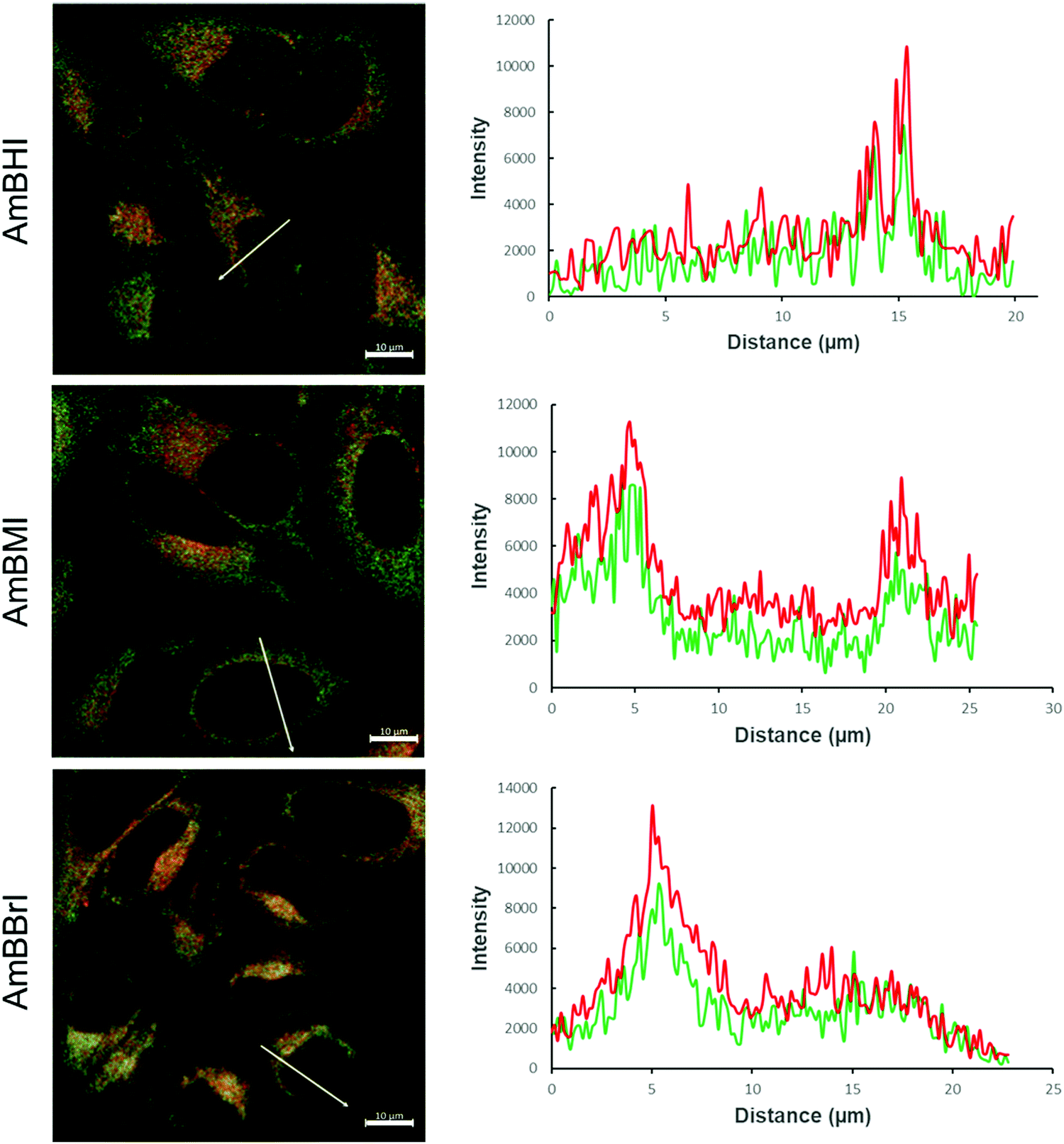 | ||
| Fig. 5 Fluorescence intensity profile of AmBHI (top), AmBMI (middle), and AmBBrI (bottom), red profile, alongside that of MitoTracker Green (green profile) of the region of interest across the cell. Scale: 10 μm. | ||
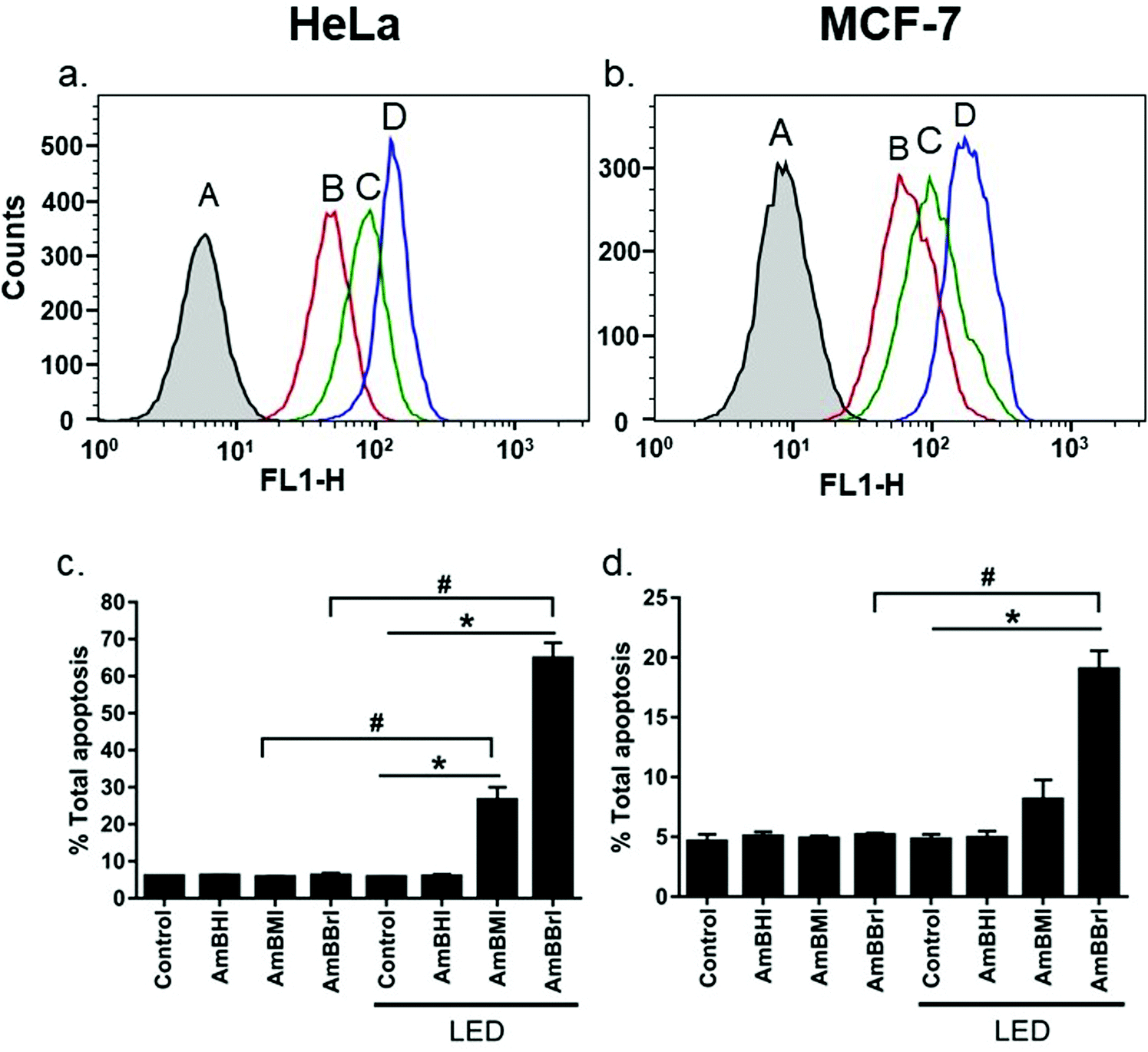 | ||
| Fig. 6 Plots of cell uptake of AmBXI analyzed by FACS (a and b), where A = control, B = AmBMI, C = AmBBrI, and D = AmBHI. The larger shift in fluorescence intensity indicates higher cellular uptake. Quantification of total apoptotic cell population. Data present mean values and error bars denote standard deviation (n = 3). Statistically significant difference was observed for irradiated dye-treated cells, compared with the irradiated control samples (*, P < 0.05) unirradiated dye-treated cells (#), as revealed by one-way ANOVA followed by Bonferroni's multiple comparisons test (c and d). | ||
| Control | AmBHI | AmBMI | AmBBrI | |
|---|---|---|---|---|
| HeLa | 0.42 ± 0.01 | 99.73 ± 0.06 | 90.57 ± 0.06 | 98.97 ± 0.50 |
| MCF-7 | 0.40 ± 0.19 | 99.93 ± 0.06 | 89.93 ± 0.01 | 99.13 ± 0.35 |
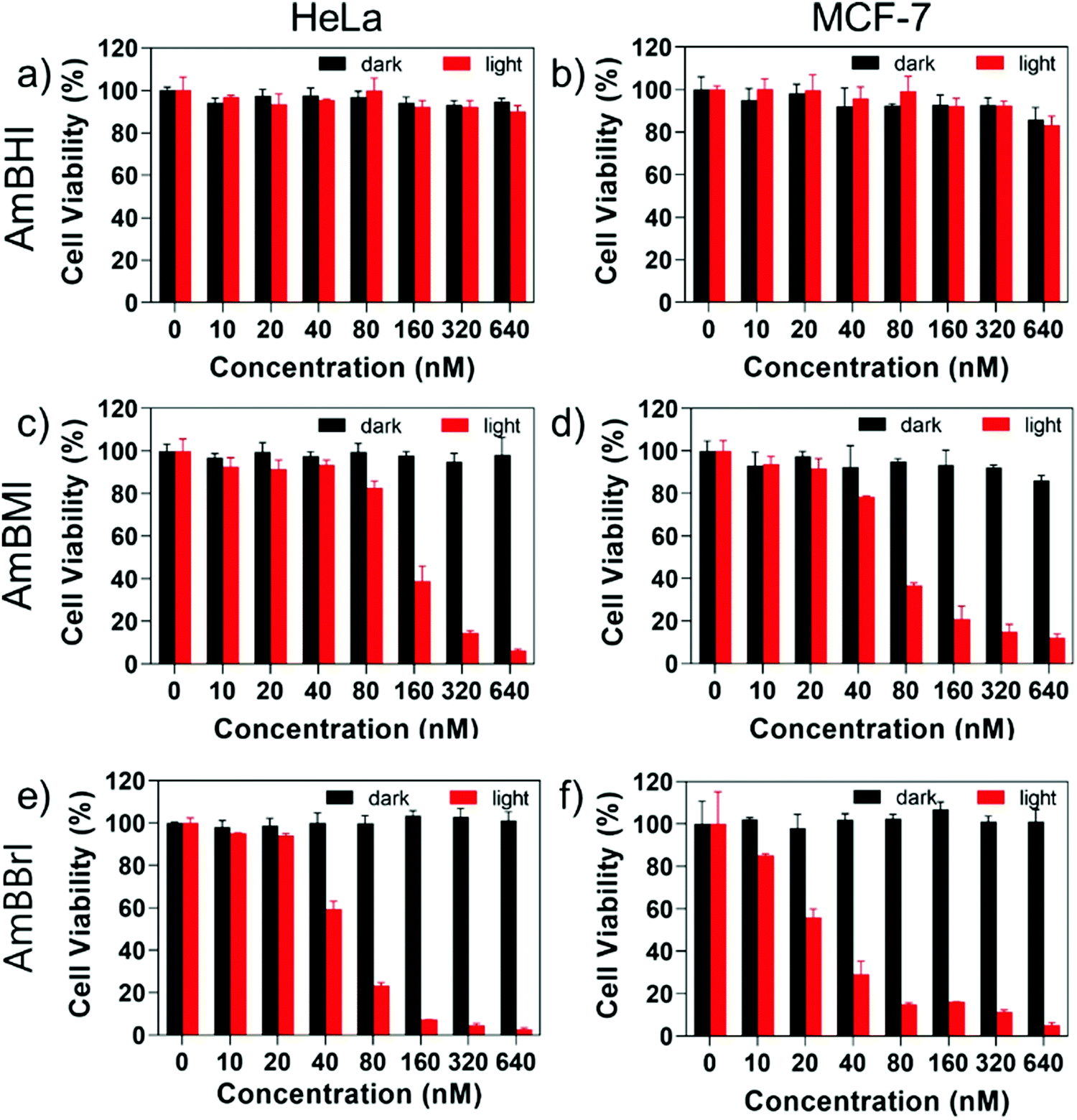 | ||
| Fig. 7 Results of dose-dependent cell proliferation (MTS) assay conducted on AmBHI, AmBMI and AmBBrI using cervical cancer (HeLa) (a, c and e) and human breast cancer (MCF-7) (b, d and f) cells without and with LED light irradiation at 690 nm for 30 min. Data represent mean values and error bars denote standard deviation (n = 3). | ||
To look deeper into the kinetics of cell death induced by AmBXI dyes, Annexin V/PI staining assay was conducted. This staining protocol is a sensitive method to quantify and differentiate between necrotic (unregulated) and apoptotic (programmed) cells. HeLa and MCF-7 cancer cells were treated with DMSO as control and AmBXI, respectively. The (early + late) apoptotic population was measured before and after LED light activation of the dyes. The observed cell population was divided into four quadrants where Q1 (PI is positive, Annexin V is negative) means necrotic cells, Q2 (PI is positive, Annexin V is positive) means late apoptotic cells, Q3 (PI is negative, Annexin V is negative) means that cells are healthy and viable, and Q4 (PI is negative, Annexin V is positive) means early apoptotic cells.
