
Differential biomolecular recognition by synthetic vs. biologically-derived components in the stone-forming process using 3D microfluidics†
Eugenia
Awuah Boadi
 a,
Samuel
Shin
a,
Farai
Gombedza
a and
Bidhan C.
Bandyopadhyay
*abc
a,
Samuel
Shin
a,
Farai
Gombedza
a and
Bidhan C.
Bandyopadhyay
*abc
aCalcium Signaling Laboratory, 151 Research Service, DC Veterans Affairs Medical Center, 50 Irving Street, NW, Washington DC 20422, USA. E-mail: bidhan.bandyopadhyay@va.gov; Fax: +202-462-2006; Tel: +202-745-8622
bDivision of Renal Diseases & Hypertension, Department of Medicine, The George Washington University, Washington DC 20037, USA
cDepartment of Biomedical Engineering, The Catholic University of America, 620 Michigan Avenue NE, Washington DC 20064, USA
First published on 27th October 2021
Abstract
Calcium phosphate (CaP) biomineralization is the hallmark of extra-skeletal tissue calcification and renal calcium stones. Although such a multistep process starts with CaP crystal formation, the mechanism is still poorly understood due to the complexity of the in vivo system and the lack of a suitable approach to simulate a truly in vivo-like environment. Although endogenous proteins and lipids are engaged with CaP crystals in such a biological process of stone formation, most in vitro studies use synthetic materials that can display differential bioreactivity and molecular recognition by the cellular component. Here, we used our in vitro microfluidic (MF) tubular structure, which is the first completely cylindrical platform, with renal tubular cellular microenvironments closest to the functional human kidney tubule, to understand the precise role of biological components in this process. We systematically evaluated the contribution of synthetic and biological components in the stone-forming process in the presence of dynamic microenvironmental cues that originated due to cellular pathophysiology, which are critical for the nucleation, aggregation, and growth of CaP crystals. Our results show that crystal aggregation and growth were enhanced by immunoglobulin G (IgG), which was further inhibited by etidronic acid due to the chelation of extracellular Ca2+. Interestingly, biogenic CaP crystals from mice urine, when applied with cell debris and non-specific protein (bovine serum albumin), exhibited a more discrete crystal growth pattern, compared to exposure to synthetic CaP crystals under similar conditions. Furthermore, proteins found on those calcium crystals from mice urine produced discriminatory effects on crystal–protein attachment. Specifically, such biogenic crystals exhibited enhanced affinity to the proteins inherent to those crystals. More importantly, a physiological comparison of crystal induction in renal tubular cells revealed that biogenic crystals are less effective at producing a sustained rise in cytosolic Ca2+ compared to synthetic crystals, suggesting a milder detrimental effect to downstream signaling. Finally, synthetic crystal-internalized cells induced more oxidative stress, inflammation, and cellular damage compared to the biogenic crystal-internalized cells. Together, these results suggest that the intrinsic nature of biogenically derived components are appropriate to generate the molecular recognition needed for spatiotemporal effects and are critical towards understanding the process of kidney stone formation.
Introduction
Calcium crystal formation within the renal tubule is the first step towards calcium nephrolithiasis (CaNL).1 Such an event takes place within the hollow tubular structure, combining interactions between various intertwining factors including fluid flow and the cellular microenvironment.2,3 Although ionic supersaturation, nucleation, growth, and aggregation are sequential events to explain stone pathogenesis, a variety of theories have been presented.4,5 All these events, however, are influenced by the presence of endogenous modulators and/or inhibitors of crystallization. In the process of kidney stone formation, the aggregation of crystals and formation of organic matrix are the two main events, where the latter serves as a binding agent generated due to oxidative stress-induced cell death. Among the idiopathic stone formers, oxidative stress-induced tubular cell death has been reported due to crystal-induced cell injury and decreased production of functional crystallization inhibitors, both of which can create a comorbid condition, such as chronic kidney disease.6 Such an injury produces cellular debris containing proteins, lipids, polysaccharides, and other cell-derived materials,7–9 which promote the subsequent accumulation of crystals for attachment to specific internal structures and ultimately accelerate the stone-growth phase.3,10 In addition, proteins are an integral part of the kidney stone formation process as they help in stone matrix formation.11 Studies on calcium phosphate (CaP) crystal formation reveal that the biochemical factors such as crystal organic matrix proteins, and cellular debris contribute towards crystal growth by creating sites for microcrystals to adhere to each other, leading to crystal development and growth.4,5,12The main problem in understanding the mechanism of CaNL is that most studies have been conducted either through the analysis of extracted stones and tissues from stone patients13 or through in vitro two-dimensional (2D) analysis of crystal internalization using artificially synthesized calcium crystals.12,14,15 The downside of such studies is that the former approach does not delve into the real kinetic information of stone formation, while the 2D analysis in the latter does not truly mimic the biological kidney tubular microenvironment and the impact of the organic matrix was not considered. The absence of all these critical factors severely limits the capability of understanding the true mechanism underlying the biological process of CaNL. Therefore, extensive research conducted over the past few years has not been successful in elucidating the mechanism by which crystals nucleate, grow, and aggregate into stones.12,13 Moreover, differences between biogenic and synthetic hydroxyapatite crystals can considerably affect bioactivity and molecular recognition.16
Since CaNL is a multistep and multifactorial process, we put forward an in vivo-like approach capable of examining each step and/or in combination with various factors associated with stone formation to tackle the complexity of such biomineralization processes. Thus, in the present study, we utilized microfluidic (MF) channels, which we have proven to have the potency of stimulating biological tubular conditions to mimic biological kidney tubular microenvironment.12,17–19 While CaP crystals are proposed to form in the loops of Henle20,21 due to incoming supersaturated filtrate from the proximal tubule, it is important to understand the microenvironmental cues involving proximal tubular (PT) cells. Moreover, PT cells are mostly vulnerable to crystal-induced injury due to oxidative stress, which generates cell debris that can help in the downstream process of stone formation. Our goal is to examine the stone-forming process in the renal tubular structure using the “MF workbench” we built within the three-dimensional (3D) MF device that enables us to achieve previously unattainable precision and control (hydrodynamic flow/pressure) over the spatiotemporal biological conditions (e.g., biological macromolecules) to understand the role of tubular microenvironmental cues. Among the biological macromolecules, we used proteins such as immunoglobulin G (IgG), commonly found within the renal tubule,22 PT cell debris as the source of biogenic lipids, and urine crystals from mice. We established a monolayer within the 3D MF using the human PT cell line (HK2),12,17 evidenced by the expressions of tight junctions and basolateral marker proteins. Here, we show the specific recognition and differential responses by the biogenic calcium crystals when compared with the synthetic one on the physiological regulation through real-time spatiotemporal Ca2+ signals and the genetic mechanism that can considerably contribute towards our understanding of the tubular microenvironment in vivo during the process of stone formation.
Materials and methods
General materials
All the reagents were purchased from commercial sources and were of the analytical grade. Hank's buffer salt solution (HBSS) without Ca2+ or Mg2+, Dulbecco's modified Eagle medium (DMEM), fetal bovine serum (FBS), Fura-2-acetoxymethyl (Fura-2-AM) ester, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA, USA). Pyr6 (N-[4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl]-3-fluoro-4-pyridinecarboxamide), 2-APB (2-aminoethoxydiphenyl borate), thapsigargin (Tg), etidronic acid monohydrate (EtA), bovine serum albumin (BSA), tetraethyl orthosilicate solution, fibronectin, Alizarin Red (3,4-dihydroxy-9,10-dioxo-2-anthracenesulfonic acid sodium; AR), Nonidet P-40, sodium deoxycholate, sodium dodecyl sulfate, and protease inhibitor were purchased from Sigma-Aldrich (St. Louis, MO, USA). IgG-free BSA was purchased from Jackson Immuno Research Inc. (West Grove, PA, USA).Ca2+ and PO43− saturation and synthesis of CaP crystals
CaP-saturated solutions were prepared by mixing solutions of 2.4 mM calcium chloride (CaCl2), 0.9 mM disodium phosphate (Na2HPO4), and 5.8 mM monosodium phosphate (NaH2PO4) in 1× HBSS.14 Preformed CaP crystals were prepared by mixing solutions of 2.9 mM CaCl2, 6.0 mM disodium phosphate (Na2HPO4), and 4.0 mM monosodium phosphate (NaH2PO4). The preformed microcrystals were prepared by first centrifuging for 5 min (8500 × g) and then agitating them for 30 min at room temperature.Assembly and coating of MF device
