
Bioinspired NO release coating enhances endothelial cells and inhibits smooth muscle cells
 a
Fan
Jia,
b
Zhi-da
Shen,
a
You-xiang
Wang,
b
Ke-feng
Ren,
*ab
Guo-sheng
Fu
*a
and
Jian
Ji
ab
a
Fan
Jia,
b
Zhi-da
Shen,
a
You-xiang
Wang,
b
Ke-feng
Ren,
*ab
Guo-sheng
Fu
*a
and
Jian
Ji
ab
Abstract
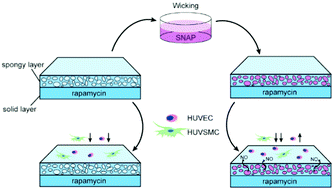
Thrombus and restenosis after stent implantation are the major complications because traditional drugs such as rapamycin delay the process of endothelialization. Nitric oxide (NO) is mainly produced by endothelial nitric oxide synthase (eNOS) on the membrane of endothelial cells (ECs) in the cardiovascular system and plays an important role in vasomotor function. It strongly inhibits the proliferation of smooth muscle cells (SMCs) and ameliorates endothelial function when ECs get hurt. Inspired by this, introducing NO to traditional stent coating may alleviate endothelial insufficiency caused by rapamycin. Here, we introduced SNAP as the NO donor, mimicking how NO affects in vivo, into rapamycin coating to alleviate endothelial damage while inhibiting SMC proliferation. Through wicking effects, SNAP was absorbed into a hierarchical coating that had an upper porous layer and a dense polymer layer with rapamycin at the bottom. Cells were cultured on the coatings, and it was observed that the injured ECs were restored while the growth of SMCs further diminished. Genome analysis was conducted to further clarify possible signaling pathways: the effect of cell growth attenuated by NO may cause by affecting cell cycle and enhancing inflammation. These findings supported the idea that introducing NO to traditional drug-eluting stents alleviates incomplete endothelialization and further inhibits the stenosis caused by the proliferation of SMCs.
- This article is part of the themed collection: Bioinspired Surfaces Engineering for Biomaterials
 Please wait while we load your content...
Please wait while we load your content...