Results are shown in Fig. 6c, d and Fig. S4 (ESI†). Under dark conditions, both HeLa and MCF-7 cancer cells treated with AmBXI dyes showed no difference from the control samples, with most of the cells remaining in Q3. However, after photoactivation, AmBMI and AmBBrI induced the cells to undergo apoptosis while AmBHI hardly did, as reflected by the emergence of appreciable cell population in Q2. For HeLa cells, AmBMI showed moderate apoptosis efficacy of 26%, whereas AmBBrI showed the highest efficacy of 65%. Consistent results were found for MCF-7 cells: total apoptosis that AmBMI and AmBBrI caused were 8% and 20%, respectively. Intriguingly, AmBBrI also caused necrosis to MCF-7 cells, as reflected by unusual cell population in Q1. This is probably due to the high concentration of the dye (0.5 μM) and a difference in the response of this cell line.
The results of phototoxicity experiments conducted on AmBXI dyes agree with the results of the 1O2 generation experiments and of the computational studies. The increase in ISC likelihood brought about by the heavy-atom effect of bromine contributed to the phototoxicity of AmBMI and AmBBrI. When BODIPY is used as the platform for PSs, the location of the substituents is known to bear more significance than the number of halogen atoms present in the structure, when it comes to the value of ϕΔ.59 In this case, bromine substitutions at the lateral 2,6-positions rendered the ISC process more likely, thus enhancing the efficiency of the generation of the cytotoxic 1O2. Notably, the introduction of one bromine atom was enough to produce a phototoxic effect. Moreover, as indicated by the results of the cell imaging experiments conducted on AmBMI and AmBBrI, biological observations may depart from predictions based on theoretical studies due to the influence of the cellular environment.
Conclusion
In developing effective PSs, light penetration depth and target specificity are just as crucial as efficient 1O2 generation. We worked around the shortcomings of BODIPY-based dyes, such as hydrophobicity, absorption in the visible region, and negligible 1O2 generation, to develop the NIR-absorbing and mitochondrion-targeting AmBXI PSs. The number of bromine atoms in AmBXI dyes can be fine-tuned to achieve a balance between cell imaging and light-controlled cell death. Theoretical studies revealed that the addition of each bromine atom rendered the ISC more likely and increased the ϕΔ value. Accordingly, the results of cell viability assays indicate that AmBXI dyes exhibited low dark cytotoxicity, while the brominated AmBMI and AmBBrI exhibited phototoxicity, with AmBBrI being characterized by lower IC50 values than AmBMI. Surprisingly, AmBXI afforded good cell imaging results, despite its low fluorescence quantum yield in aqueous solutions. Interaction between the dye and cellular biomolecules may have increased the structural rigidity of the PS, hampering nonradiative pathways. Finally, the AmBXI dyes were observed to accumulate preferentially in the mitochondria of HeLa and MCF-7 cancer cell lines. These results demonstrate the efficacy of using modular structural modifications to access desired photophysical and biological properties of PSs.Experimental Section
Materials
All reagents were obtained from commercial sources and used without additional purification unless specified. 2,4-Dimethylpyrrole, (4-dimethylamino) benzaldehyde, trifluoroacetic acid and boron trifluoride diethyl etherate (BF3·Et2O) were purchased from Sigma Aldrich (USA). 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone was bought from Alfa Aesar (USA). N-Bromosuccinimide was obtained from TCI (Japan). Triethylamine (TEA), iodomethane, sodium hydrogen carbonate (NaHCO3), magnesium sulfate (MgSO4) were procured from Daejung Chemical (South Korea). Solvents were of analytical grade and were distilled prior to use. All compounds were characterized by 1H- and 13C-NMR spectroscopy on a Bruker AM 250 spectrometer (USA) and high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) on a SYNAPT G2-Si high-definition mass spectrometer (United Kingdom).Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) resulted to orange crystals. (See Fig. S5, ESI†) (Yield: 243 mg, 9.1%) 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.05 (d, 2H, J = 9 Hz; 2Ar-H), 6.76 (d, 2H, J = 9 Hz; 2Ar-H), 5.97 (s, 2H; 2Ar-H), 3.02 (s, 6H; 2CH3), 2.55 (s, 6H; 2CH3), 1.48 ppm (s, 6H; 2CH3).