The MF devices were assembled as previously described.12,17 Briefly, to simulate an in vivo tubular structure, cylindrically shaped 3D tubes with circular cross-section microchannels were molded with a polydimethylsiloxane (PDMS) elastomer kit (DOW Corning; Midland, MI, USA) using disposable micro-sampling pipettes (DOW Corning; Midland, MI, USA). The PDMS pre-polymer and crosslinker were mixed to form a PDMS precursor. The mixture was then cured in an oven at 80 °C for 20 min and a “Y-shaped” channel was fabricated using three pieces of glass pipettes embedded into the PDMS block. This sol–gel method was widely used for the design of bioactive materials such as the formation of artificial scaffolds for in vitro mammalian cell culture using polymer silicate macroreticular composites.23 We used tetraethyl orthosilicate at 120 °C as the coating material to prevent permeation and degradation,24 as well as for cells to optimally attach to the channels. The channels were then rinsed with deionized water. Thereafter, the devices were sterilized under UV light for 30 min and further coated with fibronectin (1 mg mL−1) for 3 h at 37 °C for cellular adhesion. Collectively, the MF devices were coated with tetraethyl orthosilicate and fibronectin to primarily facilitate the adhesion of cells.25 This coating also created a more stable surface layer, enhanced wettability, and generated a more stable electroosmotic flow rate.26Identification of CaP crystal
AR staining was performed to detect the CaP crystals12 by rinsing crystals with 1× HBSS (without Ca2+ or Mg2+) and then fixing them with 3% paraformaldehyde for 15 min at room temperature, followed by washing with 1× HBSS (without Ca2+ or Mg2+). The crystals were incubated in 2% AR solution (pH 4.3, 1× HBSS) for 30 min at 37 °C. The images were captured using a Zeiss AxioVision microscope. The crystal formation index of the stained crystals were quantified using ImageJ software27,28 by measuring the areas of stained-crystal aggregate sizes; further, sizes less than 10 μm2 were excluded. CaP crystals were further confirmed by negative birefringence with a quantitative polarized light microscopy (qPLM) system (Meiji Techno America, model MT9930).29CaP induction in 3D MF device (non-cellular model)
Crystal formations in the presence or absence of BSA with IgG (1 mg mL−1), IgG-free BSA (1 mg mL−1), or EtA (10 μM) were performed in a 3D MF device. IgG-free BSA, BSA with IgG, or BSA with EtA solutions were prepared in 1× HBSS solution without Ca2+ or Mg2+. CaP-saturated solution was prepared as described above. The CaP solution alone was delivered into the first inlet, while IgG-free BSA, BSA with IgG, or BSA with EtA solutions were delivered into the second inlet, simultaneously using a different syringe pumps. Crystals formed within MF were stained in 2% AR solution (pH 4.3) and the images were captured using the Zeiss AxioVision microscope. The crystal growth index was quantified using ImageJ software as described above.Cell culture and injection of cells into MF device
The human kidney cell line (HK2; Lonza, Walkersville, MD, USA) was cultured in DMEM supplemented with 10% FBS, 2 mM glutamine, and 1% penicillin and streptomycin at 37 °C in 5% CO2. HK2 cells were directly seeded at 1× 106 cells per mL into the channel using a 25-gauge syringe pump. The device was placed into a CO2 incubator at 37 °C for 30 min to allow the cells to attach to the fibronectin-coated MF channel surface. The device was flipped on its opposite side and the cells were reseeded to ensure the entire surface of the MF channel was seeded with cells such that it mimicked the tubular structure. The cells were kept in the CO2 incubator at 37 °C with a continuous flow of supplemented media and allowed to grow within the microchannels (MF devices) until the tubular structure was achieved.Mice urine collection, urine crystal isolation, and urine protein elution from crystal
We used a common inbred strain of laboratory mouse, namely, C57BL/6, most widely used for genetically modified mice. All the mice obtained were from the Jackson Laboratory (Bar Harbor, ME, USA), which were maintained in a 12 h dark/12 h light cycle with food and water ad libitum. To induce the formation of calcium crystals in urine, these mice were treated with drinking water containing high calcium (2% CaG in 2% w/v sucrose) and/or with diuretics (0.08% acetazolamide) for 4–5 weeks.27,30 The animal protocol used in this study was designed according to the Guiding Principles in the Care and Use of Laboratory Animals (mice). All the animal procedures were performed in accordance with the recommendations for the care and use of laboratory animals as recommended by The National Institutes of Health (NIH), U.S. Department of Agriculture (USDA), and The Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Upon the completion of treatment, 24 h urine samples were collected from these mice and briefly centrifuged to obtain urine crystals and macromolecules.27,30 PT cells were isolated from the kidney cortex of these mice and then cultured for a few days as described earlier.31Biogenic crystal isolation
Biologically synthesized crystals were isolated by centrifuging mice urine and the crystals were collected from wild-type and TRPC3−/− mice as previously described.31 The crystals were solubilized with three sequential washes with HBSS. The crystals became biogenic crystals with the protein attached. To obtain biogenically synthesized crystals without the protein attached, the samples were briefly washed with RIPA buffer [150 mM NaCl2; 1% Nonidet P-40; 0.5% sodium deoxycholate; 0.1% sodium dodecyl sulfate; 50 mM Tris (pH 7.4)] containing the protease inhibitor and centrifuged to remove the crystal proteins that are attached to these crystals as previously described.11 These crystals became biogenic crystals only. Protein analyses were performed using the recovered urine macromolecules with wild-type mice PT cells as the positive control. The protein gels were characterized by staining the samples using Invitrogen SimplyBlue SafeStain (Invitrogen; Carlsbad, CA, USA) with the protocol as described by the manufacturer.Crystal induction into the 3D cellular model
Cells in the MF devices were rinsed with 1× HBSS without phenol red, Ca2+, or Mg2+, and were used to examine the effect of crystal formation by continuously delivering 1× HBSS with 2 syringe pumps to access the luminal side via the inlet tubing at a flow rate of 0.1 mL h−1 to prevent cell detachment by shear force. The appropriate crystal conditions (CaP or biogenic crystals) were delivered into the device via another inlet. A strainer screen was used to measure the number of crystals used for the MF device and were filtered to approximately 5–8 μM in size.32 In our experience, the applied crystals reduced in size when exposed to the DMEM culture media, which makes it easier for entry into the cells.14 The PT cells were lysed by glass homogenization in a buffer (1× HBSS, pH 7.4 with a protease inhibitor) and then centrifuged at 8500 × g for 7 min at 4 °C to obtain the cellular particles (debris). In the experimental setups involving the addition of cell debris and BSA (1 mg mL−1; ±IgG), the initial and final concentrations as well as pH of Ca2+ and PO43− (synthetic crystals) in the saturated CaP solutions injected into the channels and coming out of the combined tubes were measured using a Randox Calcium Assay Kit (Randox Laboratories; cat. #CA2390) and a QuantiChrom™ Phosphate Assay Kit (BioAssay Systems), respectively. Although the addition of cell debris and BSA (±IgG) altered the concentrations of saturated solutions, it did not impact the pH of the saturated crystal solutions and therefore did not alter crystal formation.Intracellular [Ca2+] measurements of HK2 cells using Fura-2 fluorescence
Ratiometric (340/380) [Ca2+]i measurements were performed as previously described.33,34 The control and the treated cells were loaded with a fluorescence Ca2+ probe (Fura-2-AM) at 37 °C for 30 min in an air–gas mixture (95% air + 5% CO2) environment. After the loaded HK2 cells were placed on an IX81 motorized inverted microscope equipped with an IX2-UCB control box (Olympus USA; Center Valley, PA, USA). For time-lapse fluorescence/ratiometric measurements, images from the IX81 microscope were fed into a C9100-02 electron-multiplier CCD camera with an A3472-07 AC adaptor (Hamamatsu, Bridgewater, NJ, USA). A Lambda-LS xenon arc lamp with a 10–2 optical filter changer (Sutter Inst.; Novato, CA, USA) was used as an illuminator, capable of a light output ranging from 340 to 380![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm with a cutoff of 700 nm. The cells were immersed in a Ca2+-free external solution and then placed in a microincubator set at 37 °C with a gas mixture of 5% CO2/95% air. Fura-2 measurements were recorded using digital microscopy imaging software (SlideBook version 5.0, 3i, Intelligent Imaging Innovations; Denver, CO, USA). Fura-2 fluorescence intensities were recorded at an emission peak absorbance at a wavelength of 500 nm with excitation peak absorbance that alternated between wavelengths of 340 and 380nm. Further, 50–150 cells were individually selected as regions of interest (background fluorescence was automatically subtracted prior to 340/380 ratio calculation and graphing). Time-lapsed measurements were set to 900–1200 time points at 1 s intervals. The capture analyses were performed offline using Slidebook™ software and further quantitated with a statistical analysis using Origin 6.1.
nm with a cutoff of 700 nm. The cells were immersed in a Ca2+-free external solution and then placed in a microincubator set at 37 °C with a gas mixture of 5% CO2/95% air. Fura-2 measurements were recorded using digital microscopy imaging software (SlideBook version 5.0, 3i, Intelligent Imaging Innovations; Denver, CO, USA). Fura-2 fluorescence intensities were recorded at an emission peak absorbance at a wavelength of 500 nm with excitation peak absorbance that alternated between wavelengths of 340 and 380nm. Further, 50–150 cells were individually selected as regions of interest (background fluorescence was automatically subtracted prior to 340/380 ratio calculation and graphing). Time-lapsed measurements were set to 900–1200 time points at 1 s intervals. The capture analyses were performed offline using Slidebook™ software and further quantitated with a statistical analysis using Origin 6.1.