:1) elution system.
:1) as elution solvents. (See Fig. S9, ESI†) (Yield: 1.3 g, 28.4%) 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 9.84 (s, 1H; Ar-H), 7.45 (s, 2H; 2Ar-H), 7.04 (s, 1H; Ar-H), 4.24 (m, 4H; 2CH2), 3.92 (m, 4H; 2CH2), 3.77 (m, 4H; 2CH2), 3.37 (s, 6H; 2CH3), 3.67 (m, 8H; 4CH2), 3.56 ppm (m, 4H; 2CH2).
:1) and CH2Cl2/MeOH (50:1) yielded the desired compounds.
:0.5) elution system afforded the final compounds.
:1206.65 [M-I] +; found: 1208.63.
:1284.56 [M-I] +; found: 1284.56.
:1362.47 [M-I]+; found: 1367.44.
:1) resulted to orange crystals. (See Fig. S5, ESI†) (Yield: 243 mg, 9.1%) 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 7.05 (d, 2H, J = 9 Hz; 2Ar-H), 6.76 (d, 2H, J = 9 Hz; 2Ar-H), 5.97 (s, 2H; 2Ar-H), 3.02 (s, 6H; 2CH3), 2.55 (s, 6H; 2CH3), 1.48 ppm (s, 6H; 2CH3).
:1) elution system.
:1) as elution solvents. (See Fig. S9, ESI†) (Yield: 1.3 g, 28.4%) 1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 9.84 (s, 1H; Ar-H), 7.45 (s, 2H; 2Ar-H), 7.04 (s, 1H; Ar-H), 4.24 (m, 4H; 2CH2), 3.92 (m, 4H; 2CH2), 3.77 (m, 4H; 2CH2), 3.37 (s, 6H; 2CH3), 3.67 (m, 8H; 4CH2), 3.56 ppm (m, 4H; 2CH2).
:1) and CH2Cl2/MeOH (50:1) yielded the desired compounds.
:0.5) elution system afforded the final compounds.
:1206.65 [M-I] +; found: 1208.63.
:1284.56 [M-I] +; found: 1284.56.
:1362.47 [M-I]+; found: 1367.44.
Methods
where m is the slope of the photodegradation curve and F is the correction factor for the difference in absorbance between sample and reference and is given as F = 1–10−A.
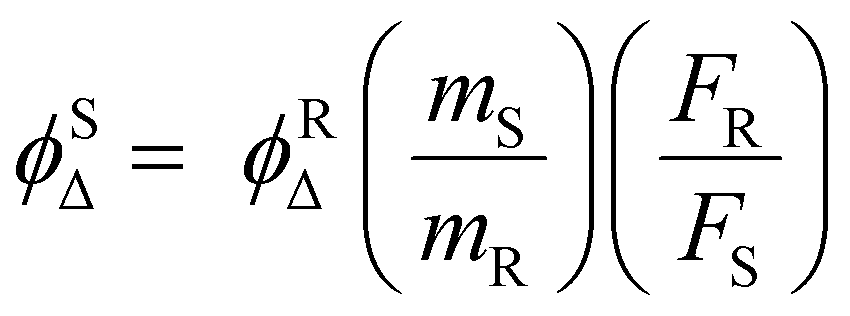
To further evaluate 1O2 generation efficiency in aqueous medium, we used water soluble 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) as alternative 1O2 quencher. Water solutions of BODIPY dyes with ABDA were prepared and experiments were conducted following the procedure described above. Higher illumination intensity was employed because, compared to DPBF, ABDA was less sensitive to singlet oxygen.64
Author information
Jaesung Yang – Department of Chemistry, Yonsei University, 1 Yonseidae-Gil, Wonju, Gangwon 26493, Korea; E-mail: jaesung.yang@yonsei.ac.kr; Ho-Joong Kim – Department of Chemistry, Chosun University, 309 Pilmun-daero, Dong-gu, Gwangju 61452, Korea; e-mail: hjkim@chosun.ac.kr; Joomin Lee – Department of Food and Nutrition, Chosun University, 309 Pilmun-daero, Dong-gu, Gwangju 61452, Korea; e-mail: joominlee@chosun.ac.kr; Isabel Wen Badon – Department of Chemistry, Chosun University, 309 Pilmun-daero, Dong-gu, Gwangju 61452, Korea; Chanwoo Kim – Department of Chemistry, Yonsei University, 1 Yonseidae-Gil, Wonju, Gangwon 26493, Korea; Jong Min Lim – Center for Molecular Spectroscopy and Dynamics, Institute for Basic Science (IBS), 145 Anam-ro, Seongbuk-gu, Seoul 02841, Korea; Duy Khuong Mai – Department of Chemistry, Chosun University, 309 Pilmun-daero, Dong-gu, Gwangju 61452, Korea; Temmy Pegarro Vales – Department of Natural Sciences, Caraga State University, KM7 NH1, Butuan City 8600, Philippines; Dongho Kang – Department of Chemistry, Yonsei University, 1 Yonseidae-Gil, Wonju, Gangwon 26493, Korea; Sung Cho – Department of Chemistry, Chonnam National University, 99 Daehak-ro, Yuseong-gu, Gwangju 61186, Korea;Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The work at Chosun University was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) (2020R1F1A1071784). The work at Yonsei University was supported by the NRF grant funded by the Korea government (MSIT) (No. 2019R1G1A109926913).References
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- M. Tarstedt, M. Gillstedt, A. M. Wennberg Larkö and J. Paoli, J. Eur. Acad. Dermatol. Venereol., 2016, 30, 420–423 CrossRef CAS PubMed.
- A. K. D'Cruz, M. H. Robinson and M. A. Biel, Head Neck, 2004, 26, 232–240 CrossRef PubMed.
- J. Usuda, S. Ichinose, T. Ishizumi, H. Hayashi, K. Ohtani, S. Maehara, S. Ono, H. Honda, N. Kajiwara, O. Uchida, H. Tsutsui, T. Ohira, H. Kato and N. Ikeda, Clin. Cancer Res., 2010, 16, 2198–2204 CrossRef CAS PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- B. W. Henderson and T. J. Dougherty, Photochem. Photobiol., 1992, 55, 145–157 CrossRef CAS PubMed.
- N. E. Aksakal, E. Tanrıverdi Eçik, H. H. Kazan, G. Yenilmez Çiftçi and F. Yuksel, Photochem. Photobiol. Sci., 2019, 18, 2012–2022 CrossRef CAS PubMed.
- E. Ruggiero, S. Alonso-deCastro, A. Habtemariam and L. Salassa, Dalton Trans., 2016, 45, 13012–13020 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- S. P. Singh and T. Gayathri, Eur. J. Org. Chem., 2014, 4689–4707 CrossRef CAS.
- J. Bañuelos, Chem. Rec., 2016, 16, 335–348 CrossRef PubMed.
- Y. H. Jung, D. Karthik, H. Lee, J. H. Maeng, K. J. Yang, S. Hwang and J. H. Kwon, ACS Appl. Mater. Interfaces, 2021, 13, 17882–17891 CrossRef CAS PubMed.
- Z. Ruan, Y. Zhao, P. Yuan, L. Liu, Y. Wang and L. Yan, J. Mater. Chem. B, 2018, 6, 753–762 RSC.
- Q. Wang, D. K. P. Ng and P.-C. Lo, J. Mater. Chem. B, 2018, 6, 3285–3296 RSC.
- H. Chen, Q. Bi, Y. Yao and N. Tan, J. Mater. Chem. B, 2018, 6, 4351–4359 RSC.