Lactate dehydrogenase (LDH) assay
LDH release in the media of treated HK2 cells was measured using a Pierce LDH cytotoxicity assay kit (Thermo Fisher Scientific) as described earlier.14 Following the internalization of crystals [control, synthetic (CaP) or biogenic (urine) crystals] within the HK2 cells, media containing HK2 cells were transferred to a 96-well plate for LDH release measurements. The reactions were done by the subtraction of absorbance at 680 nm (background) from the absorbance at 490 nm.RNA extraction and RT-PCR
Total RNAs were isolated from cells cultured in MF devices using TRIzol as previously described.12 Subsequently, DNAse treatments were performed, and the RNA concentrations were measured using a nanodrop spectrophotometer. Thereafter, reverse transcription of RNA into the cDNA was performed using a cDNA synthesis kit (Promega Madison, WI, USA) to detect amplification using gene-specific primers (Table 1) purchased from Invitrogen and Integrated DNA Technologies (Coralville, IA, USA) using the master-mix PCR amplification reagent (Promega) as described earlier.31 The T100 Thermocycler (Bio-Rad, Hercules, CA, USA) settings for PCR were set to the conditions as previously described.31| Primer | Sequence (sense, antisense) | Product size (bp) |
|---|---|---|
| hBAX1 | 5′-ATG GAC GGG TCC GGG GAG-3′ | 455 |
| 5′-ATC CAG CCC AAC AGC CGC-3′ | ||
| hBCL2 | 5′-AAG CCG GCG ACG ACT TCT-3′ | 258 |
| 5′-GGT GCC GGT TCA GGT ACT CA-3′ | ||
| hCaspase3 | 5′-TTA ATA AAG GTA TCC ATG GAG AAC ACT-3′ | 849 |
| 5′-TTA GTG ATA AAA ATA GAG TTC TTT TGT GAG-3′ | ||
| hGAPDH | 5′- TCC CTG AGC TGA ACG GGA AG-3′ | 218 |
| 5′-GGA GGA GTG GGT GTC GCT GT-3′ | ||
| hIL-6 | 5′-ATG AAC TCC TTC TCC ACA AG-3′ | 626 |
| 5′-AGA GCC CTC AGG CTG GAC TG-3′ | ||
| hMCP1 | 5′-GAT CTC AGT GCA GAG GCT CG-3′ | 155 |
| 5′-TTT GCT TGT CCA GGT GGT CC-3′ | ||
| hNOX4 | 5′-CCG GCT GCA TCA GTC TTA ACC-3′ | 220 |
| 5′-TCG GCA CAG TAC AGG CAC AA-3′ | ||
| hP22 | 5′-ATG GGG CAG ATC GAG TGG GCC ATG-3′ | 588 |
| 5′-TCACACGACCTCATCTGTCACTGG-3′ | ||
| hTNFα | 5′-GGC GTG GAG CTG AGA GAT AAC-3′ | 120 |
| 5′-GGT GTG GGT GAG GAG CAC AT-3′ |
Statistical analysis
The results were reported as the mean ± standard error of mean (SEM). Data were analyzed in Origin 6.1 software using a two-tailed t-test. Significant difference levels were set at p < 0.05 (*) or p < 0.01 (**). Details of all the statistical analysis are mentioned in the figures and figure legends.Results
IgG enhances CaP crystal formation in in vivo-like 3D MFs
To investigate the effect of IgG on CaP crystal formation, we utilized our off-the-shelf “Y-shaped” MF devices to mimic the process of CaP stone formation in in vivo-like settings (Fig. 1A). Here, we applied a saturated solution of Ca2+ and PO43− (CaP) with either IgG-free BSA (CaP + BSA; Fig. 1B) or with BSA containing IgG (CaP + BSA + IgG; Fig. 1C) to examine whether IgG can increase the formation of CaP crystals. Notably, the measurements of the final saturation levels of Ca2+ and PO43− and the pH of the luminal solutions of the MF channel have not shown much change that can impact our experimental protocol (Fig. S1, ESI†). The resulting crystals were stained with CaP-specific AR stain (AR pH 4.3), which showed a significant enhancement in CaP crystal formation by IgG-containing BSA (BSA + IgG) compared with IgG-free BSA (Fig. 1B and C). Similarly, we sought to expand our knowledge on the effect of the inhibitor of crystallization using etidronic acid (EtA; 10 μM), a calcium chelator,35,36 in the presence of IgG-containing BSA as a promoter of crystallization. We propose that with time, proteins, lipids, and fatty acids bind with CaP as calcium biomineralization progresses, thereby decreasing the availability of Ca2+ for EtA, which leads to a decrease in crystal formation. As expected, the combined CaP + IgG with EtA resulted in reduced CaP crystal formation (Fig. 1D and E). Next, we used an MF device with cultured HK2 cells to mimic the in vivo renal tubular effect by lining the entire surface of the inner channel (as shown by propidium iodide staining; Fig. S2A, ESI†). We confirmed that the formation of HK2 cellular monolayer within the MF device through the formation of a tight junction (Zonula occludens-1 expression) and polarized-distribution basolateral membrane protein (Na+/K+ ATPase; Fig. S2B, ESI†). Our results revealed that the CaP crystal aggregates in the inner channel of the MF device (detected with AR pH 4.3) have significantly compounded with each addition to the CaP only (Fig. 1F), CaP + BSA (Fig. 1G), and CaP + BSA + IgG (Fig. 1H). CaP crystal formations were then mitigated with the addition of EtA (CaP + BSA + IgG + EtA; Fig. 1I and J), reflecting a congruent pattern of CaP crystals in the non-cellular condition (Fig. 1A–E). These results indicate that IgG as an endogenous protein contributes to CaP crystal formation in 3D-cultured renal epithelial cell environment, which depends on the rise of extracellular Ca2+ levels.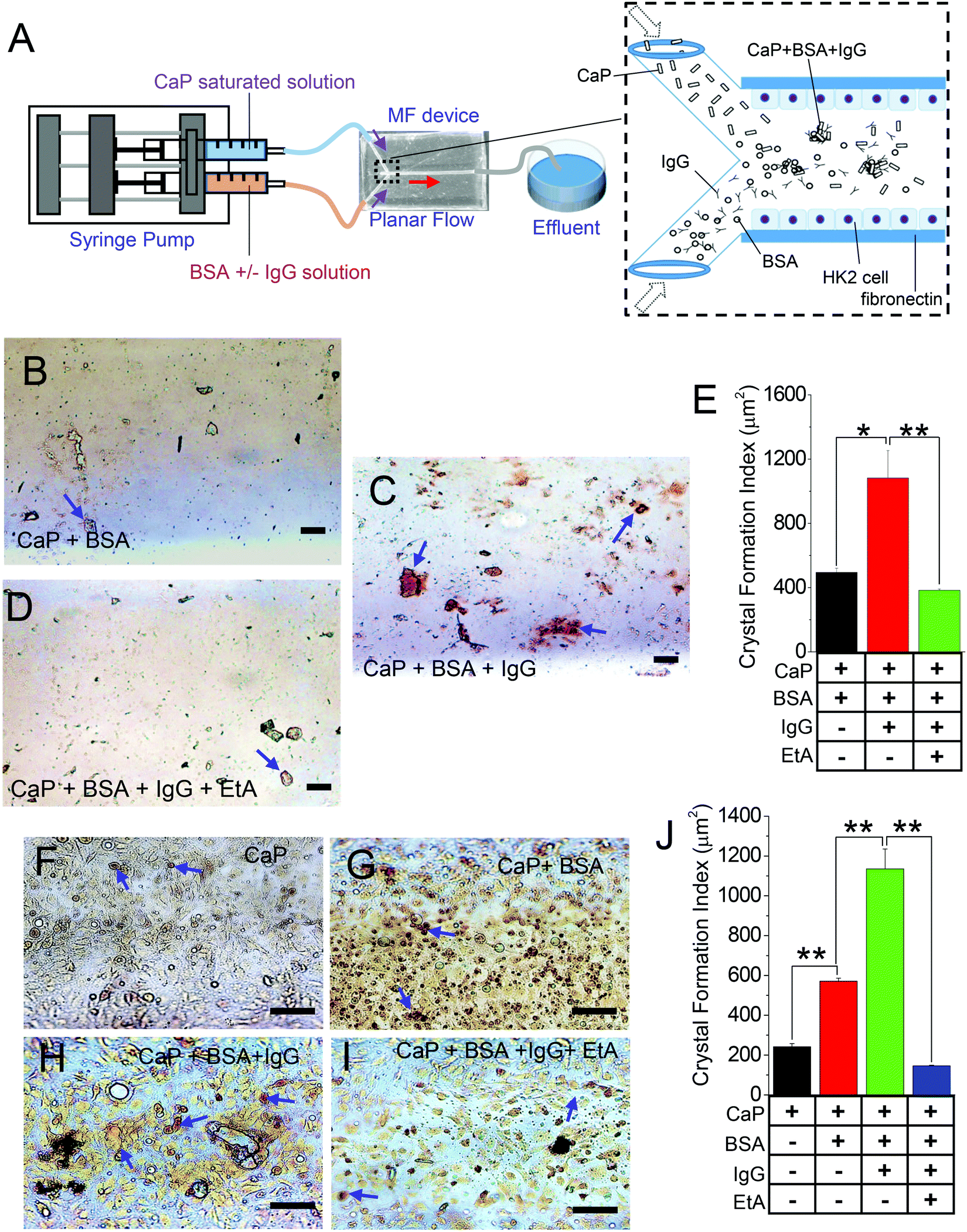 | ||
| Fig. 1 CaP crystal formation in presence of IgG in 3D MFs. (A) Schematic of the introduction of saturated CaP solution with 1× HBSS without Ca2+ or Mg2+ which was injected in one inlet, and BSA and/or IgG in 1× HBSS (without Ca2+ or Mg2+), or etidronic acid (EtA; 10 μM) which were injected in the other inlet of the 3D MF device using a syringe pump. CaP crystals were injected with (B) CaP + BSA, (C) CaP + BSA + IgG, and (D) CaP + BSA + IgG + EtA (a calcium chelator) in the MF device. The crystals were stained with alizarin red stain (AR; pH 4.3). The CaP crystals formed were visualized under a light microscope and analyzed as (E) bar graphs. Similar experiments were performed using cultured HK2 cells in the 3D MF device with (F) CaP crystals with 1× HBSS without Ca2+ or Mg2+ (CaP), (G) CaP crystals with IgG-free BSA (CaP + BSA), (H) CaP crystals with BSA containing IgG (CaP + BSA + IgG), and (I) CaP crystals with BSA containing IgG (CaP + BSA + IgG + EtA). 1 mg mL−1 BSA and IgG-free BSA were used. The crystals formed were stained with AR pH 4.3 and analyzed as (J) bar graphs. The blue arrows indicate AR pH 4.3 stained CaP crystals. Each experiment was performed n = 3 times. The bar graph quantifications are mean ± SEM; *, p < 0.05; **, p < 0.01. | ||
Cell-debris-induced rise in CaP crystal formation in cellular 3D MFs
Cell debris from apoptotic/necrotic cell death have been proposed to induce calcifications.37 Moreover, such fragments contain membranous materials that can be used as a source of biogenic lipids to augment crystal formation. Thus, to examine the effect of cell debris on CaP crystal formation, we applied cell debris (obtained from HK2 cells) alone (Fig. 2A), CaP crystals alone (Fig. 2B), and CaP with cell debris as mentioned above (Fig. 2C), all within the MF devices. The characterization and identification of CaP crystal aggregates using AR pH 4.3 stain showed larger and numerous CaP crystal formations when exposed to cell debris, indicating that cellular debris enhances CaP crystal nucleation, attachment, and growth (Fig. 2A–C). As shown with IgG, we show the effect of EtA as a calcium chelator on CaP crystal formation with the added presence of cell debris (Fig. 2D and E). EtA-inhibited CaP crystal formation within the CaP pre-incubated conditions (Fig. 2E), as seen previously with the presence of IgG (Fig. 1D); however, in comparison to the conditions of cell debris alone (Fig. 2A), more CaP formation was observed in the cell debris + CaP + EtA MF condition (Fig. 2D and E), although insignificant. | ||
| Fig. 2 Cellular debris enhances synthetic CaP crystal formation in the in vivo-like 3D MF device. (A) Cellular debris was incubated alone (cell debris), (B) CaP crystals were incubated alone (CaP), (C) CaP was incubated with cellular debris (cell debris + CaP), and (D) CaP was incubated with cellular debris and etidronic acid (10 μM) (cell debris + CaP + EtA) in the microfluidic device. All solutions were made in 1× HBSS (without Ca2+ or Mg2+). CaP crystals formed were stained with alizarin red (AR pH 4.3) and CaP crystals were visualized under a light microscope and analyzed as (E) bar graphs. The blue arrows point at the CaP crystals formed. Each experiment was performed n = 3 times. The bar graph quantifications are mean ± SEM; *, p < 0.05; **, p < 0.01. | ||
Synthetic and biogenic crystals differentially impact the formation of calcium crystals in cellular 3D MFs
Although we show that cellular debris can enhance CaP crystal formation within the in vivo-like 3D MF device, the properties and characterization of artificially formed calcium crystals would most likely deviate from calcium crystals created through biological means. It is possible that a crystal can be internalized by cells through the plasma membrane into the cytosolic space of the cells, which can alter the process of growth and aggregation of calcium crystals.12,38,39 In fact, PT cells have a greater capacity in crystal internalization than distal tubular cells due to the higher affinity of crystal binding.40 Moreover, the crystal size and shape determine the extent of internalization in various tubular cell types.41 However, most of those experiments utilized synthetic crystals and are not biogenic ones. We improved our mimetics in a 3D MF device by extracting biogenic crystals from our urine-crystal-forming mouse model (CaG treated; Fig. 3A) to show the contrasting effect between internalized CaP (Synthetic Crys.; Fig. 3B) and biogenic crystals (Bio. Crys.; Fig. 3C) into the HK2 cells cultured inside the 3D MF devices. We have confirmed crystal internalization through cytoplasmic-stained overlay images of different calcium crystals internalized in HK2 cells (Fig. S3, ESI†). Our data with confocal imaging and bright-field images of the crystals suggest that most of the synthetic CaP crystals and biogenic crystals, morphologically, were brushite42 (characterized by clear-cut platy shapes and/or finer crystallites; Fig. S3A, B and S4, ESI†). In addition, certain biogenic crystals were larger crystals with apatite-like formations42 (Fig. 3A and Fig. S4, ESI†). Moreover, the birefringence properties of different synthetic and biogenic crystals revealed that the urine crystals obtained from mice are mostly CaP crystals (Fig. S4A and B, ESI†). Additionally, BSA containing IgG was added to examine whether such proteins would increase the biogenic crystal formation (Bio. Crys. + BSA + IgG; Fig. 3D) or biogenic crystal with cell debris (Bio. Crys. + cell debris + BSA + IgG; Fig. 3E). Overall, synthetically derived CaP crystals with cell debris have significantly higher augmentations compared with that of biogenically derived crystals with cell debris (Fig. 3F). Notably, the addition of BSA containing IgG further enhanced the formation of biogenic crystals to greater effect, although not to the extent of synthetic crystals with cell debris (Fig. 3B–E). Overall, these results show that while BSA containing IgG and cell debris can augment the aggregation of biogenically derived crystals, their potential growth falls short compared with synthetically derived CaP crystals.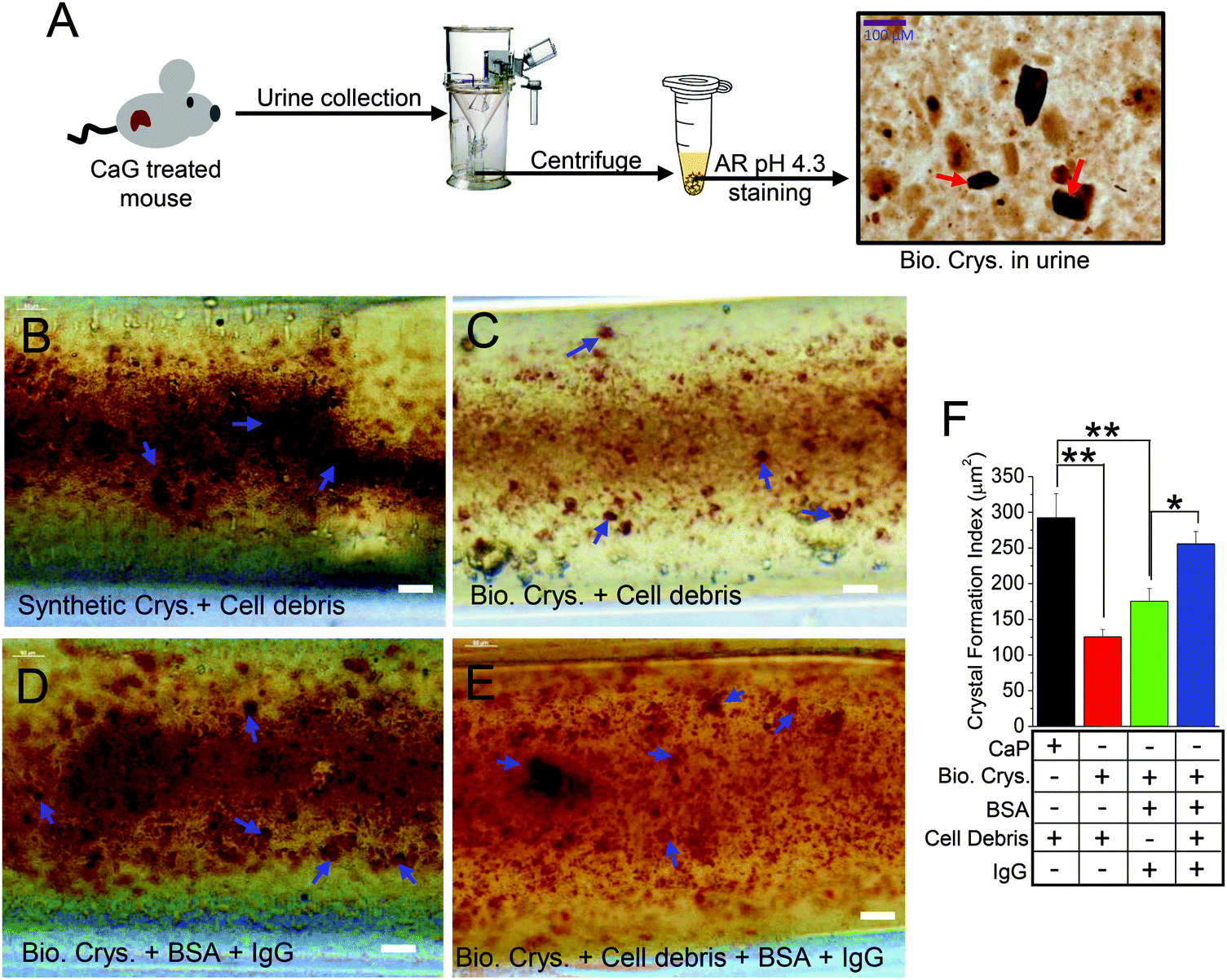 | ||
| Fig. 3 Cellular debris augments biogenic crystal formation within the in vivo-like 3D MF device. (A) Schematic of AR pH 4.3 staining of biological crystals from calcium gluconate-treated TRPC3−/− mouse urine (red arrows). Crystal internalization of HK2 cells with artificially synthetized crystals (calcium phosphate) or biogenically synthesized crystals with crystal protein matrix attached showed the impact of cellular debris and proteins on crystal aggregation and growth. (B) HK2 cells internalized with CaP were incubated with cellular debris (synthetic Crys. + cell debris), (C) HK2 cells internalized with biogenic crystals incubated with cellular debris (Bio. Crys. + cell debris), (D) HK2 cells internalized with biogenic crystals incubated with BSA containing IgG (Bio. Crys. + BSA + IgG), and (E) HK2 cells internalized with biogenic crystals incubated with cellular debris and BSA containing IgG (Bio. Crys. + cell debris + BSA + IgG). All the experiments were performed using MF devices. CaP and the calcium crystal aggregates formed were stained with alizarin red (AR pH 4.3) and visualized under a light microscope and analyzed as (F) bar graphs. Each experiment was performed n = 3 times. The bar graph quantifications are mean ± SEM; *, p < 0.05; **, p < 0.01. | ||
Matrix proteins in presence of BSA with IgG augment the growth and aggregation of biogenic crystals
Next, to provide corroborative evidence on the effect of stone matrix proteins (Mprotein) attached to those biogenic (urine) crystals, we isolated the proteins released from the biogenic crystals and found some distinct isolated band patterns compared with the proteins in crude extract of wild-type PT cells (Fig. 4A and B). Subsequently, we used those extracts to check whether those proteins can influence the growth and aggregation of biogenic crystals. We cultured HK2 cells in the MF devices forming a monolayer and then applied with biogenic crystals alone (Bio. Crys.; Fig. 4C), biogenic crystals with Mproteins alone (Bio. Crys. + Mprotein; Fig. 4D), biogenic crystals without proteins plus BSA containing IgG (Bio. Crys. + BSA; Fig. 4E), and biogenic crystals with Mproteins and BSA containing IgG (Bio. Crys. + Mprotein + BSA; Fig. 4F). Our results showed that biogenic crystals added with crystal protein induced more crystal internalization, which may be associated with increased cellular damage.14 We observed that such biogenic crystals with Mprotein enhanced crystal aggregation and growth compared with biogenic crystals alone. Significantly, the addition of BSA containing IgG aggravated the process of crystal growth under conditions of both biogenic crystal alone and biogenic crystal with protein (Fig. 4G). Together, these results suggest that although the Mprotein with biogenic crystals relatively increased the number of crystals, it needed some bulk proteins for further aggregation and crystal growth.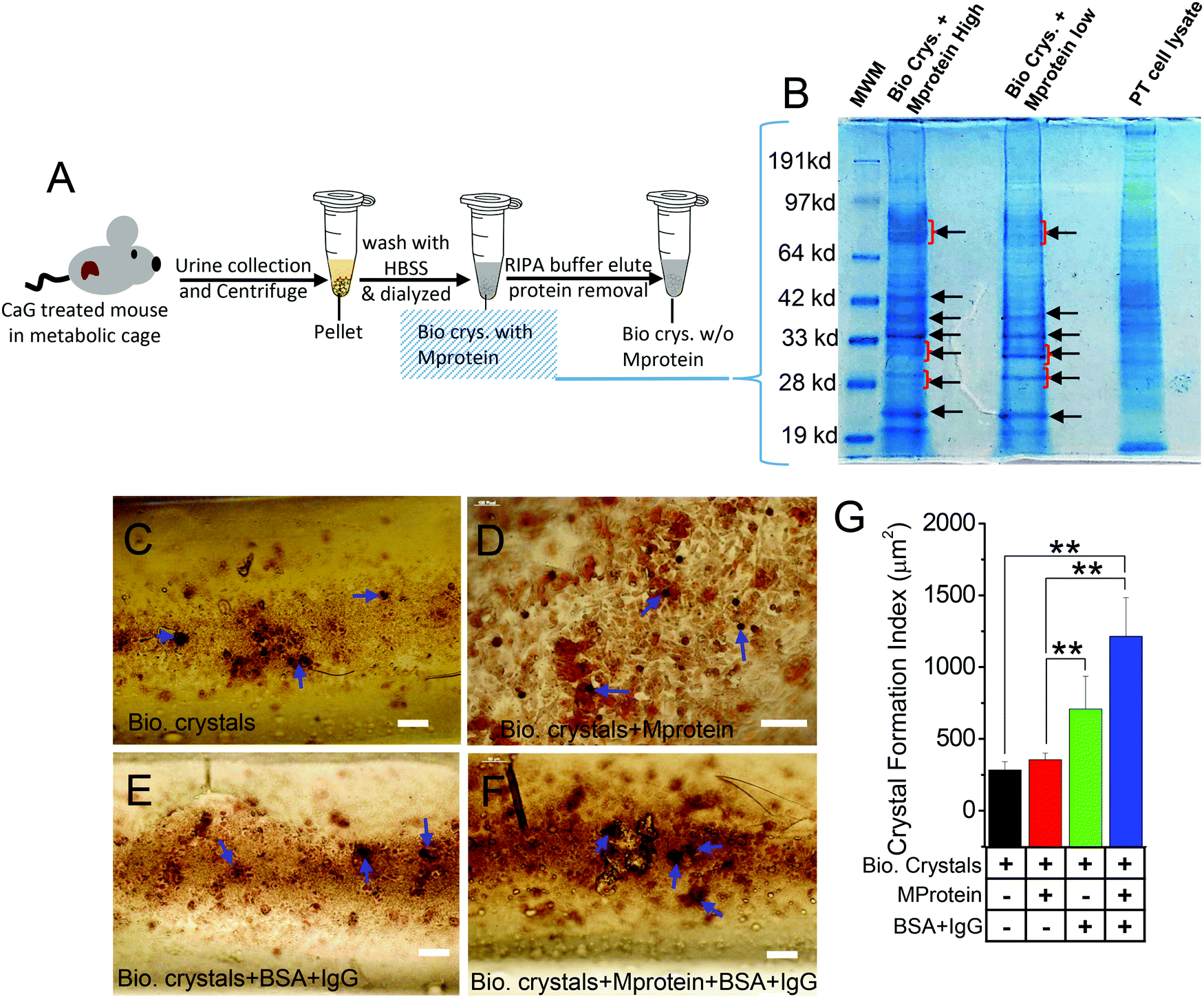 | ||
| Fig. 4 Differential stone-forming activity in response to crystal organic matrix proteins. (A) Synthesis and purification of biogenic crystals with or without protein from TRPC3−/− mice urine. (B) Protein characterization assays performed on biogenic crystal lysate with mice proximal tubule as the positive control. The black arrows show similarities in protein separation between high and low stone protein samples as well as identified stone matrix proteins (albumin, osteopontin, and apolipoprotein D) using the protein molecular weight marker (MWM) as the reference. (C) HK2 cells internalized with biogenic crystals with no organic matrix (Mprotein) attached (Bio. Crys.), (D) HK2 cells internalized with biogenic crystals with organic matrix (protein) attached (Bio. Crys. + Mprotein), (E) HK2 cells internalized with biogenic crystals with no organic matrix (protein) attached and further incubated with BSA containing IgG (Bio. Crys. + BSA + IgG), and (F) HK2 cells internalized with biogenic crystals with the organic matrix (Mprotein) attached and then incubated with BSA containing IgG (Bio. Crys. + Mprotein + BSA + IgG). (C–F) Experiments were performed using MF devices. The calcium phosphate-containing crystal aggregates formed were stained with alizarin red (AR pH 4.3) and visualized under a light microscope and analyzed as (G) bar graphs. The blue arrows indicate CaP-stained crystals. Each experiment was performed n = 3 times. The bar graph quantifications are mean ± SEM; *, p < 0.05; **, p < 0.01. The red arrows indicate crystal-internalized HK2 cells. | ||
Synthetic crystals induce more [Ca2+]i rise than biogenic crystals
Elevated levels of synthetic calcium crystals have been correlated with greater fibrosis, inflammation, and apoptosis due to Ca2+ entry induced by endoplasmic reticulum Ca2+-store depletion.14,43 We used such store-operated Ca2+ entry (SOCE) as an index of detrimental effect in crystal-internalized HK2 cells compared with the control by measuring the intracellular Ca2+ concentration ([Ca2+]i).14 To determine whether the biogenic crystals would similarly disrupt the Ca2+-signaling mechanism, we compared the [Ca2+]i mobilization due to SOCE, induced by ER-store depletion agent, namely, Tg,44 in HK2 cells (Fig. 5A–C). Here, we used biogenically produced crystals (Bio. Crys.; Fig. 5B) or artificially synthesized CaP crystals (Synthetic Crys.; Fig. 5C). As expected, the cells internalized with synthetic crystals demonstrated greater Ca2+ rise with prolonged Ca2+ entry (as shown with delayed normalization) compared with the control group (Fig. 5A–F). Notably, the rise in Ca2+ levels were insignificant in cells internalized with biogenic crystals compared with the control, while still depicting sustained Ca2+ entry (Fig. 5E and F). Prior to this experiment, when we treated those cells with an SOCE inhibitor, namely, Pyr6,45 the intracellular Ca2+ levels were significantly decreased for each group, confirming that SOCE inhibition can mitigate the effect of [Ca2+]i rise (Fig. 5A–C). Interestingly, the release of [Ca2+]i from the ER store in response to both biological and synthetic crystals were completely inhibited by Pyr6, signifying that much of the Ca2+ signaling of those crystal-internalized HK2 cells were mediated by SOCE. Since we have seen that such detrimental Ca2+ signaling can produce cellular-cytotoxicity-induced release of LDH,14 we further assessed LDH release by those cells. Our results show that biogenic crystals exhibit less cytotoxicity compared with synthetic crystals (Fig. 5G). Notably, higher [Ca2+]i rise due to synthetic CaP crystals revealed greater plasma membrane damage (LDH release) compared to those with biogenic crystals; therefore, biogenic crystals are not as potent in aggravating the stone-forming process.14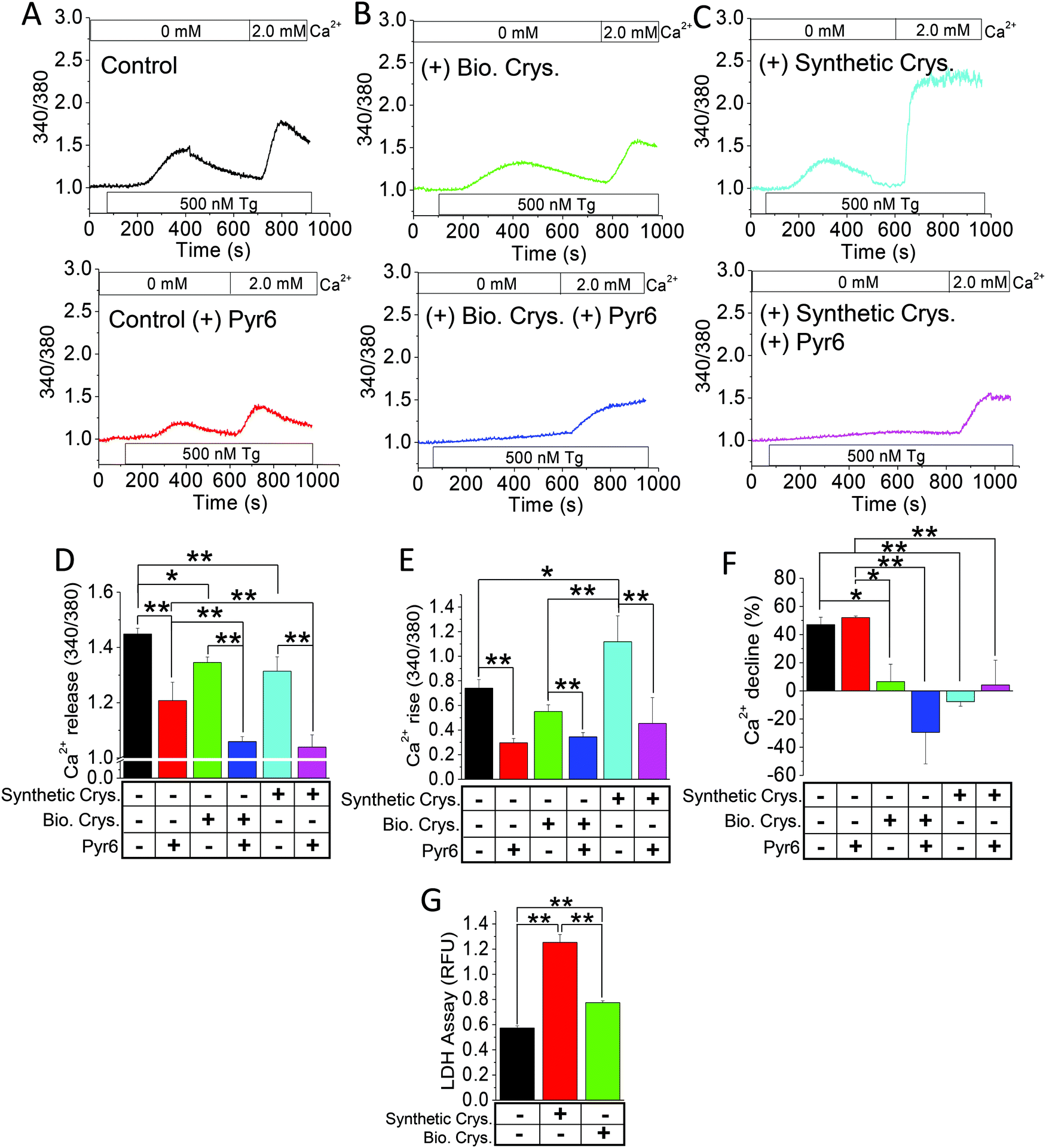 | ||
| Fig. 5 Synthetic CaP crystals induce more [Ca2+]i rise than biogenic crystals. Ratiometric fura-2 [Ca2+]i measurements were performed on HK2 cells internalized with (A) control, (B) with biogenic crystals (+Bio. Crys.), or (C) with synthetic calcium phosphate (+Synthetic Crys.) for 24 h, within a Ca2+-free cell bath. Thapsigargin (Tg) was applied to the cells with or without Pyr6 (3 μM) followed by a 2 mM Ca2+ replenishment. Bar diagrams of (D) Ca2+ release, (E) Ca2+ rise, and (F) Ca2+ decline for the Fura-2 trace graphs in A–C were assessed. (G) LDH release was assessed in the external media of HK2 cells internalized with synthetic crystals (Synthetic Crys.) or biogenic crystals (Bio. Crys.) for 24 h. Each experiment was performed n = 4 times. The bar graphs quantifications are mean + SEM for D–G; *, p < 0.05; **, p < 0.01. | ||
Discriminatory effect by synthetic vs. biogenic crystals in inducing cellular damage
Many studies have shown that the internalization of chemically synthesized CaP crystals are detrimental to produce cellular damage in PT cells,14,41,46 which are proposed to impact calcium-stone formation.14,47 To explore whether the biogenically derived crystals can produce similar genetic regulation toward cellular damage as the chemically synthesized crystals, we applied HK2 cells with CaP (Synthetic Crys.) crystals, biogenic crystals (Bio. Crys.), and biogenic crystals with Mprotein (Bio. Crys. + Mprotein). The use of biologically derived crystals was to further provide insights into calcium crystal damage and to significantly mimic the tubular microenvironment of PT. We performed the gene expression analysis on the cells internalized with these various crystals, namely, CaP, biogenic crystals, and biogenic crystals with Mprotein, in comparison to a control (non-crystal condition). Our results show the differential effect of crystal internalization on oxidative stress, inflammation, and apoptosis through gene expression profiles of known oxidative stress genes (P22 phox, and NOX4); (Fig. 6A), inflammatory genes (MCP1, IL-6, and TNFα); (Fig. 6B), and apoptotic genes (BAX/BCL2 ratio and caspase 3); (Fig. 6C). As expected, we found that all the crystal-internalized cells expressed genes that indicates more cellular damage compared to the control condition (Fig. 6A–C). In fact, artificially synthesized CaP crystals proved to induce more oxidative-stress-related apoptosis than biogenic crystals, as evident from the NOX4, MCP1, and caspase 3 expressions (Fig. 6A and C). Interestingly, biogenic crystals with Mprotein induced more oxidative damage (P22 and BAX/BCL2 ratio) and inflammation (IL-6 and TNFα) at a similar level, as evident from the artificially synthesized CaP conditions (Fig. 6A–C). Overall, these results provide significant evidence that the synthetic crystals can generate greater cellular damage to produce confounding pathological effects, whereas the biogenic crystals produce more inflammatory response rather than oxidative-stress-induced damage.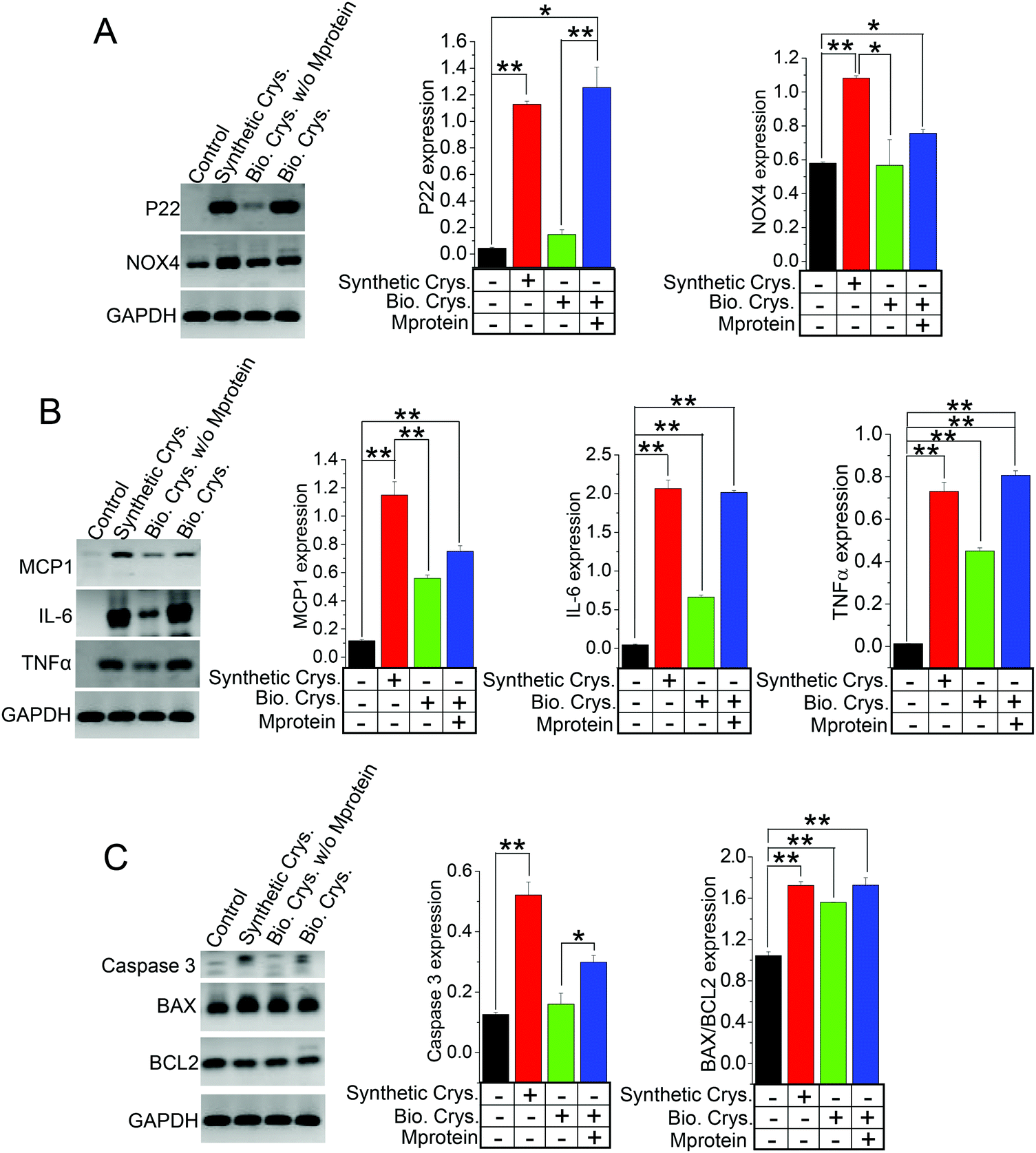 | ||
| Fig. 6 Crystal organic matrix (proteins) accelerate calcium crystal cellular damage. We show evidence of cellular damage through the gene expression analysis of HK2 cells internalized with artificially synthetized crystals (CaP) or biogenic calcium crystals with or without an organic matrix (protein). (A) Oxidative stress genes P22 phox and NOX4; (B) inflammatory genes MCP1, IL-6, and TNFα; and (C) apoptotic genes caspase 3 and BAX/BCL2. The results obtained show that the presence of an organic matrix (Mprotein) significantly induces more cellular damage by biogenic crystals. (A–C) Bar graphs are mean ± SEM; *, p < 0.05; **, p < 0.01 for three different experiments. All gene expressions are normalized to GAPDH. | ||
Discussion
In this study, our effort was directed to elucidate the mechanism by which biogenic calcium crystals discriminate between artificial crystals via biomolecular recognition. Such interactions occur in between receptor–ligand, antigen–antibody, DNA–protein, peptide–protein, small molecule–protein/peptide, etc. In particular, surface interactions that takes place during the process of stone formation from crystal nucleation, growth, and aggregation into stones are the specific chemical interaction between endogenous proteins and crystal surface. The immunoglobulins specifically bind to the crystal surface to form complex structures, which allow other sticky molecules such as lipids to bind and aggregate through weak and non-covalent interactions. We took advantage of our in vivo-like approach to examine each step by adding/subtracting the factors associated with the stone-forming process to understand the contrast between biological vs. artificial materials and tackle the complexity in the process of calcium biomineralization. Such in vivo-like MF channels are a proven system of having the potency of stimulating biological tubular conditions to mimic biological kidney tubular microenvironment.12 Furthermore, wide-ranging studies including those that we conducted previously have shown that calcium crystals (CaOx/CaP) can be internalized by the renal tubular cells, which may be crucial in the onset and development of kidney-stone formation.12,14,36Among the several factors that act as modulators/inhibitors of calcium crystal formation,12,36 proteins have been implicated as attachment sites for crystal formation, promoting the aggregation of crystals into larger multicomponent aggregates; therefore, they can strongly influence crystal binding and growth.3 In fact, upon the analysis of human kidney stones, many proteins have been found as an integral part of the core complex.2 We tested the contribution of immunoglobulins (IgG) for promoting CaP crystal formation. Significantly, such crystal formation was augmented in the presence of IgG when compared among CaP alone, CaP with BSA containing IgG, and BSA–IgG-free conditions. A broad range of molecules have been shown to act as accelerators (e.g., lipids and fatty acids) or inhibitors (e.g., calgranulin and chelators) with regard to stone formation in animal models and/or clinical settings.2,48,49 Our efforts were directed toward finding the discriminating patterns of these molecules involved in the stone-forming process, alone or in combination with other factors (e.g., cell injury). We propose that the secretion of proteins, lipids, etc. varies in time and space as the biomineralization progress, where CaP formation influenced by EtA—a fatty acid oxidizer—inhibited crystal formation. Combining CaP + BSA with EtA resulted in reduced CaP crystal formation, indicating that exogenous chemicals influence fatty acids to create microenvironmental changes to reduce stone formation. Although the effect of Ca2+ chelation by EtA may have played a role, other pathways to demonstrate such a process would, therefore, be a better approach, which has been planned for our future experiments. Our engineered 3D cellular microchannels are not only able to transport water, ions, and biomolecules but can also show real-time crystal growth and aggregation toward stone formation.12,17 Thus, to better mimic this in vivo condition that involves cellular lipids, we used cellular debris obtained from damaged PT cells. In support, another study detailing the ultrastructural analysis of kidney stones revealed cell debris embedded in calcium crystals.11,50 Our findings corroborated with these studies as we have shown here that cell debris would enhance CaP crystal formation by creating sites for microcrystals to adhere to, leading to crystal development and growth. It is possible that cell debris is likely generated by PT damage resulting from other in vivo conditions such as elevated tubular [Ca2+] and/or renal tubular acidosis.
From prior studies, stone matrix, which is predominantly composed of proteins, has been shown to enrich within the calcium stone.11 Importantly, the analysis of such matrix proteins from urine crystals has been crucial in distinguishing stone formers from non-stone formers.51 Characterization assays have shown similarities between protein separations released from biogenic crystals (samples with high and low urine protein) that is contrasted those from the PT cell protein separations. Based on the separation patterns, three of the most abundant urine proteins that have also been identified as a part of the stone matrix, namely, albumin (67 kD), osteopontin (31.56 kD), and apolipoprotein-D (29 kD) (20%, 3.1% and 2.4%, respectively, for urine protein; 6%, 3%, and 11%, respectively, for stone-matrix composition) were deduced based on band separations from the stone-protein samples (Fig. 4B).11 Comparative studies of internalizing HK2 cells with artificially synthesized calcium crystals (CaP) and urine calcium crystals obtained from CaG-treated mice (to induce hypercalciuria)30 were performed. We were able to identify that crystal aggregation was significantly increased in the presence of cell debris and/or BSA containing IgG. BSA containing IgG had the most considerable effect on crystal aggregation. It is interesting to note that crystal aggregation/growth was higher in the CaP (artificially synthesized crystals) group than all other biogenic crystal conditions (Fig. 3F). Although the reason for this outcome is unclear, this could be because of CaP crystals still undergoing crystallization/growth. Therefore, these crystals have more susceptibility toward co-aggregation in the presence of modulators such as endogenous lipids (cell debris) in contrast to crystal aggregation with biogenic crystals. Moreover, there is a distinct possibility that some form of dilution can occur with the Y-shaped MF setup that could compromise our intent to look at the effect on crystal aggregation. However, we have observed the expected outcome as hypothesized. Notably, in our cellular 3D model, because of the presence of the confluent monolayer with HK2 cells, the access of crystals to the biomaterial coating (fibronectin) on the basolateral surface of the MF channel was highly improbable.
Our approach in the utilization of biogenic crystals was to show proteins as biological modulators of crystal formation. As stated above, the urine crystal matrix (proteins) is crucial in distinguishing stone formers from non-stone formers; hence, by performing protein characterization assays, we could identify proteins by molecular weights that are synonymous with proteins that have been identified in prior studies as proteins markers for stone formation11,50 (Fig. 4B), thereby providing the validity to urine crystals utilized in this study. Numerous ethylene-glycol-treated mice and other knockout mice models exhibited prominent urinary brushite CaP crystal formation.52,53 In addition, THP-null mice, compared to wild type mice, were found to exhibit interstitial deposits of hydroxyapatite that bear a strong resemblance to renal crystals found in human kidneys having idiopathic calcium oxalate stones.52 While it is possible to induce stone formation with ethylene glycol in mice, we did not prefer for this treatment modality because of its known nephrotoxic and deleterious side-effects.54,55 We rather opted for an alternative method to elicit greater crystal formation27,30 that phenotypically has also yielded elevated brushite crystal formation in CaG mice (Fig. S3, ESI†). Interestingly, hypercalciuria was also the defining characteristic of most brushite stone formers, which was also demonstrated with our CaG-treated TRPC3−/− mice model, further supporting the clinical relevance of our study.56
Our previous study provided evidence that CaP crystals can be internalized by PT cells, which turned on SOCE channels leading to ER stress.14 Thus, from a pathophysiological standpoint, while the SOCE pathway appeared to be proportionally induced by the same Tg concentration, CaP-crystal-internalized HK2 cells exhibited greater Ca2+ entry (Fig. 5C) compared with those internalized with biogenic crystals (Fig. 5B and E). Thus, the contribution of synthetic CaP crystals toward cell debris generation seems significantly greater compared with biogenic crystals. This finding indicates that biogenic crystals may provide better translational efficacy in both in vivo or in vitro studies for calcium crystal internalization, compared with synthetic CaP crystals. By purifying those biogenic crystals through the elimination of protein matrix (Fig. 4A and B), we found—via microscopy techniques such as polarized light microscopy—that the biogenic crystals isolated from mice urine were possibly brushite/hydroxyapatite crystals (Fig. S4, ESI†), thereby making their usage in this study more relevant in investigating idiopathic calcium stone formation in humans and from our 3D MF models, as well as the fact that the crystal protein matrix significantly contributed towards crystal augmentation (Fig. 4F). While we were able to show the distinct nature of CaP, CaOx, and mixed crystals through confocal Raman spectroscopy12 and scanning electron microscopy (SEM) with energy-dispersive X-ray detection (EDS),36 our effort in getting a clean signal from biogenic crystals remains unsuccessful. Nonetheless, our biomolecular studies that involved testing the effects of crystal internalization on cellular oxidative stress, inflammation, and apoptosis (Fig. 6A–C) consolidate our findings and provide clarity on the effects of endogenous proteins to contrast the process of crystal growth, adding to the pathological process in real time.
Overall, our study is novel in its approach in examining the contrast between biogenic vs. synthetic crystals in renal stone formation in in vivo-like tubules with a continuous renal fluidic flow. Furthermore, in the present manuscript, we have shown the contribution of proteins in stone formation by clearly defining the contributions of biogenically synthesized stone-forming proteins (stone matrix on urine crystals), IgG–BSA and cell debris in CaP stone formation, and aggregation and growth in a 3D cellular environment. Simultaneously, we explored the extent of their differences in molecular recognition pattern involving biological macromolecules, which can be applicable to other cellular mechanisms.
Author contributions
E. A. B., S. S., and F. Z. performed experiments and analyzed data. wrote and edited manuscript. B. C. B. designed, performed research, interpreted, analyzed data, wrote, and edited manuscript.Institutional review board statement
The animal protocol (#01570) used in this study was designed according to the Guiding Principles in the Care and Use of Animals and has been approved on 11/18/2019 by the Institutional Animal Care and Use Committee (IACUC) and the Research and Development Committee of DC Veterans Affair Medical Center. All animal procedures were performed in accordance with the recommendations for the care and use of laboratory animals as recommended by The National Institutes of Health (NIH), U.S. Department of Agriculture (USDA) and The Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC).Data accessibility
The data that support the findings of this study are available from the corresponding author upon reasonable request.Abbreviations
| 2-APB | 2-Aminoethoxydiphenyl borate |
| BAX1 | BCL2-associated X protein |
| BC | Biogenic crystals |
| BCP | Biogenic crystals with protein |
| BSA | Bovine serum albumin |
| CaP | Calcium phosphate |
| CaNL | Calcium nephrolithiasis |
| EtA | Etidronic acid |
| Fura-2-AM | Fura-2-acetoxymethyl ester |
| IgG | Immunoglobulin G |
| MCP1 | Monocyte chemoattractant protein-1 |
| MF | Microfluidic |
| PT | Proximal tubular |
| ROS | Reactive oxygen species |
| HK2 | Human kidney 2 |
| 2-APB | 2-Aminoethoxydiphenyl borate |
| LDH | Lactate dehydrogenase |
| NOX | NAPDH oxidase |
| IL | Interleukin |
| SOCE | Store-operated Ca2+ entry |
| TNFα | Tumor necrosis factor alpha |
Conflicts of interest
The authors declare no competing financial and/or non-financial interests in relation to the work described in this manuscript.Acknowledgements
We acknowledge the helps of Dr. Zhihong Nie from University of Maryland College Park for MF Device and Dr. Christopher Raub from The Catholic University of America for polarized light microscopy. We also acknowledge the funding support from National Institute of Diabetes and Digestive and Kidney Diseases (DK102043) and National Institute of Biomedical Imaging and Bioengineering (EB021483) to BCB. These funding agencies were not involved in the preparation of the article, study design, collection, analyses, and interpretation of data, writing of the report, or decision to submit this article for publication.References
- C. F. Verkoelen and A. Verhulst, Kidney Int., 2007, 72, 13–18 CrossRef CAS PubMed.
- K. P. Aggarwal, S. Narula, M. Kakkar and C. Tandon, BioMed Res. Int., 2013, 2013, 292953 Search PubMed.
- V. N. Ratkalkar and J. G. Kleinman, Clin. Rev. Bone Miner. Metab., 2011, 9, 187–197 CrossRef CAS PubMed.
- C. F. Verkoelen, J. Am. Soc. Nephrol., 2006, 17, 1673–1687 CrossRef CAS.
- A. Verhulst, M. Asselman, V. P. Persy, M. S. J. Schepers, M. F. Helbert, C. F. Verkoelen and M. E. De Broe, J. Am. Soc. Nephrol., 2003, 14, 107–115 CrossRef CAS PubMed.
- S. R. Khan, J. Urol., 2013, 189, 803–811 CrossRef CAS PubMed.
- H. H. Dorian, P. Rez and G. W. Drach, J. Urol., 1996, 156, 1833–1837 CrossRef CAS PubMed.
- F. Grases, A. Costa-Bauzá and A. Conte, Scanning Microsc., 1993, 7, 1067–1073 CAS.
- S. R. Khan and R. L. Hackett, J. Urol., 1993, 150, 239–245 CrossRef CAS PubMed.
- D. J. Kok and S. R. Khan, Kidney Int., 1994, 46, 847–854 CrossRef CAS PubMed.
- J. A. Wesson, A. M. Kolbach-Mandel, B. R. Hoffmann, C. Davis and N. S. Mandel, Urolithiasis, 2019, 47, 521–532 CrossRef CAS PubMed.
- F. Gombedza, S. Evans, S. Shin, E. Awuah Boadi, Q. Zhang, Z. Nie and B. C. Bandyopadhyay, Sci. Rep., 2019, 9, 875 CrossRef PubMed.
- S. R. Khan, M. S. Pearle, W. G. Robertson, G. Gambaro, B. K. Canales, S. Doizi, O. Traxer and H.-G. Tiselius, Nat. Rev. Dis. Primers, 2016, 2, 16008 CrossRef PubMed.
- F. C. Gombedza, S. Shin, Y. L. Kanaras and B. C. Bandyopadhyay, Cell Death Discovery, 2019, 5, 124 CrossRef PubMed.
- H.-G. Tiselius, Urol. Res., 2011, 39, 231–243 CrossRef PubMed.
- T. Leventouri, Biomaterials, 2006, 27, 3339–3342 CrossRef CAS PubMed.
- Z. Wei, P. K. Amponsah, M. Al-Shatti, Z. Nie and B. C. Bandyopadhyay, Lab Chip, 2012, 12, 4037–4040 RSC.
- J. Wan, W. D. Ristenpart and H. A. Stone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16432–16437 CrossRef CAS PubMed.
- J. Wan, A. M. Forsyth and H. A. Stone, Integr. Biol. Quant. Biosci. Nano Macro, 2011, 3, 972–981 CAS.
- V. Sepe, G. Adamo, A. La Fianza, C. Libetta, M. G. Giuliano, G. Soccio and A. Dal Canton, Am. J. Kidney Dis. Off. J. Natl. Kidney Found., 2006, 48, 706–711 CrossRef CAS.
- D. A. Bushinsky, J. Clin. Invest., 2003, 111, 602–605 CrossRef CAS.
- K. M. K. Sand, M. Bern, J. Nilsen, H. T. Noordzij, I. Sandlie and J. T. Andersen, Front. Immunol., 2014, 5, 682 Search PubMed.
- A. Beganskiene, R. Raudonis, S. Z. Jokhadar, U. Batista and A. Kareiva, J. Phys. Conf. Ser., 2007, 93, 012050 CrossRef.
- A. R. Abate, D. Lee, T. Do, C. Holtze and D. A. Weitz, Lab Chip, 2008, 8, 516–518 RSC.
- R. M. Lafrenie, S. M. Bernier and K. M. Yamada, J. Cell. Physiol., 1998, 175, 163–173 CrossRef CAS PubMed.
- J. H. L. Beal, A. Bubendorfer, T. Kemmitt, I. Hoek and W. Mike Arnold, Biomicrofluidics, 2012, 6, 36503 CrossRef PubMed.
- E. Awuah Boadi, S. Shin, S. Yeroushalmi, B.-E. Choi, P. Li and B. C. Bandyopadhyay, Int. J. Mol. Sci., 2021, 22, 3050 CrossRef PubMed.
- I. Lau, A. Potluri, C.-L. Ibeh, R. S. Redman, E. Paal and B. C. Bandyopadhyay, Arch. Oral Biol., 2017, 82, 99–108 CrossRef CAS PubMed.
- K. Li, S. O. Correa, P. Pham, C. B. Raub and X. Luo, Biofabrication, 2017, 9, 034101 CrossRef CAS PubMed.
- S. Shin, C.-L. Ibeh, E. Awuah Boadi, B.-E. Choi, S. K. Roy and B. C. Bandyopadhyay, Genes Dis., 2021 DOI:10.1016/j.gendis.2021.04.006.
- C.-L. Ibeh, A. J. Yiu, Y. L. Kanaras, E. Paal, L. Birnbaumer, P. A. Jose and B. C. Bandyopadhyay, J. Cell Sci., 2019, 132, jcs225268 CrossRef CAS PubMed.
- X.-Y. Sun, J.-M. Ouyang and M. Xu, CrystEngComm, 2016, 18, 5463–5473 RSC.
- A. J. Yiu, C.-L. Ibeh, S. K. Roy and B. C. Bandyopadhyay, Am. J. Physiol.: Cell Physiol., 2017, 313, C27–C41 CrossRef PubMed.
- B. C. Bandyopadhyay, W. D. Swaim, A. Sarkar, X. Liu and I. S. Ambudkar, J. Biol. Chem., 2012, 287, 30305–30316 CrossRef CAS PubMed.
- M. D. Francis, R. G. Russell and H. Fleisch, Science, 1969, 165, 1264–1266 CrossRef CAS PubMed.
- E. A. Boadi, N. J. Deems, C. B. Raub and B. C. Bandyopadhyay, Cryst. Growth Des., 2019, 19, 6636–6648 CrossRef PubMed.
- E. Canet-Soulas, L. Bessueille, L. Mechtouff and D. Magne, Front. Cell Dev. Biol., 2021, 9, 622736 CrossRef PubMed.
- S. Chaiyarit and V. Thongboonkerd, J. Proteome Res., 2012, 11, 3269–3280 CrossRef CAS PubMed.
- X.-Y. Sun, Q.-Z. Gan and J.-M. Ouyang, Cell Death Discovery, 2015, 1, 15055 CrossRef CAS PubMed.
- C. F. Verkoelen, B. G. van der Boom, D. J. Kok, A. B. Houtsmuller, P. Visser, F. H. Schröder and J. C. Romijn, Kidney Int., 1999, 55, 1426–1433 CrossRef CAS PubMed.
- X.-Y. Sun, Q.-Z. Gan and J.-M. Ouyang, Sci. Rep., 2017, 7, 41949 CrossRef CAS PubMed.
- T. V. Fadeeva and O. A. Golovanova, Russ. J. Inorg. Chem., 2019, 64, 847–856 CrossRef CAS.
- R. L. Patterson, D. B. van Rossum and D. L. Gill, Cell, 1999, 98, 487–499 CrossRef CAS PubMed.
- G. S. Bird, W. I. DeHaven, J. T. Smyth and J. W. Putney, Methods, 2008, 46, 204–212 CrossRef CAS PubMed.
- H. Schleifer, B. Doleschal, M. Lichtenegger, R. Oppenrieder, I. Derler, I. Frischauf, T. N. Glasnov, C. O. Kappe, C. Romanin and K. Groschner, Br. J. Pharmacol., 2012, 167, 1712–1722 CrossRef CAS PubMed.
- S. R. Khan, Transl. Androl. Urol., 2014, 3, 256–276 Search PubMed.
- K. Aihara, K. J. Byer and S. R. Khan, Kidney Int., 2003, 64, 1283–1291 CrossRef CAS PubMed.
- S. R. Khan, P. A. Glenton, R. Backov and D. R. Talham, Kidney Int., 2002, 62, 2062–2072 CrossRef CAS PubMed.
- E. M. Worcester and F. L. Coe, N. Engl. J. Med., 2010, 363, 954–963 CrossRef CAS PubMed.
- A. M. Kolbach-Mandel, N. S. Mandel, B. R. Hoffmann, J. G. Kleinman and J. A. Wesson, Urolithiasis, 2017, 45, 337–346 CrossRef CAS PubMed.
- K. J. Bergsland, J. K. Kelly, B. J. Coe and F. L. Coe, Am. J. Physiol. Renal Physiol., 2006, 291, F530–F536 CrossRef CAS PubMed.
- Y. Liu, L. Mo, D. S. Goldfarb, A. P. Evan, F. Liang, S. R. Khan, J. C. Lieske and X.-R. Wu, Am. J. Physiol. Renal Physiol., 2010, 299, F469–F478 CrossRef PubMed.
- S. R. Khan and P. A. Glenton, J. Urol., 2010, 184, 1189–1196 CrossRef CAS PubMed.
- V. Poldelski, A. Johnson, S. Wright, V. D. Rosa and R. A. Zager, Am. J. Kidney Dis. Off. J. Natl. Kidney Found., 2001, 38, 339–348 CrossRef CAS PubMed.
- R. W. Tyl, L. C. Fisher, M. F. Kubena, M. A. Vrbanic and P. E. Losco, Fundam. Appl. Toxicol. Off. J. Soc. Toxicol., 1995, 27, 155–166 CrossRef CAS.
- A. E. Krambeck, S. E. Handa, A. P. Evan and J. E. Lingeman, J. Urol., 2010, 184, 1367–1371 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb01213d |
| This journal is © The Royal Society of Chemistry 2022 |