- P. Majumdar, X. Yuan, S. Li, B. Le Guennic, J. Ma, C. Zhang, D. Jacquemin and J. Zhao, J. Mater. Chem. B, 2014, 2, 2838–2854 RSC.
- D. Khuong Mai, B. Kang, T. Pegarro Vales, I. W. Badon, S. Cho, J. Lee, E. Kim and H.-J. Kim, Molecules, 2020, 25, 3340 CrossRef PubMed.
- V.-N. Nguyen, Y. Yim, S. Kim, B. Ryu, K. M. K. Swamy, G. Kim, N. Kwon, C.-Y. Kim, S. Park and J. Yoon, Angew. Chem., Int. Ed., 2020, 59, 8957–8962 CrossRef CAS PubMed.
- A. J. Sánchez-Arroyo, E. Palao, A. R. Agarrabeitia, M. J. Ortiz and D. García-Fresnadillo, Phys. Chem. Chem. Phys., 2017, 19, 69–72 RSC.
- M. Bröring, R. Krüger, S. Link, C. Kleeberg, S. Köhler, X. Xie, B. Ventura and L. Flamigni, Chemistry, 2008, 14, 2976–2983 CrossRef PubMed.
- Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC.
- T. Uppal, X. Hu, F. R. Fronczek, S. Maschek, P. Bobadova-Parvanova and M. G. H. Vicente, Chem. – Eur. J., 2012, 18, 3893–3905 CrossRef CAS PubMed.
- X.-D. Jiang, R. Gao, Y. Yue, G.-T. Sun and W. Zhao, Org. Biomol. Chem., 2012, 10, 6861–6865 RSC.
- T. Bura and R. Ziessel, Org. Lett., 2011, 13, 3072–3075 CrossRef CAS PubMed.
- S. Zhu, J. Zhang, G. Vegesna, F.-T. Luo, S. A. Green and H. Liu, Org. Lett., 2011, 13, 438–441 CrossRef CAS PubMed.
- D. G. Rudmann, Toxicol. Pathol., 2013, 41, 310–314 CrossRef PubMed.
- C. H. Wong, K. W. Siah and A. W. Lo, Biostatistics, 2018, 20, 273–286 CrossRef PubMed.
- A. Lin, C. J. Giuliano, A. Palladino, K. M. John, C. Abramowicz, M. L. Yuan, E. L. Sausville, D. A. Lukow, L. Liu, A. R. Chait, Z. C. Galluzzo, C. Tucker and J. M. Sheltzer, Sci. Transl. Med., 2019, 11, eaaw8412 CrossRef CAS PubMed.
- J. Neuzil, L. F. Dong, J. Rohlena, J. Truksa and S. J. Ralph, Mitochondrion, 2013, 13, 199–208 CrossRef CAS PubMed.
- T. A. Tabish and M. R. Hamblin, Biomater. Biosyst., 2021, 3, 100023 CrossRef.
- H. M. McBride, M. Neuspiel and S. Wasiak, Curr. Biol., 2006, 16, R551–560 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- F. Sivandzade, A. Bhalerao and L. Cucullo, Bio-Protoc., 2019, 9, e3128 Search PubMed.
- B. Sui, S. Tang, A. W. Woodward, B. Kim and K. D. Belfield, Eur. J. Org. Chem., 2016, 2851–2857 CrossRef CAS.
- T. Gao, H. He, R. Huang, M. Zheng, F.-F. Wang, Y.-J. Hu, F.-L. Jiang and Y. Liu, Dyes Pigm., 2017, 141, 530–535 CrossRef CAS.
- K. Kirakci, J. Zelenka, M. Rumlová, J. Cvačka, T. Ruml and K. Lang, Biomater. Sci., 2019, 7, 1386–1392 RSC.
- D. Chen, J. Zhang, Y. Tang, X. Huang, J. Shao, W. Si, J. Ji, Q. Zhang, W. Huang and X. Dong, J. Mater. Chem. B, 2018, 6, 4522–4530 RSC.
- Q. Hu, M. Gao, G. Feng and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 14225–14229 CrossRef CAS PubMed.
- J. Sun, X. Li, K. Du and F. Feng, Chem. Commun., 2018, 54, 9194–9197 RSC.
- Y. Zhang, L. Wang, Q. Rao, Y. Bu, T. Xu, X. Zhu, J. Zhang, Y. Tian and H. Zhou, Sens. Actuators, B, 2020, 321, 128460 CrossRef CAS.
- D. P. Murale, M. M. Haque, S. C. Hong, S.-Y. Jang, J. H. Lee, S. J. An and J.-S. Lee, Dyes Pigm., 2021, 196, 109830 CrossRef CAS.
- O. Karaman, T. Almammadov, M. Emre Gedik, G. Gunaydin, S. Kolemen and G. Gunbas, ChemMedChem, 2019, 14, 1879–1886 CrossRef CAS PubMed.
- H. Kang, Y. Si, Y. Liu, X. Zhang, W. Zhang, Y. Zhao, B. Yang, Y. Liu and Z. Liu, J. Phys. Chem. A, 2018, 122, 5574–5579 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- Z. Lou, Y. Hou, K. Chen, J. Zhao, S. Ji, F. Zhong, Y. Dede and B. Dick, J. Phys. Chem. C, 2018, 122, 185–193 CrossRef CAS.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS PubMed.
- H. Chong, E. Fron, Z. Liu, S. Boodts, J. Thomas, J. N. Harvey, J. Hofkens, W. Dehaen, M. Van der Auweraer and M. Smet, Chem. – Eur. J., 2017, 23, 4687–4699 CrossRef CAS PubMed.
- C. Dolan, A. Byrne, C. Long, K. Czamara, A. Kaczor, M. Baranska and T. E. Keyes, RSC Adv., 2017, 7, 43743–43754 RSC.
- Y.-L. Chen, S.-W. Li, Y. Chi, Y.-M. Cheng, S.-C. Pu, Y.-S. Yeh and P.-T. Chou, ChemPhysChem, 2005, 6, 2012–2017 CrossRef CAS PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- Y. Dong, P. Kumar, P. Maity, I. Kurganskii, S. Li, A. Elmali, J. Zhao, D. Escudero, H. Wu, A. Karatay, O. F. Mohammed and M. Fedin, Phys. Chem. Chem. Phys., 2021, 23, 8641–8652 RSC.
- Q. Lin and D. Buccella, J. Mater. Chem. B, 2018, 6, 7247–7256 RSC.
- N. Dorh, S. Zhu, K. B. Dhungana, R. Pati, F.-T. Luo, H. Liu and A. Tiwari, Sci. Rep., 2015, 5, 18337 CrossRef CAS PubMed.
- L. Biasutto, L. F. Dong, M. Zoratti and J. Neuzil, Mitochondrion, 2010, 10, 670–681 CrossRef CAS PubMed.
- G. Bagkos, K. Koufopoulos and C. Piperi, Med. Hypotheses, 2014, 83, 175–181 CrossRef CAS PubMed.
- H. He, D. W. Li, L. Y. Yang, L. Fu, X. J. Zhu, W. K. Wong, F. L. Jiang and Y. Liu, Sci. Rep., 2015, 5, 13543 CrossRef PubMed.
- R. Prieto-Montero, A. Prieto-Castañeda, R. Sola-Llano, A. R. Agarrabeitia, D. García-Fresnadillo, I. López-Arbeloa, A. Villanueva, M. J. Ortiz, S. de la Moya and V. Martínez-Martínez, Photochem. Photobiol., 2020, 96, 458–477 CrossRef CAS PubMed.
- M. L. Agazzi, M. B. Ballatore, E. Reynoso, E. D. Quiroga and E. N. Durantini, Eur. J. Med. Chem., 2017, 126, 110–121 CrossRef CAS PubMed.
- X. Shi, L. Tan, J. Xing, F. Cao, L. Chen, Z. Luo and Y. Wang, J. Appl. Polym. Sci., 2013, 128, 1995–2002 CAS.
- J. Zhang, M. Yang, C. Li, N. Dorh, F. Xie, F.-T. Luo, A. Tiwari and H. Liu, J. Mater. Chem. B, 2015, 3, 2173–2184 RSC.
- T. Papalia, G. Siracusano, I. Colao, A. Barattucci, M. C. Aversa, S. Serroni, G. Zappalà, S. Campagna, M. T. Sciortino, F. Puntoriero and P. Bonaccorsi, Dyes Pigm., 2014, 110, 67–71 CrossRef CAS.
- T. Entradas, S. Waldron and M. Volk, J. Photochem. Photobiol., B, 2020, 204, 111787 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02393d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |